Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs

Abstract
:1. Idiopathic Pulmonary Fibrosis
2. IPF Symptoms
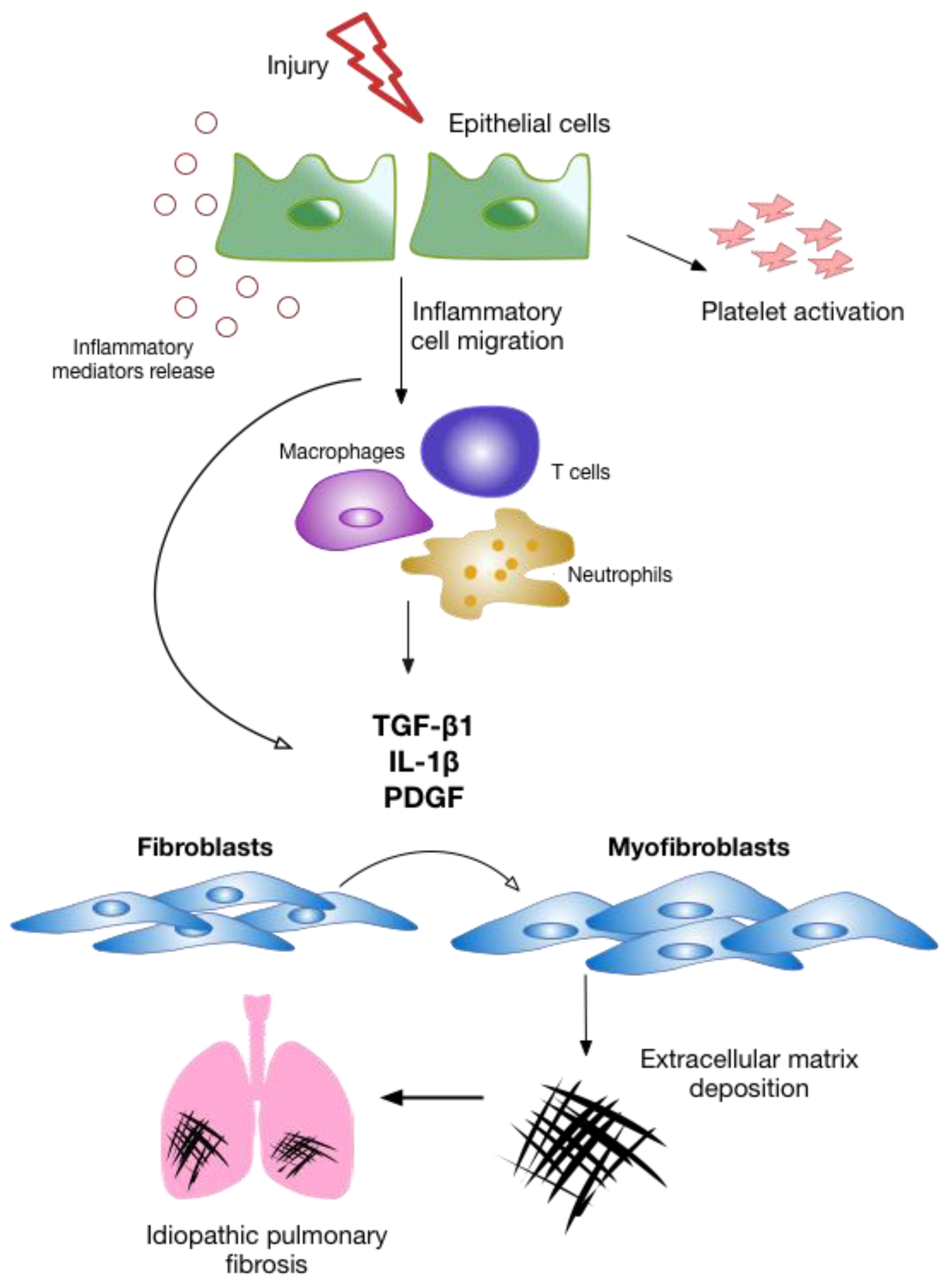
3. The Pathogenesis of IPF
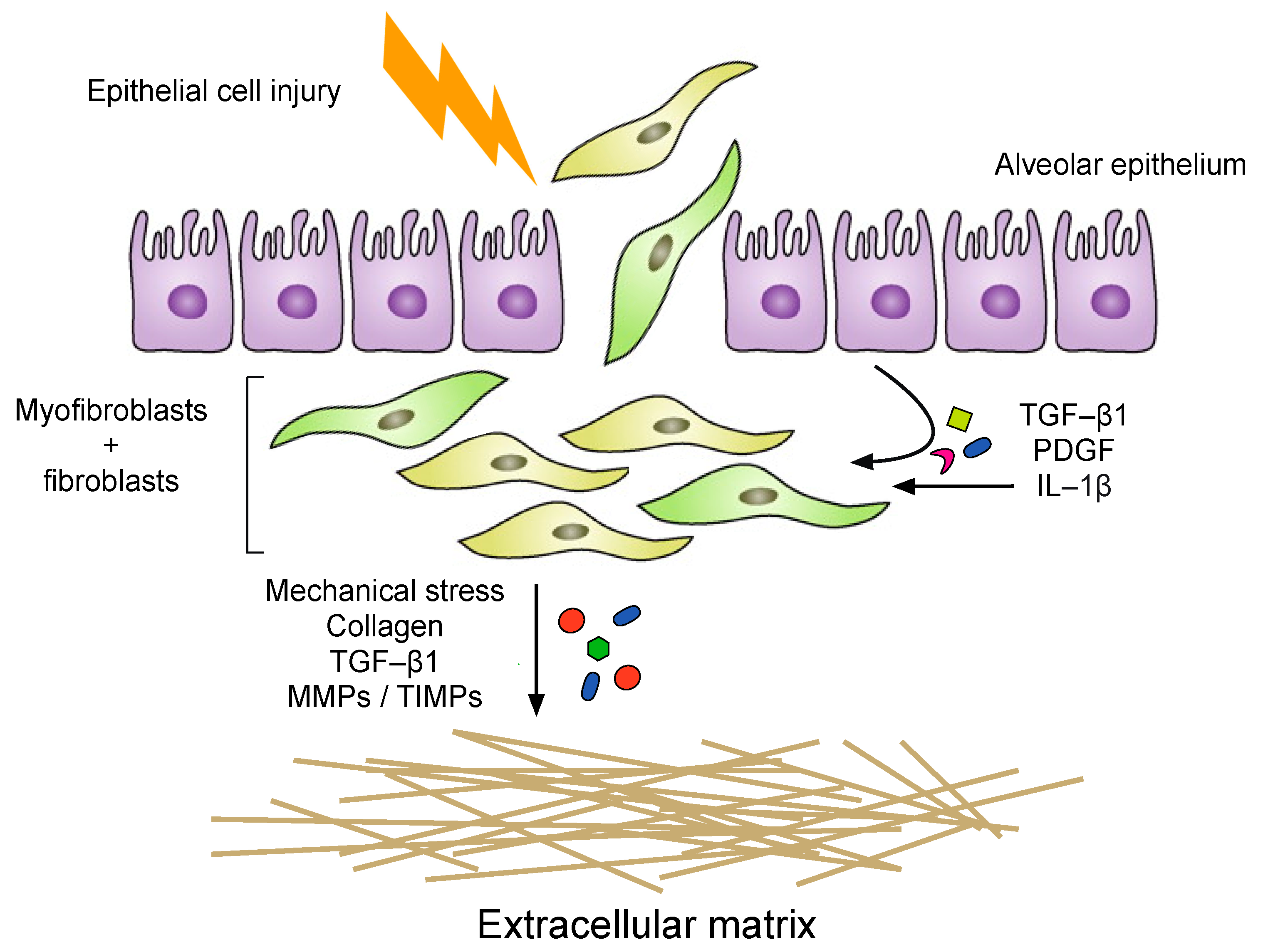
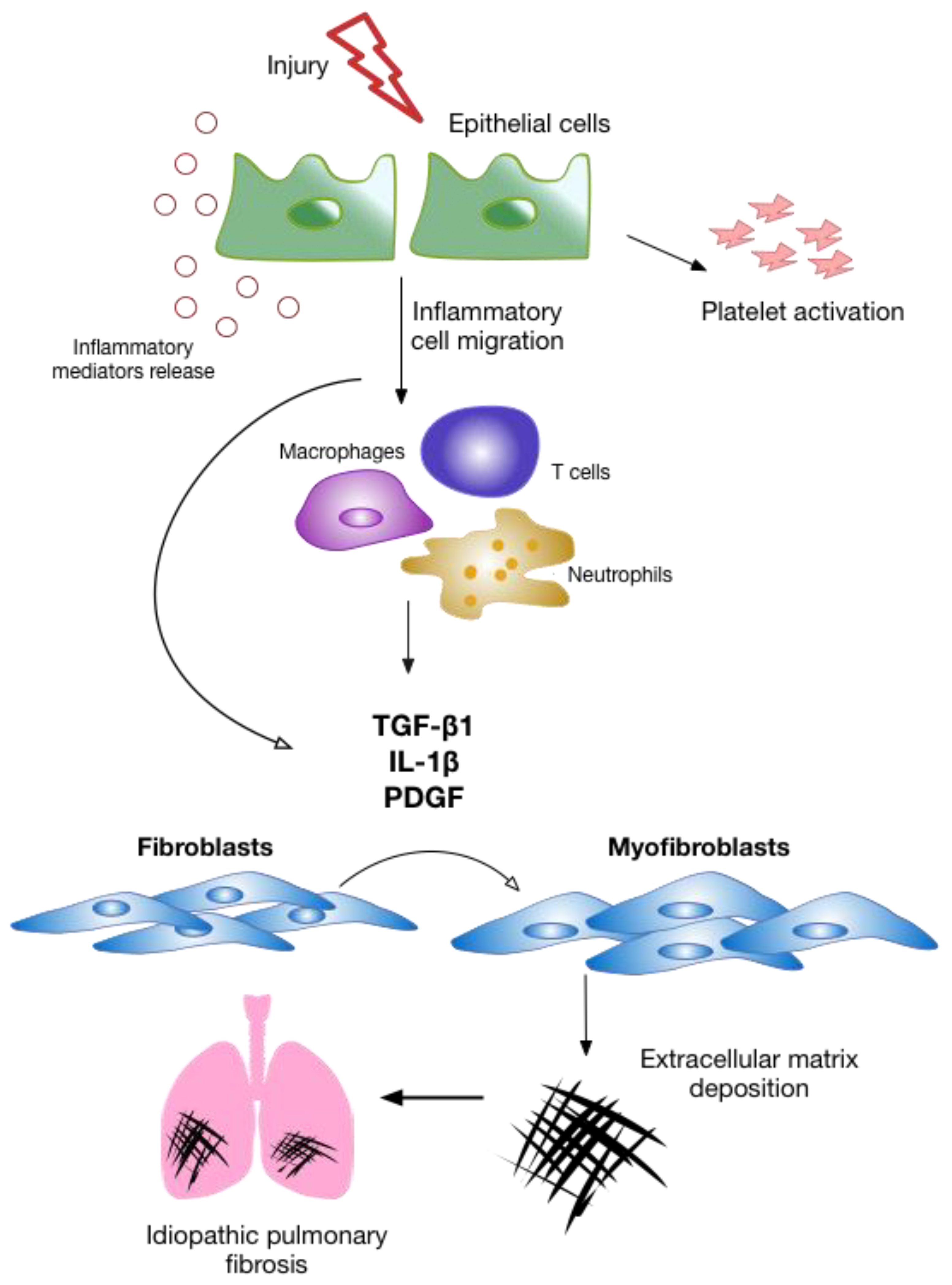
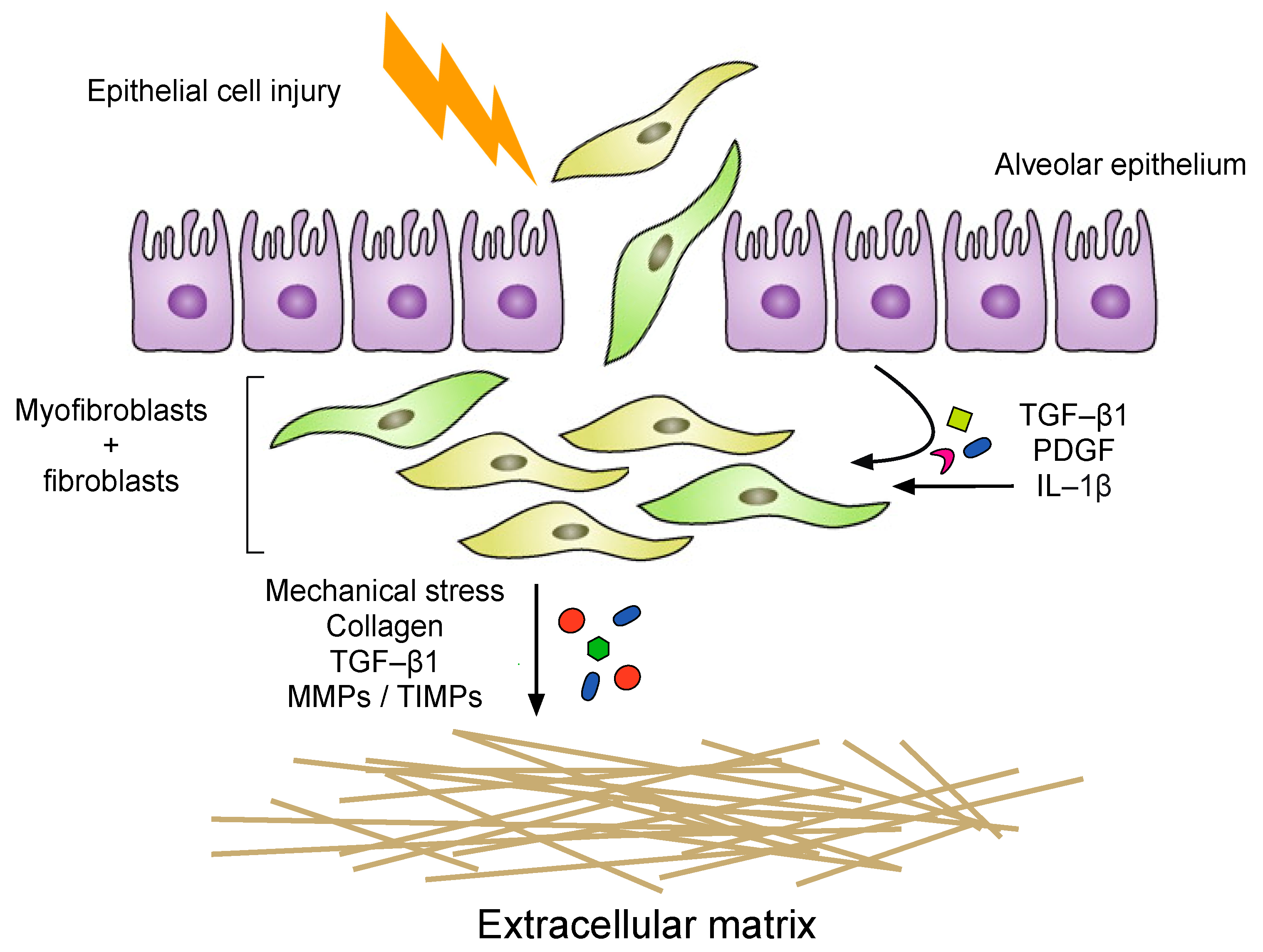
4. Alveolar Epithelial Injury
5. Inflammation
6. The Fibrotic Response and ECM Remodeling Phase
7. Genetic Studies in IPF
8. Gene Expression Studies in IPF
9. Current Pharmacological Strategies for the Treatment of IPF
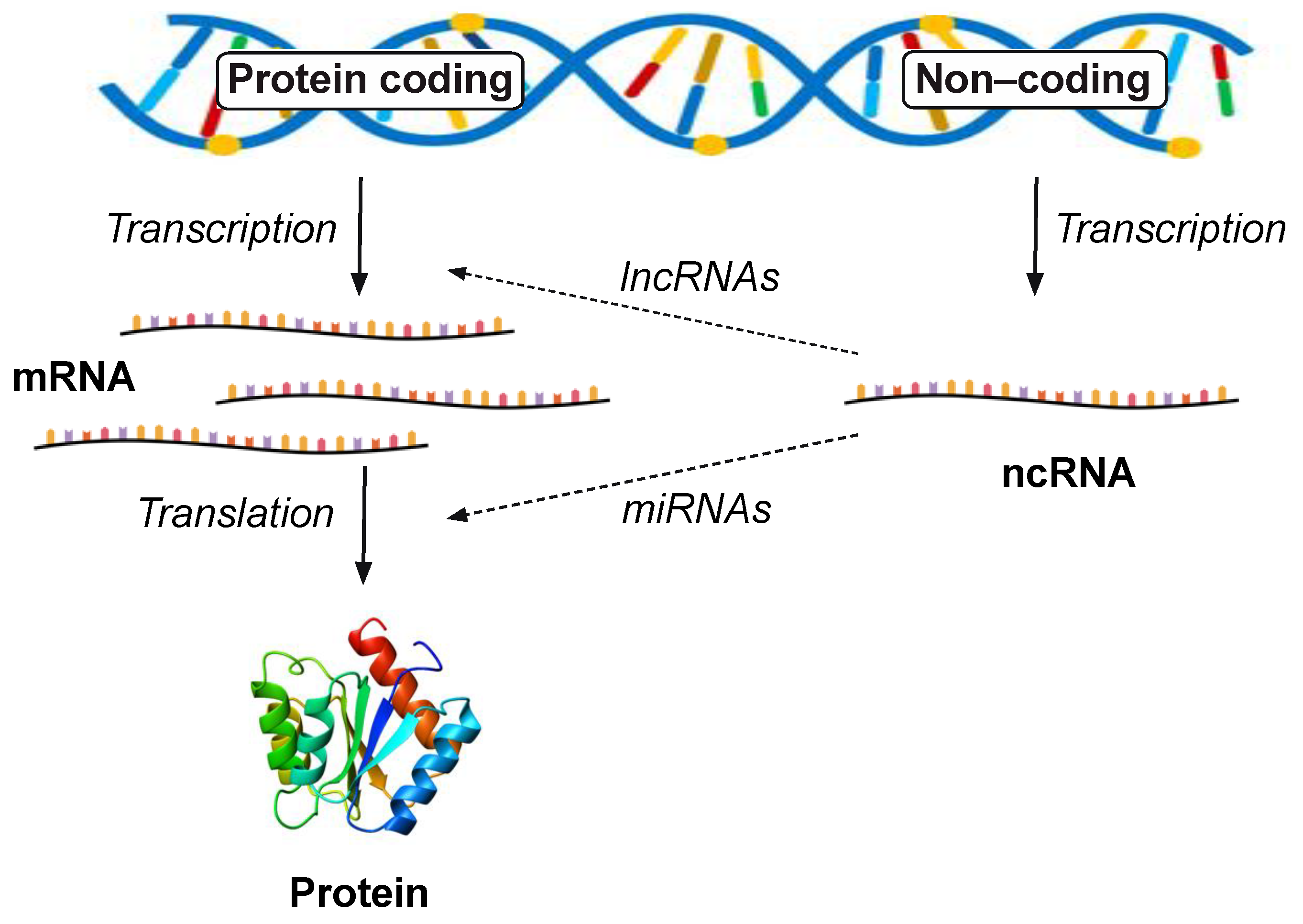
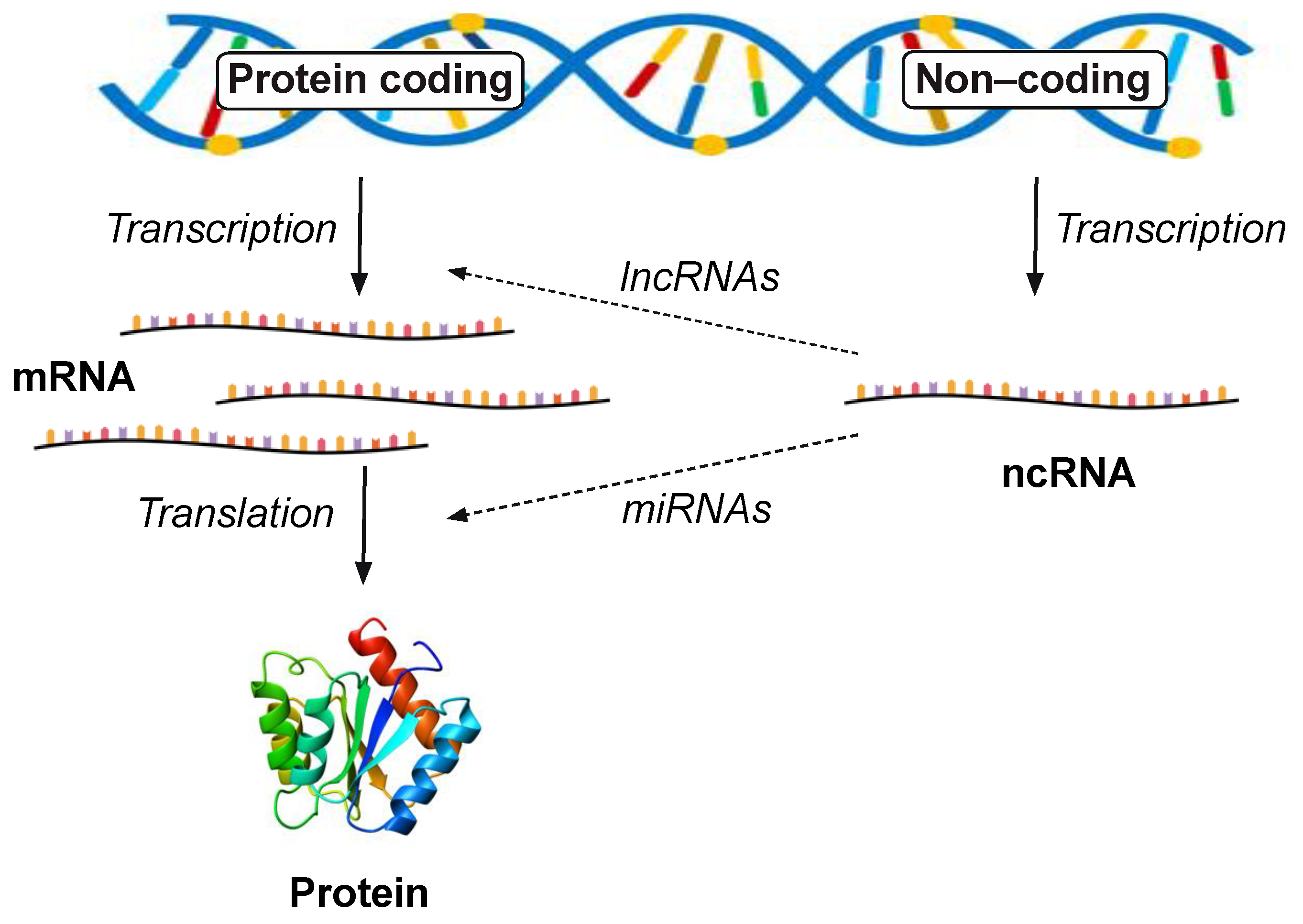
10. Non-Coding RNAs
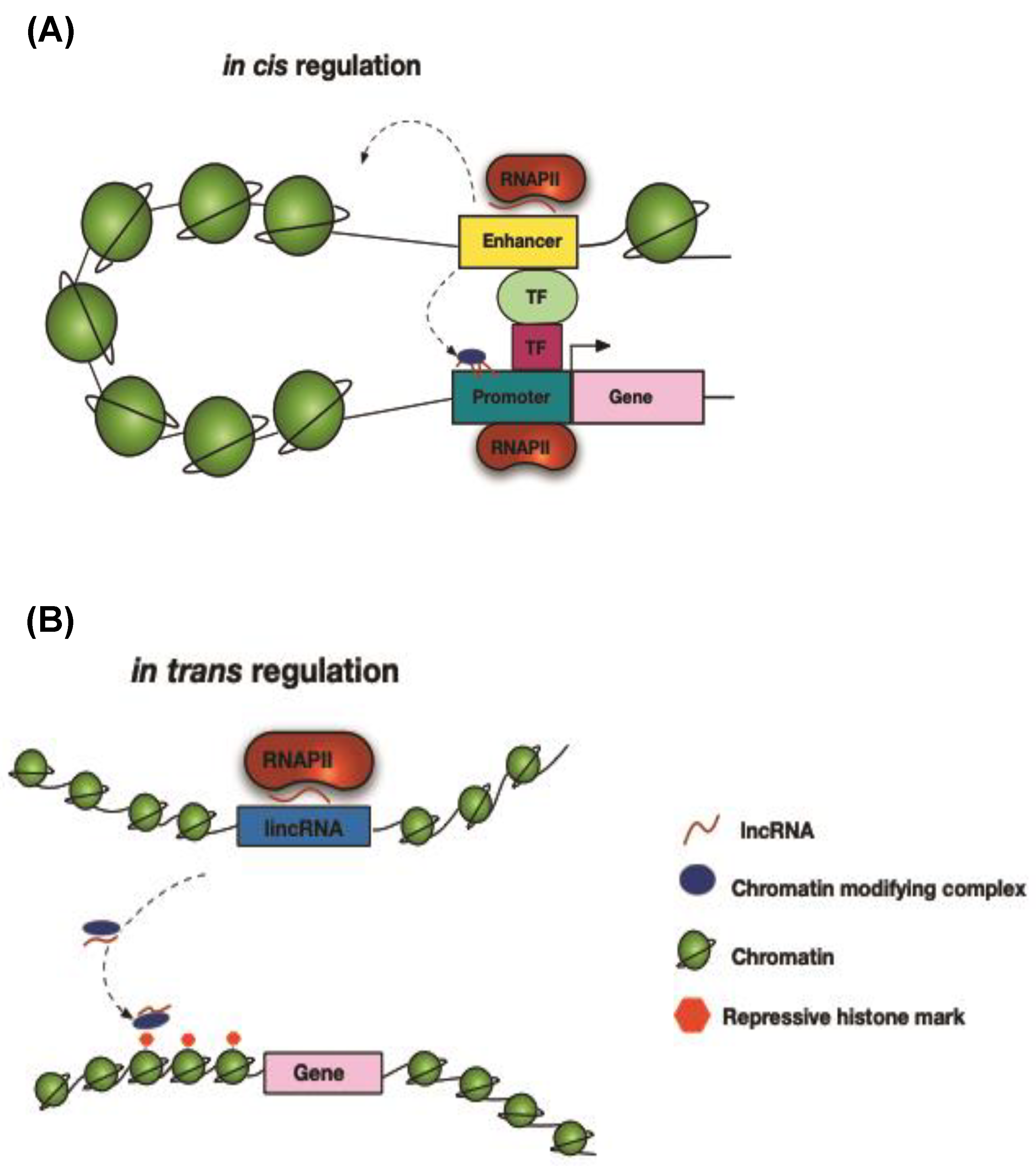
11. Long Non-Coding RNAs
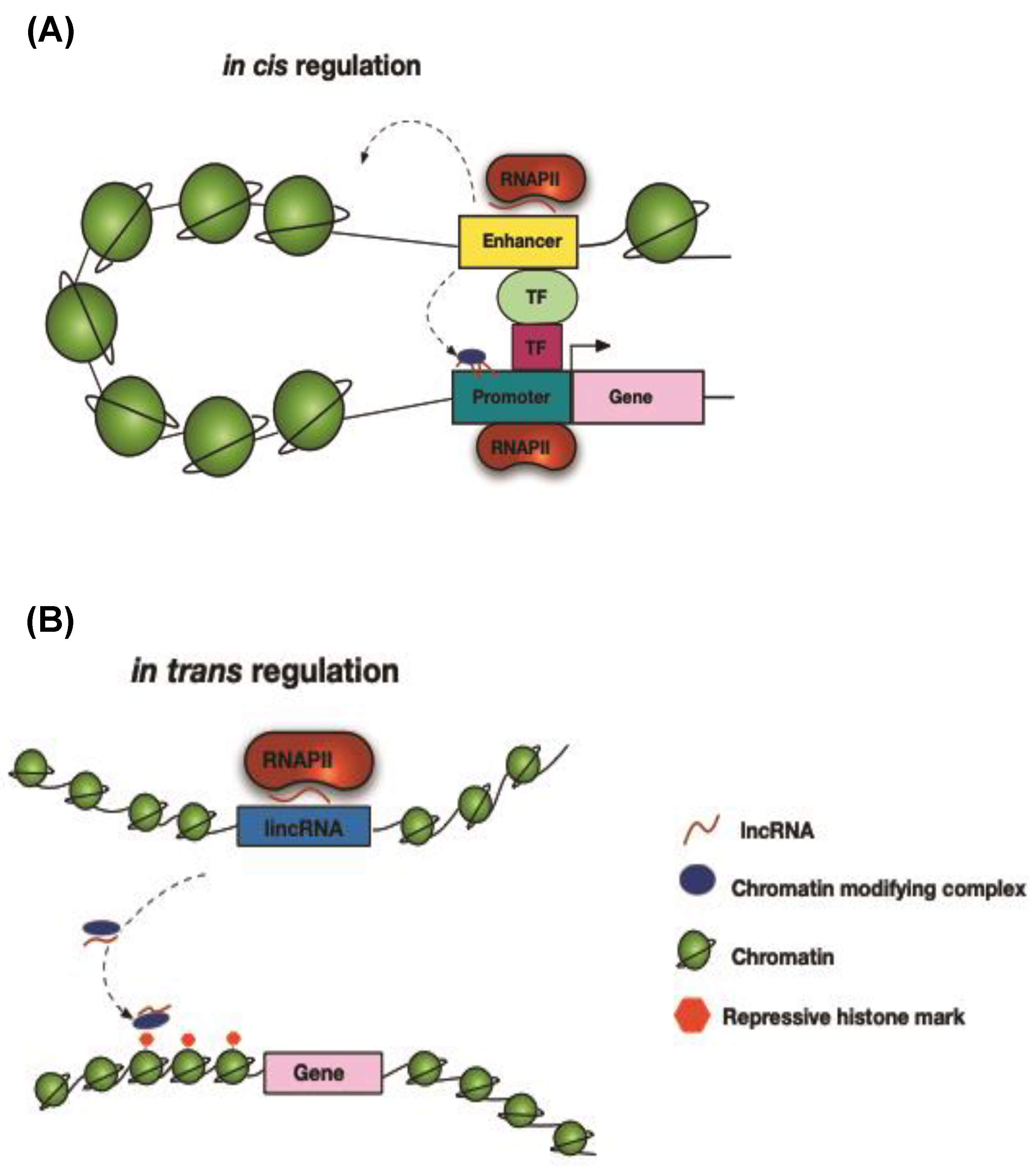
12. Chromatin Modifications
13. Transcriptional Regulation
14. Post-Transcriptional Regulation
15. Long Non-Coding RNAs and the Regulation of Lung Fibrosis
16. Long Non-Coding RNAs AJ005396 and S69206
17. Long Non-Coding RNAs MRAK088388 and MRAK081523
18. Long-Non-Coding RNAs CD99P1 and n341773
19. Long Non-Coding RNAs uc.77 and 2700086A05Rik
20. Long Non-Coding RNA CHRF
21. Long Non-Coding RNA H19
22. Long Non-Coding RNA AP003419.16
23. Lon Non-Coding RNA NONMMUT065582 (PFAR)
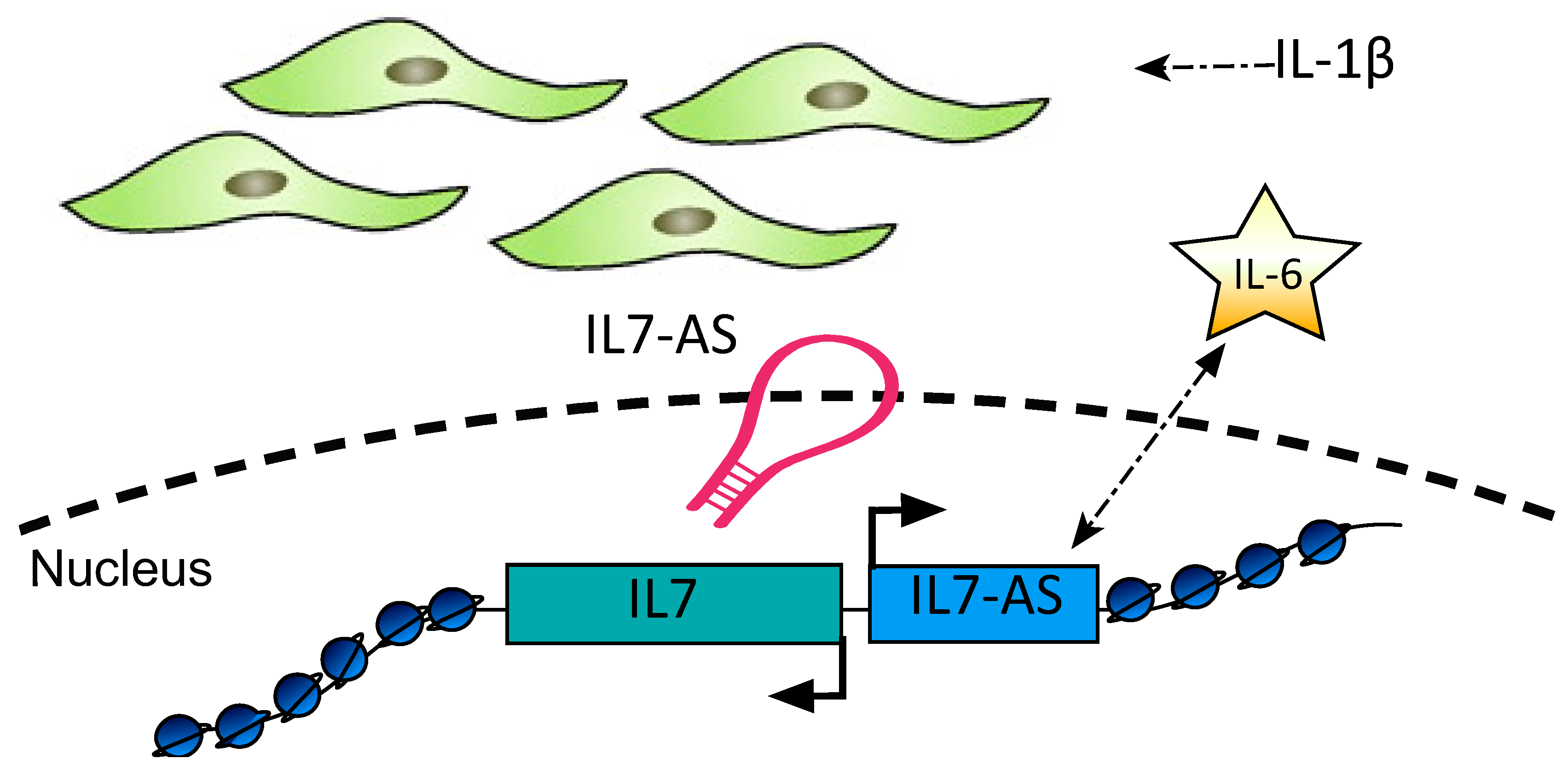
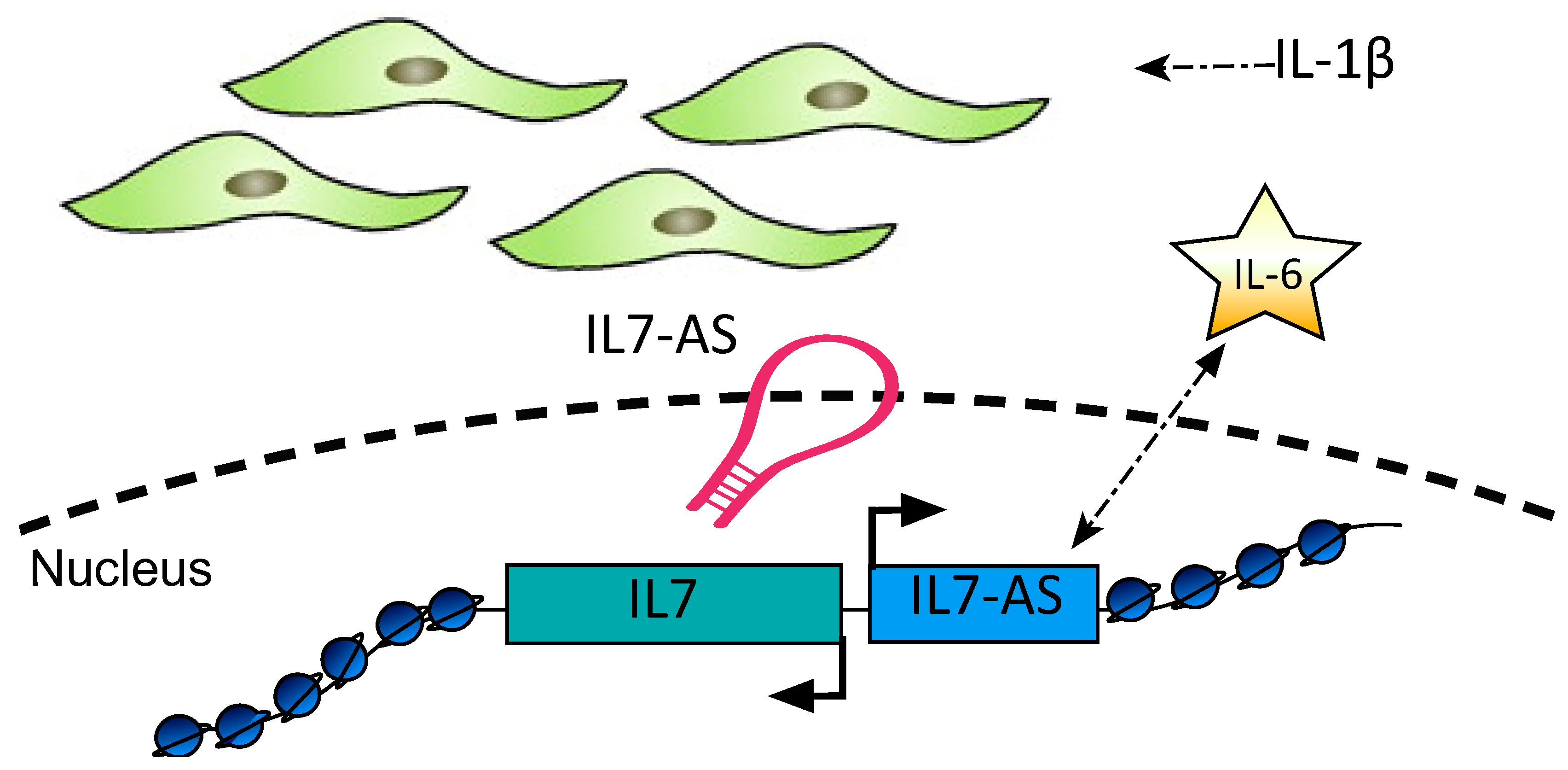
24. Long Non-Coding RNAs IL7AS and MIR3142GHG
25. Long Non-Coding RNAs LINC01140 and LINC0090
26. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- MacNeal, K.; Schwartz, D.A. The Genetic and Environmental Causes of Pulmonary Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 120–125. [Google Scholar] [CrossRef]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogen. Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; Davies, H.R.H.R.; Spagnolo, P.; Luppi, F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst. Rev. 2003, 3. [Google Scholar] [CrossRef]
- Davies, H.R.H.R.; Richeldi, L.; Walters, E.H. Immunomodulatory agents for idiopathic pulmonary fibrosis. Cochrane Database of Syst. Rev. 2003, 158, 003134. [Google Scholar]
- King, T.E., Jr.; Schwarz, M.I.; Brown, K.; Tooze, J.A.; Colby, T.V.; Waldron, J.A.; Flint, A.; Thurlbeck, W.; Cherniack, R.M. Idiopathic pulmonary fibrosis: Relationship between histopathologic features and mortality. Am. J. Respir Crit. Care Med. 2001, 15, 1025–1032. [Google Scholar] [CrossRef]
- Gross, T.J.; Hunninghake, G.W. Medical progress: Idiopathic pulmonary fibrosis. N. Eng. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef]
- Katzenstein, A.L.; Myers, J.L. Idiopathic pulmonary fibrosis: Clinical relevance of pathologic classification. Am. J. Respir. Crit. Care Med. 1998, 157, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Balestro, E.; Calabrese, F.; Turato, G.; Lunardi, F.; Bazzan, E.; Marulli, G.; Biondini, D.; Rossi, E.; Sanduzzi, A.; Rea, F.; et al. Immune Inflammation and Disease Progression in Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0154516. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Tooze, J.A.; Schwarz, M.I.; Brown, K.R.; Cherniack, R.M. Predicting survival in idiopathic pulmonary fibrosis: Scoring system and survival model. Am. J. Respir. Crit. Care Med. 2001, 164, 1171–1181. [Google Scholar] [CrossRef]
