Impact of HLA-DR Antigen Binding Cleft Rigidity on T Cell Recognition

 , ,
, ,  and
and 
Abstract

1. Introduction
2. Results
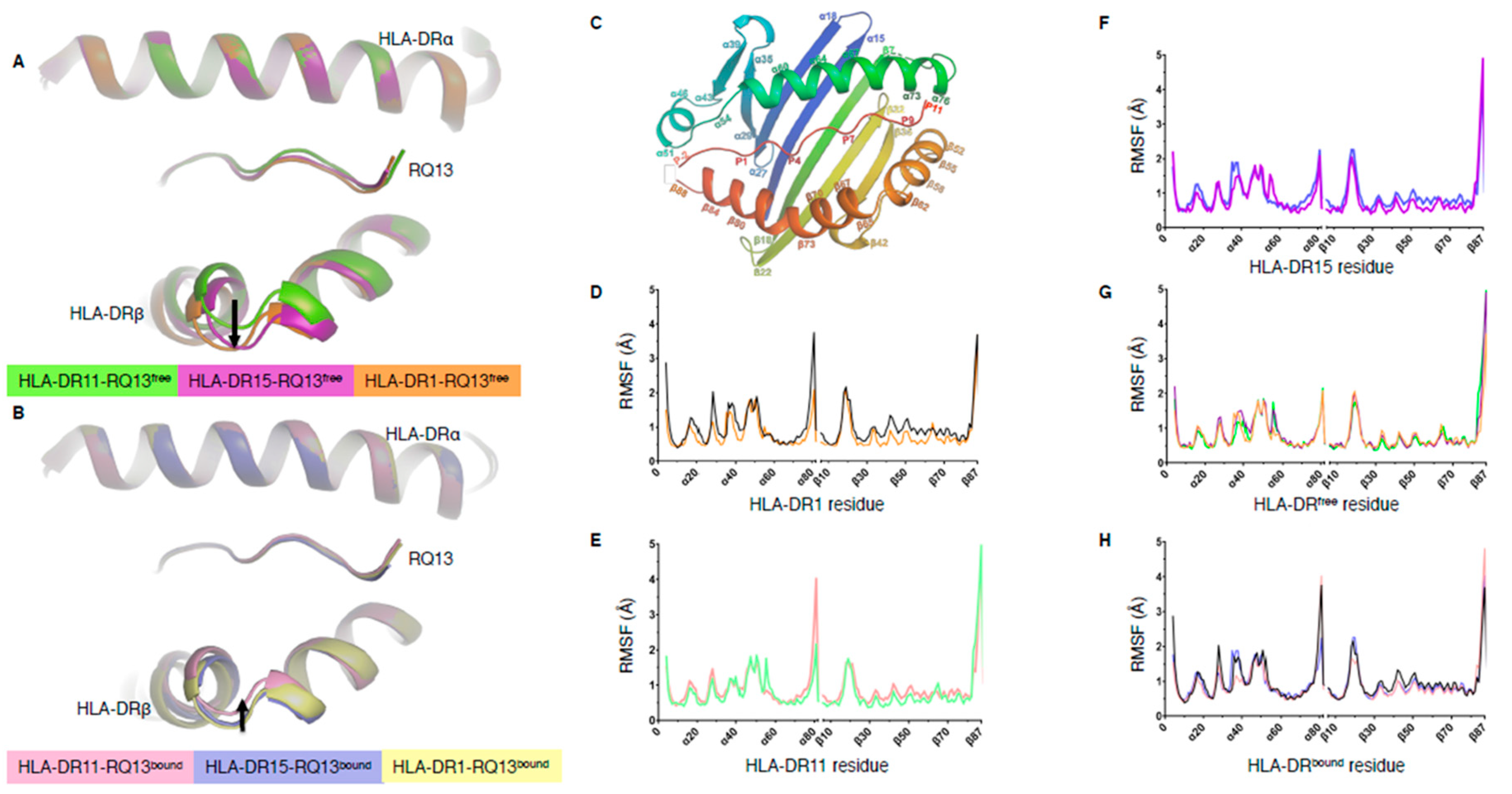
2.1. Peptide Binding Cleft Rigidity of HLA-DR-RQ13 Complexes Revealed by Molecular Dynamics
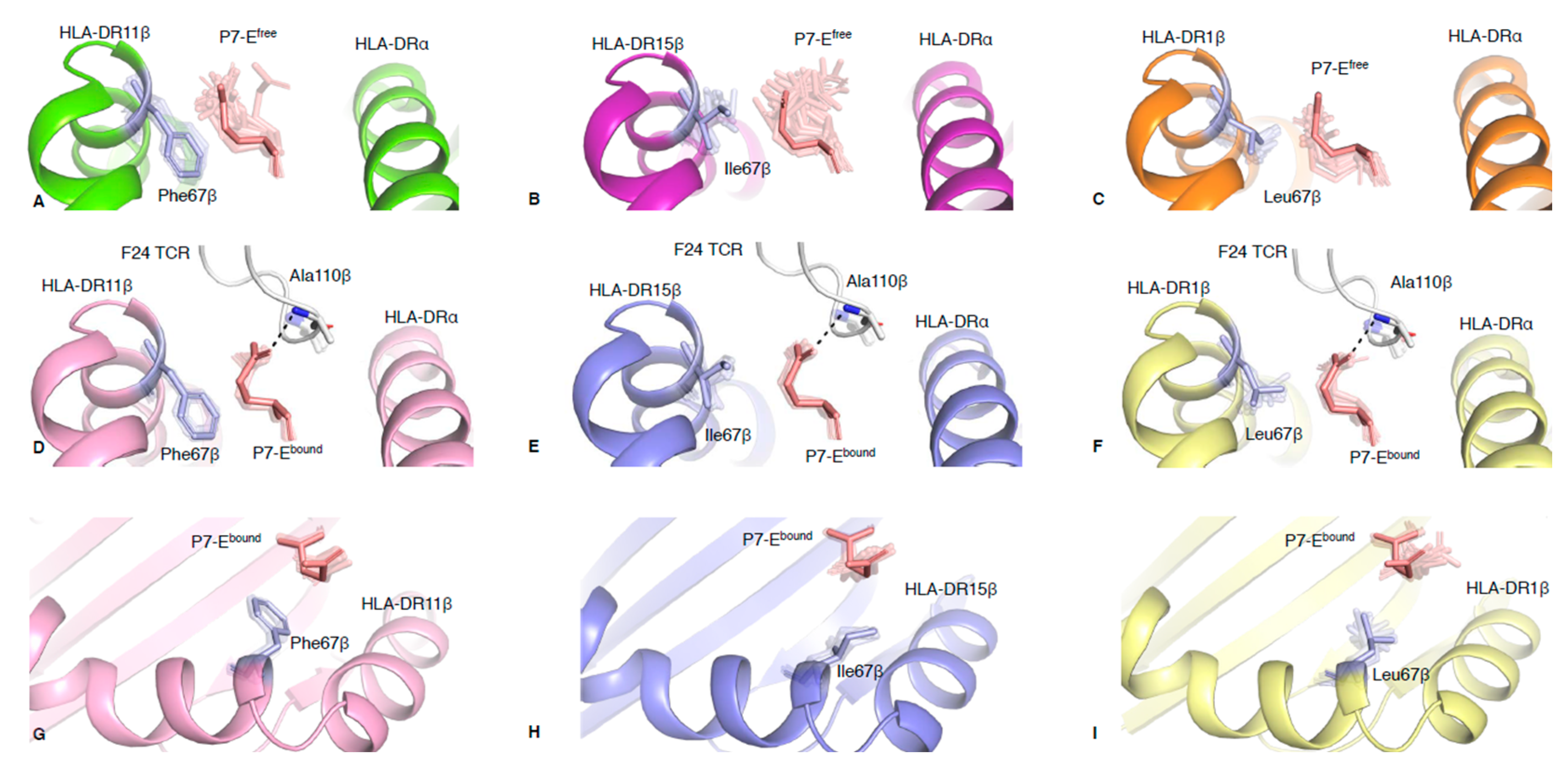
2.2. Polymorphic Residue HLA-DRβ67 Dictates Peptide Binding Cleft Opening
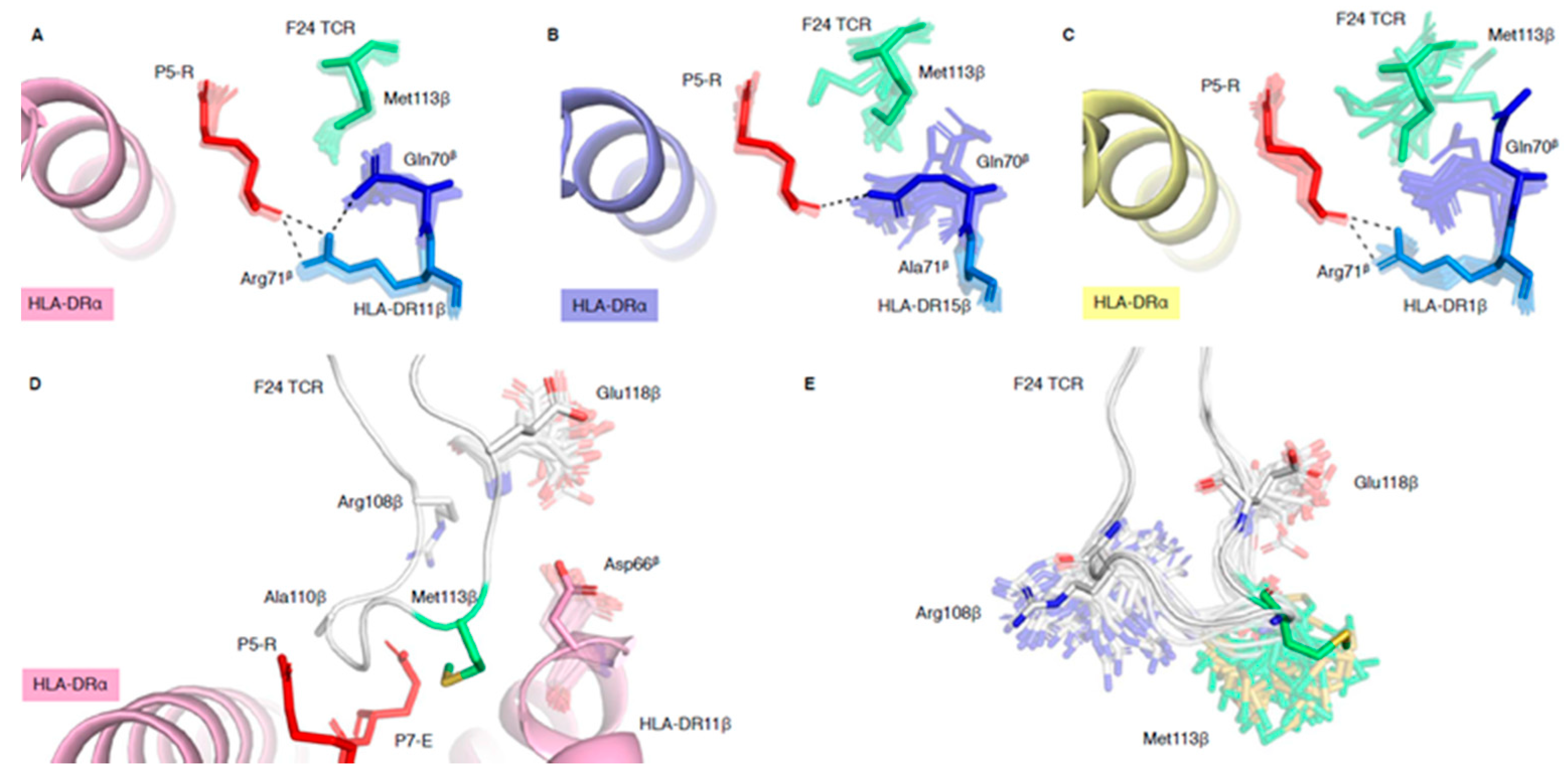
2.3. HLA-DR Polymorphisms Destabilise an Intricate TCR Peg-Notch Interaction
2.4. Structural Rigidity Is a Shared Feature of pHLA-DR Complexes
2.5. HLA-DR1 Displays High Stability and Low HLA-DM Susceptibility Despite Its Open Cleft Conformation
3. Discussion
4. Methods
4.1. HLA-DR-CLIP Expression, Purification, and Peptide Loading
4.2. F24 TCR Production and Surface Plasmon Resonance
4.3. Thermal Stability Assay
4.4. Fluorescence Polarization Assay
4.5. Statistical Analysis for Fluorescence Polarization Assay
4.6. Computational Resources
4.7. Ensemble Refinement
4.8. Atomic Coordinates, Modelling, and Graphics
4.9. MD Systems Setup and Simulation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, N.; Hartig, H.; Dzhagalov, I.; Draper, D.; He, Y.W. The role of apoptosis in the development and function of T lymphocytes. Cell Res. 2005, 15, 749. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, P.A.; Dushek, O. Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 2011, 11, 47. [Google Scholar] [CrossRef]
- Bodmer, W.F.; Albert, E.; Bodmer, J.G.; Dausset, J.; Kissmeyer-Nielsen, F.; Mayr, W.; Payne, R.; Van Rood, J.J.; Trnka, Z.; Walford, R.L. Nomenclature for factors of the HLA system 1984. Immunogenetics 1984, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Stern, L.J.; Brown, J.H.; Jardetzky, T.S.; Gorga, J.C.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 1994, 368, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef]
- Pos, W.; Sethi, D.K.; Call, M.J.; Schulze, M.S.; Anders, A.K.; Pyrdol, J.; Kai, W. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell 2012, 151, 1557–1568. [Google Scholar] [CrossRef]
- Dornmair, K.; McConnell, H.M. Refolding and reassembly of separate alpha and beta chains of class II molecules of the major histocompatibility complex leads to increased peptide-binding capacity. Proc. Natl. Acad. Sci. USA 1990, 87, 4134–4138. [Google Scholar] [CrossRef] [PubMed]
