Comprehensive Analysis of GABAA-A1R Developmental Alterations in Rett Syndrome: Setting the Focus for Therapeutic Targets in the Time Frame of the Disease

 , , and
, , and 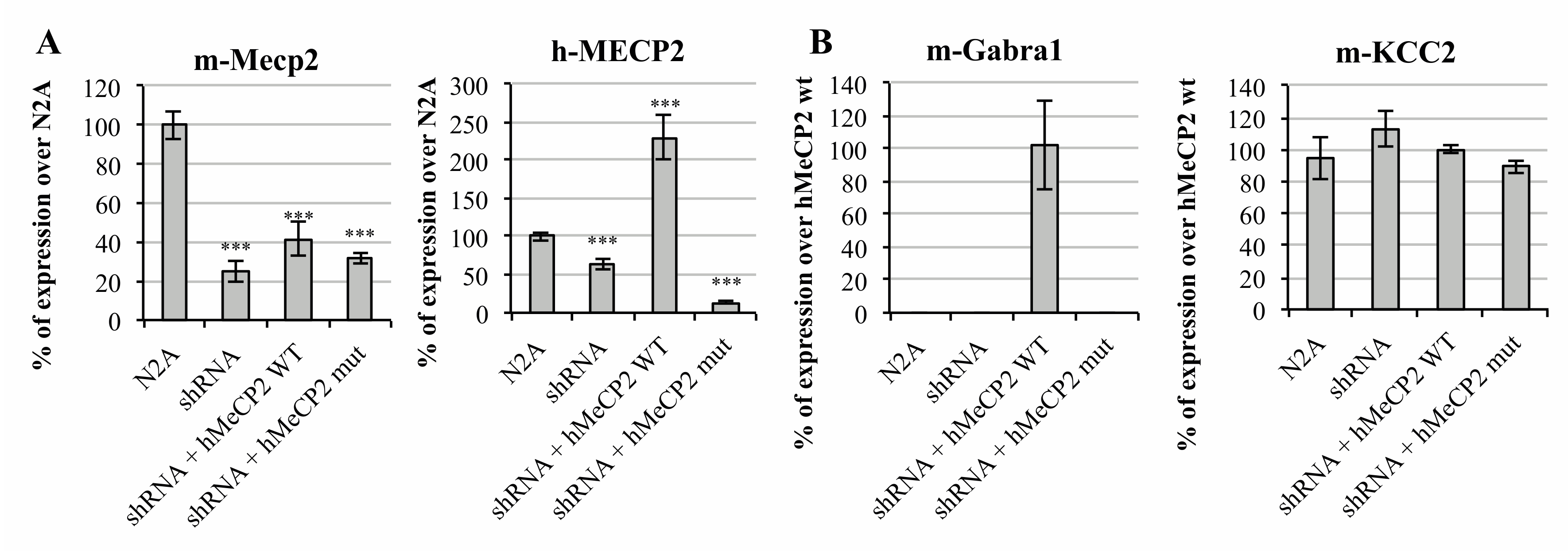{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
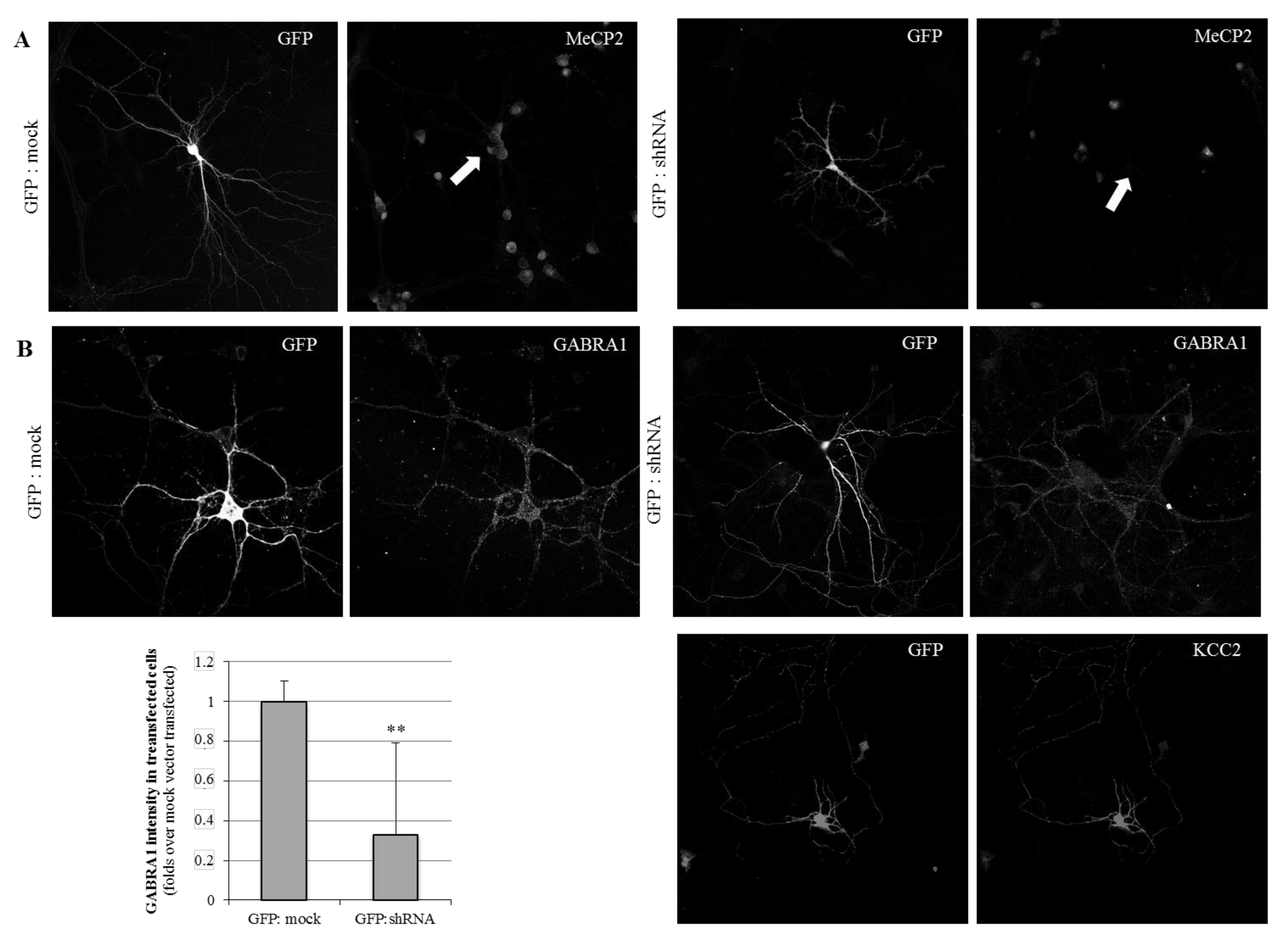
2.1. MeCP2 Levels Are Associated with GABAA Receptors’ Expression in Cellular Models
2.2. Neurodevelopmental Changes of GABAA-A1R and KCC2 in a Rett Syndrome Mouse Model Point Towards the Importance of Pre-Symptomatic Versus Symptomatic Manifestations
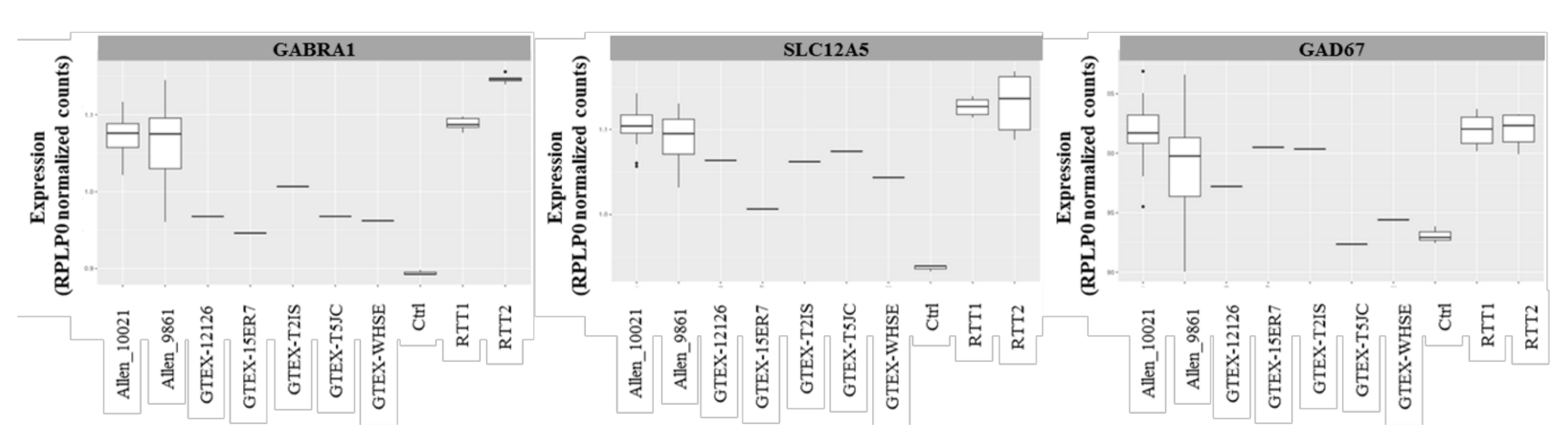
2.3. Transcriptomic Profile of the GABAergic Pathway in Post-Mortem Brain of Rett Syndrome Patients Shows a Generalized GABAergic Pathway Upregulation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Samples Utilization
4.2. Mouse Colony
4.3. Plasmids and Mutagenesis
4.4. RNA Extraction and qRT-PCR
4.5. Western Blotting and ICC
4.6. RNAseq Data Analysis
4.7. Data Availability Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leonard, H.; Cobb, S.; Downs, J. Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol. 2017, 13, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Rastegar, M. Rett syndrome and MeCP2. Neuromol. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, F.; Coort, S.L.; Cirillo, E.; Smeets, E.; Evelo, C.T.; Curfs, L.M. Rett syndrome-biological pathways leading from MECP2 to disorder phenotypes. Orphanet J. Rare Dis. 2016, 11, 158. [Google Scholar] [PubMed]
- Bedogni, F.; Rossi, R.L.; Galli, F.; Gigli, C.C.; Gandaglia, A.; Kilstrup-Nielsen, C.; Landsberger, N. Rett syndrome and the urge of novel approaches to study MeCP2 functions and mechanisms of action. Neurosci. Biobehav. Rev. 2014, 46, 187–201. [Google Scholar] [CrossRef]
- Gonzales, M.L.; LaSalle, J.M. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 2010, 12, 127–134. [Google Scholar] [CrossRef]
- Jung, B.P.; Jugloff, D.G.; Zhang, G.; Logan, R.; Brown, S.; Eubanks, J.H. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 2003, 55, 86–96. [Google Scholar] [CrossRef]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef]
- Chapleau, C.A.; Calfa, G.D.; Lane, M.C.; Albertson, A.J.; Larimore, J.L.; Kudo, S.; Armstrong, D.L.; Percy, A.K.; Pozzo-Miller, L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 2009, 35, 219–233. [Google Scholar] [CrossRef]