- Oldham, J.M.; Noth, I. Idiopathic pulmonary fibrosis: Early detection and referral. Respir. Med. 2014, 108, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Meltzer, E.B.; Noble, P.W. Idiopathic pulmonary fibrosis. Orphanet J. Rare Dis. 2008, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Strieter, R.M. What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, and fibrosis. Proc. Am. Thorac. Soc. 2008, 15, 305–310. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Desmoulière, A. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Boil. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniades, H.N.; Bravo, A.M.; Avila, E.R.; Galanopoulos, T.; Neville-Golden, J.; Maxwell, M.; Selman, M. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J. Clin. Investig. 1990, 86, 1055–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lee, T.C.; Guillemin, B.; Yu, M.C.; Rom, W.N. Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J. Immunol. 1993, 150, 4188–4199. [Google Scholar] [PubMed]
- Sahin, H.; Wasmuth, H.E. Chemokines in tissue fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthwick, L.A.; Wynn, T.A.; Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1832, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primer. 2017, 3, 17074. [Google Scholar] [CrossRef]
- Shannon, J.M.; Hyatt, B.A. Epithelial-Mesenchymal Interactions in the Developing Lung. Annu. Rev. Physiol. 2004, 66, 625–645. [Google Scholar] [CrossRef]
- Chambers, R.C. Abnormal wound healing responses in pulmonary fibrosis: Focus on coagulation signalling. Eur. Respir. Rev. 2008, 17, 130–137. [Google Scholar] [CrossRef]
- Selman, M. Role of Epithelial Cells in Idiopathic Pulmonary FibrosisFrom Innocent Targets to Serial Killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.T.; Spiteri, M.A. Growth factors in idiopathic pulmonary fibrosis: Relative roles. Respir. Res. 2002, 3, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, M.; Barth, K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci. Rep. 2017, 37, BSR20171301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Brown, K.K.; Bradford, W.Z.; Starko, K.; Noble, P.W.; Schwartz, D.A.; King, T.E. A Placebo-Controlled Trial of Interferon Gamma-1b in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2004, 350, 125–133. [Google Scholar] [CrossRef]
- King, E.T.; Albera, C.; Bradford, W.Z.; Costabel, U.; Hormel, P.; Lancaster, L.; Noble, P.W.; Sahn, A.S.; Szwarcberg, J.; Thomeer, M.; et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): A multicentre, randomised, placebo-controlled trial. Lancet 2009, 374, 222–228. [Google Scholar] [CrossRef]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-Dose Acetylcysteine in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242. [Google Scholar] [CrossRef] [Green Version]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, E.T.; Lasky, A.J.; Martinez, F.J. The Idiopathic Pulmonary Fibrosis Clinical Research Network Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez, F.J.; De Andrade, J.A.; Anstrom, K.J.; King, T.E.; Raghu, G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2093–2101. [Google Scholar] [CrossRef] [Green Version]