- Sadegh-Nasseri, S.; Germain, R.N. A role for peptide in determining MHC class II structure. Nature 1991, 353, 167–170. [Google Scholar] [CrossRef]
- Sadegh-Nasseri, S.; Germain, R.N. How MHC class II molecules work: Peptide-dependent completion of protein folding. Immunol. Today 1992, 13, 43–46. [Google Scholar] [CrossRef]
- Reich, Z.; Altman, J.D.; Boniface, J.J.; Lyons, D.S.; Kozono, H.; Ogg, G.; Morgan, C.; Davis, M.M. Stability of empty and peptide-loaded class II major histocompatibility complex molecules at neutral and endosomal pH: Comparison to class I proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 2495. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.K.; Zarutskie, J.A.; Rushe, M.M.; Lomakin, A.; Natarajan, S.K.; Sadegh-Nasseri, S.; Benedek, G.B.; Stern, L.J. Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J. Biol. Chem. 2000, 275, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Lazarski, C.A.; Chaves, F.A.; Jenks, S.A.; Wu, S.; Richards, K.A.; Weaver, J.M.; Sant, J.A. The kinetic stability of MHC class II: Peptide complexes is a key parameter that dictates immunodominance. Immunity 2005, 23, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Fodor, J.; Riley, B.T.; Borg, N.A.; Buckle, A.M. Previously hidden dynamics at the TCR-Peptide-MHC interface revealed. J. Immunol. 2018, 200, 4134–4145. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.; Farenc, C.; Mukhopadhyay, M.; Jayasinghe, D.; Decroos, A.; Benati, D.; Tan, L.L.; Ciacchi, L.; Reid, H.H.; Rossjohn, J.; et al. CD4(+) T cell-mediated HLA class II cross-restriction in HIV controllers. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef]
- Furnham, N.; Blundell, T.L.; DePristo, M.A.; Terwilliger, T.C. Is one solution good enough? Nat. Struct. Mol. Biol. 2006, 13, 184–185. [Google Scholar] [CrossRef]
- Levin, E.J.; Kondrashov, D.A.; Wesenberg, G.E.; Phillips, G.N., Jr. Ensemble refinement of protein crystal structures: Validation and application. Structure 2007, 15, 1040. [Google Scholar] [CrossRef]
- Burnley, B.T.; Afonine, P.V.; Adams, P.D.; Gros, P. Modelling dynamics in protein crystal structures by ensemble refinement. eLife. 2012, 1. [Google Scholar] [CrossRef]
- Buckle, A.M.; Borg, N.A. Integrating experiment and theory to understand TCR-pMHC dynamics. Front. Immunol. 2018, 9, 2898. [Google Scholar] [CrossRef]