- Kang, S.K.; Kim, S.T.; Johnston, M.V.; Kadam, S.D. Temporal- and Location-Specific Alterations of the GABA Recycling System in Mecp2 KO Mouse Brains. J. Cent. Nerv. Syst. Dis. 2014, 6, 21–28. [Google Scholar] [CrossRef]
- Maezawa, I.; Jin, L.W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef]
- Na, E.S.; Nelson, E.D.; Kavalali, E.T.; Monteggia, L.M. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology 2013, 38, 212–219. [Google Scholar] [CrossRef]
- Calfa, G.; Li, W.; Rutherford, J.M.; Pozzo-Miller, L. Excitation/inhibition imbalance and impaired synaptic inhibition in hippocampal area CA3 of Mecp2 knockout mice. Hippocampus 2015, 25, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.T.; Armstrong, J.; Roche, A.; Ortez, C.; Perez, A.; Maria del Mar, O.C.; Pereira, A.; Sanmartí, F.; Ormazábal, A.; Artuch, R.; et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS ONE 2013, 8, e68851. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Moss, S.J. GABA(A) Receptor Dynamics and Constructing GABAergic Synapses. Front. Mol. Neurosci. 2008, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- McKernan, R.M.; Whiting, P.J. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996, 19, 139–143. [Google Scholar] [CrossRef]
- Cicek, S.S. Structure-Dependent Activity of Natural GABA(A) Receptor Modulators. Molecules 2018, 23, 1512. [Google Scholar] [CrossRef]
- Blue, M.E.; Naidu, S.; Johnston, M.V. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp. Neurol. 1999, 156, 345–352. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Di Lucente, J.; Lin, Y.-C.; Lien, C.-C.; Rogawski, M.A.; Maezawa, I.; Jin, L.-W. Defective GABAergic neurotransmission in the nucleus tractus solitarius in Mecp2-null mice, a model of Rett syndrome. Neurobiol. Dis. 2018, 109, 25–32. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Zak, J.D.; Liu, H. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J. Neurophysiol. 2010, 103, 2470–2481. [Google Scholar] [CrossRef][Green Version]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.N.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Ure, K.; Lu, H.; Wang, W.; Ito-Ishida, A.; Wu, Z.; He, L.J.; Sztainberg, Y.; Chen, W.; Tang, J.; Zoghbi, H.Y. Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. Elife 2016, 5, e14198. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.; Guy, J.; Bird, A. Reversibility of functional deficits in experimental models of Rett syndrome. Biochem. So. Trans. 2010, 38, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.M.; Baker, S.A.; Zoghbi, H.Y. MECP2 disorders: From the clinic to mice and back. J. Clin. Investig. 2015, 125, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar]
- El-Khoury, R.; Panayotis, N.; Matagne, V.; Ghata, A.; Villard, L.; Roux, J.-C. GABA and glutamate pathways are spatially and developmentally affected in the brain of Mecp2-deficient mice. PLoS ONE 2014, 9, e92169. [Google Scholar] [CrossRef]
- Zhang, W.; Peterson, M.; Beyer, B.; Frankel, W.N.; Zhang, Z.-W. Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J. Neurosci. 2014, 34, 2754–2763. [Google Scholar] [CrossRef]
- Meng, X.; Wang, W.; Lu, H.; He, L.J.; Chen, W.; Chao, E.S.; Fiorotto, M.L.; Tang, B.; Herrera, J.A.; Seymour, M.L.; et al. Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife 2016, 5, e14199. [Google Scholar] [CrossRef]
- Côme, E.; Heubl, M.; Schwartz, E.J.; Poncer, J.C.; Lévi, S. Reciprocal Regulation of KCC2 Trafficking and Synaptic Activity. Front. Cell Neurosci. 2019, 13, 48. [Google Scholar] [CrossRef]
- Heubl, M.; Zhang, J.; Pressey, J.C.; Al Awabdh, S.; Renner, M.; Gomez-Castro, F.; Moutkine, I.; Eugène, E.; Russeau, M.; Kahle, K.T.; et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl(-)-sensitive WNK1 kinase. Nat. Commun. 2017, 8, 1776. [Google Scholar] [CrossRef]
- Simon, J.; Wakimoto, H.; Fujita, N.; Lalande, M.; Barnard, E.A. Analysis of the set of GABA(A) receptor genes in the human genome. J. Biol. Chem. 2004, 279, 41422–41435. [Google Scholar] [CrossRef] [PubMed]
- Enoch, M.-A.; Baghal, B.; Yuan, Q.; Goldman, D. A factor analysis of global GABAergic gene expression in human brain identifies specificity in response to chronic alcohol and cocaine exposure. PLoS ONE 2013, 8, e64014. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, A.; Shen, K.; Gottlieb, A.; Limon, A. Human brain transcriptome analysis finds region- and subject-specific expression signatures of GABAAR subunits. Commun. Biol. 2019, 2, 153. [Google Scholar] [CrossRef] [PubMed]