- Wick, G.; Grundtman, C.; Mayerl, C.; Wimpissinger, T.-F.; Feichtinger, J.; Zelger, B.; Sgonc, R.; Wolfram, D. The Immunology of Fibrosis. Annu. Rev. Immunol. 2013, 31, 107–135. [Google Scholar] [CrossRef] [Green Version]
- Antoniades, H.N.; Neville-Golden, J.; Galanopoulos, T.; Kradin, R.L.; Valente, A.J.; Graves, D.T. Expression of monocyte chemoattractant protein 1 mRNA in human idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1992, 89, 5371–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huaux, F.; Gharaee-Kermani, M.; Liu, T.; Morel, V.; McGarry, B.; Ullenbruch, M.; Kunkel, S.L.; Wang, J.; Xing, Z.; Phan, S.H. Role of Eotaxin-1 (CCL11) and CC Chemokine Receptor 3 (CCR3) in Bleomycin-Induced Lung Injury and Fibrosis. Am. J. Pathol. 2005, 167, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-U.; Cheong, H.S.; Shim, E.-Y.; Bae, D.-J.; Chang, H.S.; Uh, S.-T.; Kim, Y.H.; Park, J.-S.; Lee, B.; Shin, H.D.; et al. Gene profile of fibroblasts identify relation of CCL8 with idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Investig. 2001, 107, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, U.; Whitsett, J.A.; Wert, S.E.; Tichelaar, J.W.; Bry, K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. American Journal of Respiratory Cell and Molecular Biology. Am. Thorac. Soc. 2005, 32, 311–318. [Google Scholar]
- Cantin, A.M.; North, S.L.; Fells, A.G.; Hubbard, R.C.; Crystal, R.G. Oxidant-mediated epithelial cell injury in idiopathic pulmonary fibrosis. J. Clin. Investig. 1987, 79, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Strausz, J.; Müller-Quernheim, J.; Steppling, H.; Ferlinz, R. Oxygen Radical Production by Alveolar Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Am. Rev. Respir. Dis. 1990, 141, 124–128. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.N.; Ju, H.R.; Han, S.H.; Im, J.Y.; Kang, H.S.; Lee, T.H.; Bae, Y.S.; Ha, K.S.; Lee, Z.W.; et al. Requirement of hydrogen peroxide generation in TGF-beta 1 signal transduction in human lung fibroblast cells: Involvement of hydrogen peroxide and Ca2+ in TGF-beta 1-induced IL-6 expression. J. Immunol. 2000, 15, 2190–2197. [Google Scholar] [CrossRef] [Green Version]