- Chen, S.; Li, Y.; Depontieu, F.R.; Sidney, J.; Salay, T.M.; Engelhard, V.H.; Hunt, D.F.; Sette, A.; Rosenberg, S.A.; McMiller, T. Structural basis for the presentation of tumor-associated MHC class II-restricted phosphopeptides to CD4+ T cells. J. Mol. Biol. 2010, 399, 596. [Google Scholar]
- Gunther, S.; Schlundt, A.; Sticht, J.; Roske, Y.; Heinemann, U.; Wiesmuller, K.H.; Jung, G.; Falk, K.; Rötzschke, O.; Freund, C. Bidirectional binding of invariant chain peptides to an MHC class II molecule. Proc. Natl. Acad. Sci. USA 2010, 107, 22219. [Google Scholar] [CrossRef]
- Painter, C.A.; Negroni, M.P.; Kellersberger, K.A.; Zavala-Ruiz, Z.; Evans, J.E.; Stern, L.J. Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proc. Natl. Acad. Sci. USA 2011, 108, 19329. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.S.; Anders, A.K.; Sethim, D.K.; Call, M.J. Disruption of hydrogen bonds between major histocompatibility complex class II and the peptide N-terminus is not sufficient to form a human leukocyte antigen-DM receptive state of major histocompatibility complex class II. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Trenh, P.; Guce, A.; Wieczorek, M.; Lange, S.; Sticht, J.; Jiang, W.; Bylsma, M.; Mellins, E.D.; Freund, C. Susceptibility to HLA-DM protein is determined by a dynamic conformation of major histocompatibility complex class II molecule bound with peptide. J. Biol. Chem. 2014, 289, 23449. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Sticht, J.; Stolzenberg, S.; Gunther, S.; Wehmeyer, C.; El Habre, Z.; Alvaro-Benito, M.; Noé, F.; Freund, C. MHC class II complexes sample intermediate states along the peptide exchange pathway. Nat. Commun. 2016, 7, 13224. [Google Scholar] [CrossRef] [PubMed]
- MacLachlan, B.J.; Dolton, G.; Papakyriakou, A.; Greenshields-Watson, A.; Mason, G.H.; Schauenburg, A.; Besneux, M.; Szomolay, B.; Elliott, T.; Sewell, A.K.; et al. Human leukocyte antigen (HLA) class II peptide flanking residues tune the immunogenicity of a human tumor-derived epitope. J. Biol. Chem. 2019, 294, 20246. [Google Scholar] [CrossRef]
- Zavala-Ruiz, Z.; Strug, I.; Anderson, M.W.; Gorski, J.; Stern, L.J. A polymorphic pocket at the P10 position contributes to peptide binding specificity in class II MHC proteins. Chem. Biol. 2004, 11, 1395. [Google Scholar] [CrossRef]
- Ghosh, P.; Amaya, M.; Mellins, E.; Wiley, D.C. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 1995, 378, 457. [Google Scholar] [CrossRef]