- Ricceri, L.; de Filippis, B.; Laviola, G. Mouse models of Rett syndrome: From behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav. Pharmacol. 2008, 19, 501–517. [Google Scholar] [CrossRef]
- Moore, Y.E.; Conway, L.C.; Wobst, H.J.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Developmental Regulation of KCC2 Phosphorylation Has Long-Term Impacts on Cognitive Function. Front. Mol. Neurosci. 2019, 12, 173. [Google Scholar] [CrossRef]
- Tang, X.; Drotar, J.; Li, K.; Clairmont, C.D.; Brumm, A.S.; Sullins, A.J.; Wu, H.; Liu, X.S.; Wang, J.; Gray, N.S.; et al. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 2019, 11, eaau0164. [Google Scholar] [CrossRef]
- Lozovaya, N.; Nardou, R.; Tyzio, R.; Chiesa, M.; Pons-Bennaceur, A.; Eftekhari, S.; Bui, T.-T.; Billon-Grand, M.; Rasero, J.; Bonifazi, P.; et al. Early alterations in a mouse model of Rett syndrome: The GABA developmental shift is abolished at birth. Sci. Rep. 2019, 9, 9276. [Google Scholar] [CrossRef]
- Renthal, W.; Boxer, L.D.; Hrvatin, S.; Li, E.; Silberfeld, A.; Nagy, M.A.; Griffith, E.C.; Vierbuchen, T.; Greenberg, M.E. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci. 2018, 21, 1670–1679. [Google Scholar] [CrossRef]
- Cosentino, L.; Vigli, D.; Franchi, F.; Laviola, G.; De Filippis, B. Rett syndrome before regression: A time window of overlooked opportunities for diagnosis and intervention. Neurosci. Biobehav. Rev. 2019, 107, 115–135. [Google Scholar] [CrossRef]
- Cortelazzo, A.; De Felice, C.; Guy, J.; Timperio, A.M.; Zolla, L.; Guerranti, R.; Leoncini, S.; Signorini, C.; Durand, T.; Hayek, J. Brain protein changes in Mecp2 mouse mutant models: Effects on disease progression of Mecp2 brain specific gene reactivation. J. Proteom. 2020, 210, 103537. [Google Scholar] [CrossRef]
- Cortelazzo, A.; De Felice, C.; De Filippis, B.; Ricceri, L.; Laviola, G.; Leoncini, S.; Signorini, C.; Pescaglini, M.; Guerranti, R.; Timperio, A.M.; et al. Persistent Unresolved Inflammation in the Mecp2-308 Female Mutated Mouse Model of Rett Syndrome. Mediat. Inflamm. 2017, 2017, 9467819. [Google Scholar] [CrossRef] [PubMed]
- Cortelazzo, A.; De Felice, C.; Guerranti, R.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Zollo, G.; Landi, C.; Valacchi, G.; Ciccoli, L.; et al. Subclinical inflammatory status in Rett syndrome. Mediat. Inflamm. 2014, 2014, 480980. [Google Scholar] [CrossRef] [PubMed]