- Vayalil, P.K.; Iles, K.E.; Choi, J.; Yi, A.-K.; Postlethwait, E.M.; Liu, R.-M. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L1281–L1292. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinder, B.W.; Brown, K.K.; Schwarz, M.I.; Ix, J.H.; Kervitsky, A.; King, T.E. Baseline BAL Neutrophilia Predicts Early Mortality in Idiopathic Pulmonary Fibrosis. Chest 2008, 133, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Baroni, G.S.; D’Ambrosio, L.; Curto, P.; Casini, A.; Mancini, R.; Jezequel, A.M.; Benedetti, A. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology 1996, 23, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L.; Doody, J.; Massagué, J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 1999, 397, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, M.G.; Donaldson, D.D.; Cheever, A.W.; Wynn, T.A. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2–dominated inflammatory response. J. Clin. Investig. 1999, 104, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, A.T.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [Green Version]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.L.; Carruthers, A.M.; Mustelin, T.; Murray, A.L. Matrix regulation of idiopathic pulmonary fibrosis: The role of enzymes. Fibrogenesis Tissue Repair 2013, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wipff, P.-J.; Rifkin, D.B.; Meister, J.-J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, A.; Selman, M.; Kaminski, N. Approaching the degradome in idiopathic pulmonary fibrosis☆. Int. J. Biochem. Cell Boil. 2008, 40, 1141–1155. [Google Scholar] [CrossRef]
- Kulkarni, T.; O’Reilly, P.; Antony, V.B.; Gaggar, A.; Thannickal, V.J. Matrix Remodeling in Pulmonary Fibrosis and Emphysema. Am. J. Respir. Cell Mol. Boil. 2016, 54, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef]
- Coghlan, A.M.; Shifren, A.; Huang, H.J.; Russell, T.D.; Mitra, R.D.; Zhang, Q.; Wegner, D.J.; Cole, F.S.; Hamvas, A. Sequencing of idiopathic pulmonary fibrosis-related genes reveals independent single gene associations. BMJ Open Respir. Res. 2014, 1, e000057. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldán, J.; Cheng, N.-S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [Green Version]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant Protein A2 Mutations Associated with Pulmonary Fibrosis Lead to Protein Instability and Endoplasmic Reticulum Stress*. J. Boil. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spagnolo, P.; Cottin, V. Genetics of idiopathic pulmonary fibrosis: From mechanistic pathways to personalised medicine. J. Med. Genet. 2017, 54, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Ingersoll, R.G.; Markin, C.; Xie, M.; Vulto, I.; Greider, C.W.; Loyd, J.E.; Alder, J.K.; et al. Telomerase Mutations in Families with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Chen, J.J.-L.; Lancaster, L.; Danoff, S.; Su, S.-C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [Green Version]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Steele, M.P.; Schwartz, D.A. Molecular Mechanisms in Progressive Idiopathic Pulmonary Fibrosis. Annu. Rev. Med. 2013, 64, 265–276. [Google Scholar] [CrossRef]
- Vukmirovic, M.; Kaminski, N. Impact of Transcriptomics on Our Understanding of Pulmonary Fibrosis. Front. Med. 2018, 5, 87. [Google Scholar] [CrossRef]