- Chen, S.; Li, Y.; Depontieu, F.R.; McMiller, T.L.; English, A.M.; Shabanowitz, J.; Kos, F.; Sidney, J.; Sette, A.; Rosenberg, S.A.; et al. Structure-based design of altered MHC class II-restricted peptide ligands with heterogeneous immunogenicity. J. Immunol. 2013, 191, 5097. [Google Scholar] [CrossRef]
- Scally, S.W.; Petersen, J.; Law, S.C.; Dudek, N.L.; Nel, H.J.; Loh, K.L.; Wijeyewickrema, L.C.; Eckle, S.B.; van Heemst, J.; Pike, R.N.; et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 2013, 210, 2569. [Google Scholar] [CrossRef]
- Gerstner, C.; Dubnovitsky, A.; Sandin, C.; Kozhukh, G.; Uchtenhagen, H.; James, E.A.; Rönnelid, J.; Ytterberg, A.J.; Pieper, J.; Reed, E.; et al. Functional and structural characterization of a novel HLA-DRB1*04:01-restricted alpha-enolase t cell epitope in rheumatoid arthritis. Front. Immunol. 2016, 7, 494. [Google Scholar] [CrossRef]
- Pieper, J.; Dubnovitsky, A.; Gerstner, C.; James, E.A.; Rieck, M.; Kozhukh, G.; Tandre, K.; Pellegrino, S.; Gebe, J.A.; Rönnblom, L.; et al. Memory T cells specific to citrullinated alpha-enolase are enriched in the rheumatic joint. J. Autoimmun. 2018, 92, 47. [Google Scholar] [CrossRef] [PubMed]
- Ting, Y.T.; Petersen, J.; Ramarathinam, S.H.; Scally, S.W.; Loh, K.L.; Thomas, R.; Suri, A.; Baker, D.G.; Purcell, A.W.; Reid, H.H.; et al. The interplay between citrullination and HLA-DRB1 polymorphism in shaping peptide binding hierarchies in rheumatoid arthritis. J. Biol. Chem. 2018, 293, 3236. [Google Scholar] [CrossRef] [PubMed]
- Scally, S.W.; Law, S.C.; Ting, Y.T.; Heemst, J.V.; Sokolove, J.; Deutsch, A.J.; Clemens, E.B.; Moustakas, A.K.; Papadopoulos, G.K.; Van Der Woude, D.; et al. Molecular basis for increased susceptibility of indigenous North Americans to seropositive rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1915. [Google Scholar] [CrossRef] [PubMed]
- Parry, C.S.; Gorski, J.; Stern, L.J. Crystallographic structure of the human leukocyte antigen DRA, DRB3*0101: Models of a directional alloimmune response and autoimmunity. J. Mol. Biol. 2007, 371, 435. [Google Scholar] [CrossRef]
- Dai, S.; Crawford, F.; Marrack, P.; Kappler, J.W. The structure of HLA-DR52c: Comparison to other HLA-DRB3 alleles. Proc. Natl. Acad. Sci. USA 2008, 105, 11893. [Google Scholar] [CrossRef]