- Soto, D.; Olivella, M.; Grau, C.; Armstrong, J.; Alcon, C.; Gasull, X.; Gómez de Salazar, M.; Gratacòs-Batlle, E.; Ramos-Vicente, D.; Fernández-Dueñas, V.; et al. Rett-like Severe Encephalopathy Caused by a De Novo GRIN2B Mutation Is Attenuated by D-serine Dietary Supplement. Biol. Psychiatry 2018, 83, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, T.; Villard, L.; De Roux, N.; Bourdon, V.; Fontés, M.; Beldjord, C.; Tardieu, M.; Jonveaux, P.; Chelly, J. Spectrum of MECP2 mutations in Rett syndrome. Genet Test 2002, 6, 1–6. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oyarzabal, A.; Xiol, C.; Castells, A.A.; Grau, C.; O’Callaghan, M.; Fernández, G.; Alcántara, S.; Pineda, M.; Armstrong, J.; Altafaj, X.; et al. Comprehensive Analysis of GABAA-A1R Developmental Alterations in Rett Syndrome: Setting the Focus for Therapeutic Targets in the Time Frame of the Disease. Int. J. Mol. Sci. 2020, 21, 518. https://doi.org/10.3390/ijms21020518
Oyarzabal A, Xiol C, Castells AA, Grau C, O’Callaghan M, Fernández G, Alcántara S, Pineda M, Armstrong J, Altafaj X, et al. Comprehensive Analysis of GABAA-A1R Developmental Alterations in Rett Syndrome: Setting the Focus for Therapeutic Targets in the Time Frame of the Disease. International Journal of Molecular Sciences. 2020; 21(2):518. https://doi.org/10.3390/ijms21020518
Chicago/Turabian StyleOyarzabal, Alfonso, Clara Xiol, Alba Aina Castells, Cristina Grau, Mar O’Callaghan, Guerau Fernández, Soledad Alcántara, Mercè Pineda, Judith Armstrong, Xavier Altafaj, and et al. 2020. "Comprehensive Analysis of GABAA-A1R Developmental Alterations in Rett Syndrome: Setting the Focus for Therapeutic Targets in the Time Frame of the Disease" International Journal of Molecular Sciences 21, no. 2: 518. https://doi.org/10.3390/ijms21020518
APA StyleOyarzabal, A., Xiol, C., Castells, A. A., Grau, C., O’Callaghan, M., Fernández, G., Alcántara, S., Pineda, M., Armstrong, J., Altafaj, X., & García-Cazorla, A. (2020). Comprehensive Analysis of GABAA-A1R Developmental Alterations in Rett Syndrome: Setting the Focus for Therapeutic Targets in the Time Frame of the Disease. International Journal of Molecular Sciences, 21(2), 518. https://doi.org/10.3390/ijms21020518