- Nance, T.; Smith, K.S.; Anaya, V.; Richardson, R.; Ho, L.; Pala, M.; Mostafavi, S.; Battle, A.; Feghali-Bostwick, C.; Rosen, G.; et al. Transcriptome Analysis Reveals Differential Splicing Events in IPF Lung Tissue. PLoS ONE 2014, 9, e92111. [Google Scholar] [CrossRef] [Green Version]
- DePianto, D.J.; Chandriani, S.; Abbas, A.R.; Jia, G.; N’Diaye, E.N.; Caplazi, P.; Kauder, S.E.; Biswas, S.; Karnik, S.K.; Ha, C.; et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 2015, 70, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.S.; Kass, D.; Loh, K.; Glackin, C.; Borczuk, A.C.; Greenberg, S. Gene Expression Profiling of Pulmonary Fibrosis Identifies Twist1 as an Antiapoptotic Molecular “Rectifier” of Growth Factor Signaling. Am. J. Pathol. 2009, 175, 2351–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, K.; Gibson, K.F.; Lindell, K.O.; Richards, T.J.; Zhang, Y.; Dhir, R.; Bisceglia, M.; Gilbert, S.; Yousem, S.A.; Song, J.W.; et al. Gene Expression Profiles of Acute Exacerbations of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Bailey, N.W.; Yang, J.; Steel, M.P.; Groshong, S.; Kervitsky, L.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A. Molecular Phenotypes Distinguish Patients with Relatively Stable from Progressive Idiopathic Pulmonary Fibrosis (IPF). PLoS ONE 2009, 4, e5134. [Google Scholar] [CrossRef] [PubMed]
- Hadjicharalambous, M.R.; Roux, B.T.; Csomor, E.; Feghali-Bostwick, C.A.; Murray, L.A.; Clarke, D.L.; Lindsay, M.A. Long intergenic non-coding RNAs regulate human lung fibroblast function: Implications for idiopathic pulmonary fibrosis. Sci. Rep. 2019, 9, 6020. [Google Scholar] [CrossRef] [Green Version]
- Plantier, L.; Renaud, H.; Respaud, R.; Marchand-Adam, S.; Crestani, B. Transcriptome of Cultured Lung Fibroblasts in Idiopathic Pulmonary Fibrosis: Meta-Analysis of Publically Available Microarray Datasets Reveals Repression of Inflammation and Immunity Pathways. Int. J. Mol. Sci. 2016, 17, 2091. [Google Scholar] [CrossRef] [Green Version]
- Renzoni, E.A.; Abraham, D.J.; Howat, S.; Shi-Wen, X.; Sestini, P.; Bou-Gharios, G.; Wells, A.U.; Veeraraghavan, S.; Nicholson, A.G.; Denton, C.P.; et al. Gene expression profiling reveals novel TGFbeta targets in adult lung fibroblasts. Respir. Res. 2004, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.V.; Luna, L.G.; Cotter, J.; Talbert, J.; Leach, S.M.; Kidd, R.; Turner, J.; Kummer, N.; Kervitsky, L.; Brown, K.K.; et al. The Peripheral Blood Transcriptome Identifies the Presence and Extent of Disease in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e37708. [Google Scholar] [CrossRef] [Green Version]