- Yin, L.; Crawford, F.; Marrack, P.; Kappler, J.W.; Dai, S. T-cell receptor (TCR) interaction with peptides that mimic nickel offers insight into nickel contact allergy. Proc. Natl. Acad. Sci. USA 2012, 109, 18517. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Martin, R.; Mariuzza, R.A. Structural basis for the binding of an immunodominant peptide from myelin basic protein in different registers by two HLA-DR2 proteins. J. Mol. Biol. 2000, 304, 177. [Google Scholar] [CrossRef]
- Lang, H.L.; Jacobsen, H.; Ikemizu, S.; Andersson, C.; Harlos, K.; Madsen, L.; Hjorth, P.; Sondergaard, L.; Svejgaard, A.; Wucherpfennig, K.; et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 2002, 3, 940. [Google Scholar] [CrossRef]
- Harndahl, M.; Rasmussen, M.; Order, G.; Dalgaard Pedersen, I.; Sorensen, M.; Nielsen, M.; Buus, S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol. 2012, 42, 1405–1416. [Google Scholar] [CrossRef]
- Yin, L.; Calvo-Calle, J.M.; Dominguez-Amorocho, O.; Stern, L.J. HLA-DM constrains epitope selection in the human CD4 T cell response to vaccinia virus by favoring the presentation of peptides with longer HLA-DM-mediated half-lives. J. Immunol. 2012, 189, 3983. [Google Scholar] [CrossRef]
- Lamb, J.R.; Eckels, D.D.; Lake, P.; Woody, J.N.; Green, N. Human T-cell clones recognize chemically synthesized peptides of influenza haemagglutinin. Nature 1982, 300, 66. [Google Scholar] [CrossRef] [PubMed]
- Denzin, L.K.; Cresswell, P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell 1995, 82, 155. [Google Scholar] [CrossRef]
- Sherman, M.A.; Weber, D.A.; Jensen, P.E. DM enhances peptide binding to class II MHC by release of invariant chain-derived peptide. Immunity 1995, 3, 197. [Google Scholar] [CrossRef]
- Pos, W.; Sethi, D.K.; Wucherpfennig, K.W. Mechanisms of peptide repertoire selection by HLA-DM. Trends Immunol. 2013, 34, 495. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.; De Riva, A.; Hadjinicolaou, A.V.; Jiang, W.; Hou, T.; Mellins, E.D. On the perils of poor editing: Regulation of peptide loading by HLA-DQ and H2-A molecules associated with celiac disease and type 1 diabetes. Expert Rev. Mol. Med. 2012, 14. [Google Scholar] [CrossRef]
- Fallang, L.E.; Roh, S.; Holm, A.; Bergseng, E.; Yoon, T.; Fleckenstein, B.; Bandyopadhyay, A.; Mellins, E.D.; Sollid, L.M. Complexes of two cohorts of CLIP peptides and HLA-DQ2 of the autoimmune DR3-DQ2 haplotype are poor substrates for HLA-DM. J. Immunol. 2008, 181, 5451. [Google Scholar] [CrossRef]
- Ferrante, A.; Anderson, M.W.; Klug, C.S.; Gorski, J. HLA-DM mediates epitope selection by a “compare-exchange” mechanism when a potential peptide pool is available. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Ferrante, A.; Gorski, J. Cutting edge: HLA-DM-mediated peptide exchange functions normally on MHC class II-peptide complexes that have been weakened by elimination of a conserved hydrogen bond. J. Immunol. 2010, 184, 1153. [Google Scholar] [CrossRef]
- Joshi, R.V.; Zarutskie, J.A.; Stern, L.J. A three-step kinetic mechanism for peptide binding to MHC class II proteins. Biochemistry 2000, 39, 3751. [Google Scholar] [CrossRef][Green Version]
- Narayan, K.; Chou, C.L.; Kim, A.; Hartman, I.Z.; Dalai, S.; Khoruzhenko, S.; Sadegh-Nasseri, S. HLA-DM targets the hydrogen bond between the histidine at position beta81 and peptide to dissociate HLA-DR-peptide complexes. Nat. Immunol. 2007, 8, 92. [Google Scholar] [CrossRef]
- Roche, P.A.; Cresswell, P. High-affinity binding of an influenza hemagglutinin-derived peptide to purified HLA-DR. J. Immunol. 1990, 144, 1849. [Google Scholar] [PubMed]
- Zhou, Z.; Callaway, K.A.; Weber, D.A.; Jensen, P.E. Cutting edge: HLA-DM functions through a mechanism that does not require specific conserved hydrogen bonds in class II MHC-peptide complexes. J. Immunol. 2009, 183, 4187. [Google Scholar] [CrossRef] [PubMed]
- Sant, A.J.; Chaves, F.A.; Jenks, S.A.; Richards, K.A.; Menges, P.; Weaver, J.M.; Sant, A.J. The relationship between immunodominance, DM editing, and the kinetic stability of MHC class II: Peptide complexes. Immunol. Rev. 2005, 207, 261. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Stern, L.J. Measurement of peptide binding to MHC class II molecules by fluorescence polarization. Curr. Protoc. Immunol. 2014, 106, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Stern, L.J. A novel method to measure HLA-DM-susceptibility of peptides bound to MHC class II molecules based on peptide binding competition assay and differential IC(50) determination. J. Immunol. Methods 2014, 406, 21. [Google Scholar] [CrossRef][Green Version]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernandez, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528. [Google Scholar] [CrossRef]
- Sondergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., 3rd. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Allomorph | Resolution (Å) | Peptide Sequence | Peptide Name | Rfree (PDB) | Rfree (Refine) | Rfree (Ensemble) | ΔRFree | Ensemble Size | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 3L6F | HLA-DRB1*01:01 | 2.10 | APPAYEKL(SEP)AEQSPP | MART-1 | 0.249 | 0.311 | 0.214 | 0.098 | 56 | [19] |