- Hilberg, F.; Roth, G.J.; Krššák, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, N.I.; Roth, G.J.; Hilberg, F.; Muller-Quernheim, J.; Prasse, A.; Zissel, G.; Schnapp, A.; Park, J.E. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur. Respir. J. 2007, 29, 976–985. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and Anti-inflammatory Activity of the Tyrosine Kinase Inhibitor Nintedanib in Experimental Models of Lung Fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Hostettler, E.K.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spagnolo, P.; Maher, T.; Richeldi, L. Idiopathic pulmonary fibrosis: Recent advances on pharmacological therapy. Pharmacol. Ther. 2015, 152, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 291, 367–373. [Google Scholar]
- Raghu, G.; Johnson, W.C.; Lockhart, D.; Mageto, Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: Results of a prospective, open-label Phase II study. Am. J. Respir. Crit. Care Med. 1999, 159, 1061–1069. [Google Scholar] [CrossRef]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, E.T.; Lancaster, L.; Sahn, A.S.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Hadjicharalambous, M.R.; Lindsay, M.A. Long Non-Coding RNAs and the Innate Immune Response. Non-Coding RNA 2019, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Necsulea, A.; Kaessmann, H. Evolutionary dynamics of coding and non-coding transcriptomes. Nat. Rev. Genet. 2014, 15, 734–748. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H. Long non-coding RNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2016, 18, 558–576. [Google Scholar] [CrossRef] [Green Version]
- Pontier, D.B.; Gribnau, J. Xist regulation and function explored. Qual. Life Res. 2011, 130, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Froberg, J.E.; Yang, L.; Lee, J.T. Guided by RNAs: X-inactivation as a model for lncRNA function. J. Mol. Boil. 2013, 425, 3698–3706. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.T.; Bartolomei, M.S. X-Inactivation, Imprinting, and Long Noncoding RNAs in Health and Disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Kugel, J.F.; Goodrich, J.A. Non-coding RNAs: Key regulators of mammalian transcription. Trends Biochem. Sci. 2012, 37, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjari, M.; Salavaty, A. HOTAIR: An oncogenic long non-coding RNA in different cancers. Cancer Boil. Med. 2015, 12, 1–9. [Google Scholar]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Kanduri, C. Long noncoding RNAs: Lessons from genomic imprinting. Biochim. et Biophys. Acta (BBA)—Bioenergy 2016, 1859, 102–111. [Google Scholar] [CrossRef]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Boil. 2013, 14, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Beltran, M.; Puig, I.; Pena, C.; Garcia, J.M.; Alvarez, A.B.; Pena, R.; Bonilla, F.; De Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genome Res. 2008, 22, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Podbevšek, P.; Fasolo, F.; Bon, C.; Cimatti, L.; Reißer, S.; Carninci, P.; Bussi, G.; Zucchelli, S.; Plavec, J.; Gustincich, S. Structural determinants of the SINE B2 element embedded in the long non-coding RNA activator of translation AS Uchl. Sci. Rep. 2018, 8, 3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.-H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Shi, Z.-M.; Chang, Y.-N.; Hu, Z.-M.; Qi, H.-X.; Hong, W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene 2014, 547, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Shah, A.; Shan, G. Long Non-coding RNAs in the Cytoplasm. Genom. Proteom. Bioinform. 2016, 14, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Pandit, K.V.; Milosevic, J. MicroRNA regulatory networks in idiopathic pulmonary fibrosis. Biochem. Cell Boil. 2015, 93, 129–137. [Google Scholar] [CrossRef]
- Cao, G.; Zhang, J.; Wang, M.; Song, X.; Liu, W.; Mao, C.; Lv, C. Differential expression of long non-coding RNAs in bleomycin-induced lung fibrosis. Int. J. Mol. Med. 2013, 32, 355–364. [Google Scholar] [CrossRef]
- Song, X.; Cao, G.; Jing, L.; Lin, S.; Wang, X.; Zhang, J.; Wang, M.; Liu, W.; Lv, C. Analysing the relationship between lncRNA and protein-coding gene and the role of lncRNA as ceRNA in pulmonary fibrosis. J. Cell. Mol. Med. 2014, 18, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yang, Y.; Liu, L. Interaction of long noncoding RNAs and microRNAs in the pathogenesis of idiopathic pulmonary fibrosis. Physiol. Genom. 2015, 47, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Chen, J.; Qian, W.; Kang, J.; Wang, J.; Jiang, L.; Qiao, L.; Chen, W.; Zhang, J. Integrated long non-coding RNA analyses identify novel regulators of epithelial-mesenchymal transition in the mouse model of pulmonary fibrosis. J. Cell. Mol. Med. 2016, 20, 1234–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Han, L.; Yan, W.; Ji, X.; Han, R.; Yang, J.; Yuan, J.; Ni, C. miR-489 inhibits silica-induced pulmonary fibrosis by targeting MyD88 and Smad3 and is negatively regulated by lncRNA CHRF. Sci. Rep. 2016, 6, 30921. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; He, R.; An, J.; Deng, P.; Huang, L.; Yang, W. The effect of H19-miR-29b interaction on bleomycin-induced mouse model of idiopathic pulmonary fibrosis. Biochem. Biophys. Res. Commun. 2016, 479, 417–423. [Google Scholar] [CrossRef]
- Lu, Q.; Guo, Z.; Xie, W.; Jin, W.; Zhu, D.; Chen, S.; Ren, T. The lncRNA H19 Mediates Pulmonary Fibrosis by Regulating the miR-196a/COL1A1 Axis. Inflammmation 2018, 41, 896–903. [Google Scholar] [CrossRef]
- Hao, X.; Du, Y.; Qian, L.; Li, D.; Liu, X. Upregulation of long noncoding RNA AP003419.16 predicts high risk of aging-associated idiopathic pulmonary fibrosis. Mol. Med. Rep. 2017, 16, 8085–8091. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sun, J.; Chen, Y.; Su, W.; Shan, H.; Li, Y.; Wang, Y.; Zheng, N.; Shan, H.; Liang, H. lncRNA PFAR Promotes Lung Fibroblast Activation and Fibrosis by Targeting miR-138 to Regulate the YAP1-Twist Axis. Mol. Ther. 2018, 26, 2206–2217. [Google Scholar] [CrossRef] [Green Version]
- Hadjicharalambous, M.R.; Roux, B.T.; Feghali-Bostwick, C.A.; Murray, L.A.; Clarke, D.L.; Lindsay, M.A. Long Non-coding RNAs Are Central Regulators of the IL-1β-Induced Inflammatory Response in Normal and Idiopathic Pulmonary Lung Fibroblasts. Front. Immunol. 2018, 9, 2906. [Google Scholar] [CrossRef]