| 3PDO | HLA-DRB1*01:01 | 1.95 | KPVSKMRMATPLLMQALPM | CLIP102–120 | 0.240 | 0.417 | 0.225 | 0.192 | 50 | [20] |
| 3QXA | HLA-DRB1*01:01 | 2.71 | KPVSKMRMATPLLMQALPM | CLIP102–120 | 0.235 | 0.411 | 0.238 | 0.173 | 30 | [21] |
| 4I5B | HLA-DRB1*01:01 | 2.12 | VVKQNCLKLATK | HA308–319 | 0.237 | 0.329 | 0.215 | 0.114 | 45 | [22] |
| 4OV5 | HLA-DRB1*01:01 | 2.20 | GSDARFLRGYHLYA | HLA-A2104–117 | 0.239 | 0.327 | 0.241 | 0.002 | 34 | [23] |
| 4X5W | HLA-DRB1*01:01 | 1.34 | KPVSKWRMATPLLMQALPM | CLIP102–120-W107 | 0.170 | 0.210 | 0.166 | 0.004 | 110 | [24] |
| 6CQJ | HLA-DRB1*01:01 | 2.75 | RFYKTLRAEQASQ | HIVRQ13 | 0.249 | 0.376 | 0.268 | 0.108 | 30 | [14] |
| 6HBY | HLA-DRB1*01:01 | 1.95 | ARRPPLAELAALNLSGSRL | 5T4 tumour | 0.241 | 0.427 | 0.258 | 0.169 | 42 | [25] |
| 1T5W | HLA-DRB1*01:01 | 2.40 | AAYSDQATPLLLSPR | MIG1448–460 | 0.255 | 0.356 | 0.229 | 0.127 | 39 | [26] |
| 1A6A | HLA-DRB1*03:01 | 2.75 | KPVSKMRMATPLLMQALPM | CLIP102–120 | 0.325 | 0.372 | 0.297 | 0.028 | 38 | [27] |
| 4IS6 | HLA-DRB1*04:01 | 2.50 | WNRQLYPEWTEAQRLD | GP100 | 0.298 | 0.400 | 0.269 | 0.131 | 30 | [28] |
| 4MCY | HLA-DRB1*04:01 | 2.30 | SAVRL-CIT-SSVPGVR | Vimentin66–78-Cit | 0.225 | 0.314 | 0.235 | 0.079 | 30 | [29] |
| 4MCZ | HLA-DRB1*04:01 | 2.41 | GVYAT-CIT-SSAVRLR | Vimentin59–71-Cit | 0.231 | 0.407 | 0.236 | 0.005 | 24 | [29] |
| 4MD0 | HLA-DRB1*04:01 | 2.19 | GVYAT-CIT-SSAV-CIT-L-CIT | Vimentin59–71-Cit | 0.208 | 0.304 | 0.226 | 0.018 | 39 | [29] |
| 4MD4 | HLA-DRB1*04:01 | 1.95 | ATEY-CIT-V-CIT-VNSAYQDK | Aggrecan89–103-Cit | 0.209 | 0.266 | 0.197 | -0.29 | 42 | [29] |
| 5JLZ | HLA-DRB1*04:01 | 1.99 | TSKGLF(CIR)AAVPSGAS | αEnolasse26–40-Cit | 0.243 | 0.389 | 0.267 | 0.024 | 32 | [30] |
| 5LAX | HLA-DRB1*04:01 | 2.60 | TSKGLFRAAVPSGAS | αEnolase26–40 | 0.270 | 0.385 | 0.298 | 0.028 | 20 | [30] |
| 5NI9 | HLA-DRB1*04:01 | 1.33 | KRIAKAVNEKSCNCL | αEnolase326–340 | 0.176 | 0.329 | 0.184 | 0.008 | 62 | [31] |
| 5NIG | HLA-DRB1*04:01 | 1.35 | K-CIT-IAKAVNEKSCNCL | αEnolase326–340-Cit | 0.181 | 0.656 | 0.178 | 0.003 | 92 | [31] |
| 6BIJ | HLA-DRB1*04:01 | 2.10 | GGY-CIT-A-CIT-PAKAAAT | Fibrinogen69–81-Cit | 0.239 | 0.289 | 0.220 | 0.069 | 36 | [32] |
| 6BIL | HLA-DRB1*04:01 | 2.40 | GGYRA-CIT-PAKAAAT | Fibrinogen69–81-Cit | 0.243 | 0.305 | 0.227 | 0.078 | 34 | [32] |