- Arun, G.; Diermeier, S.D.; Spector, D.L. Therapeutic Targeting of Long Non-Coding RNAs in Cancer. Trends Mol. Med. 2018, 24, 257–277. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LncRNA | Stimuli | Function | Research Model | Reference |
|---|---|---|---|---|
| AJ005396, S69206 | Bleomycin | N/A | Sprague-Dawley (SD) rats | [130] |
| MRAK088388, MRAK081523 | Bleomycin | Act as ceRNA for miR-29b-3p and let-7i-5p to regulate N4bp2 and Plxna4 expression | Sprague-Dawley (SD) rats | [131] |
| CD99P1, n341773 | N/A | CD99P1 regulates proliferation and α-SMA expression n341773 regulates collagen expression | Human lung tissue, LL29 human lung fibroblasts | [132] |
| uc.77, 2700086A05Rik | Paraquat | Demonstrate regulation Zeb2 and Hoxa3 gene as well as induce expression of several EMT markers and cell morphology | BALB/c mouse model, A549 human lung epithelial cells and primary bronchial epithelial cells | [133] |
| lncRNA CHRF | Silica PMA TGF-β1 | Negatively regulates miR-489 expression to promote silica-induced fibrosis | C57BL/6 mouse model, mouse macrophages (RAW 264.7) and fibroblasts (NIH3T3), human monocytes (THP-1) and fibroblasts (MRC-5) | [134] |
| LncRNA H19 | Bleomycin | Regulates COL1A1 and ACTA2 expression and interacts with miR-29b | C57BL/6 mouse model, NIH3T3 mouse fibroblast cells | [135] |
| LncRNA H19 | Bleomycin TGF-β1 | Regulates COL1A1 expression by sponging miR-196a | C57BL/6 mouse model, human fibroblast (MRC-5) and kidney (HEK-293T) cell lines | [136] |
| AP003419.16 | TGF-β1 | Elevates the expression of RPS6KB2 | Human venous blood, A549 human lung epithelial cells | [137] |
| PFAR | Bleomycin TGF-β1 | Promotes fibrogenesis by modulating miR-138 expression | C57BL/6 mouse model, primary mouse fibroblasts | [138] |
| Il7AS MIR3142HG | IL-1β | Il7AS regulates Il-6 and MIR3142HG regulates IL-8 and CCL2 mRNA expression and protein release via a NF-κB-dependent pathway | Human primary lung fibroblasts | [139] |
| LINC01140 LINC00960 | IL-1β PDGF-AB | Both lncRNAs regulate proliferation and LINC01140 negatively regulates IL-6 release | IPF lung biopsies, human primary lung fibroblasts | [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadjicharalambous, M.R.; Lindsay, M.A. Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 524. https://doi.org/10.3390/ijms21020524
Hadjicharalambous MR, Lindsay MA. Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs. International Journal of Molecular Sciences. 2020; 21(2):524. https://doi.org/10.3390/ijms21020524
Chicago/Turabian StyleHadjicharalambous, Marina R., and Mark A. Lindsay. 2020. "Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs" International Journal of Molecular Sciences 21, no. 2: 524. https://doi.org/10.3390/ijms21020524
APA StyleHadjicharalambous, M. R., & Lindsay, M. A. (2020). Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs. International Journal of Molecular Sciences, 21(2), 524. https://doi.org/10.3390/ijms21020524