| 6BIN | HLA-DRB1*04:01 | 2.50 | QYM-CIT-ADQAAGGLR | Collagen1237–1249-Cit | 0.238 | 0.319 | 0.232 | 0.087 | 34 | [32] |
| 6BIV | HLA-DRB1*04:01 | 2.90 | ETVCP-CIT-TTQQSPE | LL3786–98-Cit | 0.248 | 0.469 | 0.266 | 0.204 | 12 | [32] |
| 6NIX | HLA-DRB1*04:01 | 2.10 | GIAGFKGEQGPKGEP | Collagen259–273 | 0.235 | 0.344 | 0.226 | 0.118 | 42 | [NA] |
| 4MDI | HLA-DRB1*04:02 | 2.00 | SAVRL-CIT-SSVPGVR | Vimentin66–78-Cit | 0.203 | 0.290 | 0.206 | 0.003 | 50 | [29] |
| 4MDJ | HLA-DRB1*04:02 | 1.70 | SAVRLRSSVPGVR | Vimentin66–78 | 0.188 | 0.287 | 0.190 | 0.097 | 70 | [29] |
| 4MD5 | HLA-DRB1*04:04 | 1.65 | SAVRL-CIT-SSVPGVR | Vimentin66–78-Cit | 0.186 | 0.322 | 0.200 | 0.014 | 70 | [29] |
| 6BIX | HLA-DRB1*04:04 | 2.20 | ETVCP-CIT-TTQQSPE | LL3786–98-Cit | 0.213 | 0.407 | 0.217 | 0.190 | 27 | [32] |
| 6BIY | HLA-DRB1*04:04 | 2.05 | DIFERIASEASRL | Histone70–82 | 0.232 | 0.305 | 0.224 | 0.082 | 56 | [32] |
| 6BIZ | HLA-DRB1*04:04 | 2.10 | DIFE-CIT-IASEAS-CIT-L | Histone70–84-Cit74-Cit81 | 0.228 | 0.322 | 0.213 | 0.108 | 32 | [32] |
| 6BIR | HLA-DRB1*04:05 | 2.30 | SSLNL-CIT-ETNLDSL | Vimentin418–431-Cit423 | 0.237 | 0.322 | 0.225 | 0.098 | 39 | [32] |
| 6CPN | HLA-DRB1*11:01 | 2.00 | RFYKTLRAEQASQ | HIVRQ13 | 0.236 | 0.359 | 0.235 | 0.124 | 42 | [14] |
| 6ATZ | HLA-DRB1*14:02 | 2.70 | GGYRA-CIR-PAKAAT | Fibrinogen | 0.240 | 0.334 | 0.242 | 0.092 | 34 | [33] |
| 6ATF | HLA-DRB1*14:02 | 1.90 | GVYATRSSAVRLR | Vimentin59–71 | 0.202 | 0.251 | 0.201 | 0.001 | 60 | [33] |
| 6ATI | HLA-DRB1*14:02 | 1.98 | GVYAT-CIR-SSAVRLR | Vimentin59–71Cit64 | 0.233 | 0.306 | 0.235 | 0.002 | 50 | [33] |
| 6CPO | HLA-DRB1*15:02 | 2.40 | RFYKTLRAEQASQ | HIVRQ13 | 0.246 | 0.332 | 0.243 | 0.089 | 30 | [14] |
| 2Q6W | HLA-DRB3*01:01 | 2.25 | AWRSDEALPLGS | Integrin | 0.265 | 0.353 | 0.249 | 0.105 | 34 | [34] |
| 3C5J | HLA-DRB3*01:01 | 1.80 | QVIILNHPGQISA | Tu elongation factor | 0.227 | 0.307 | 0.216 | 0.091 | 67 | [35] |
| 4H26 | HLA-DRB3*03:01 | 2.50 | QWIRVNIPKRI | Synthetic peptide | 0.270 | 0.410 | 0.264 | 0.146 | 24 | [36] |
| 1FV1 | HLA-DRB5*01:01 | 1.90 | NPVVHFFKNIVTPRTPPPSQ | MBP119–236 | 0.267 | 0.342 | 0.252 | 0.090 | 43 | [37] |
| 1H15 | HLA-DRB5*01:01 | 3.10 | GGVYHFVKKHVHES | DNA Pol628–641 | 0.310 | 0.347 | 0.280 | 0.067 | 12 | [38] |
| pHLA Complex | Tm (°C) |
|---|---|
| HLA-A2-M1 | 58.7 ± 0.3 |
| HLA-DR1-CLIP | 71.5 ± 0.3 |
| HLA-DR1-RQ13 | 81.3 ± 0.5 |
| HLA-DR1-HA306–318 | 81.5 ± 0.3 |
| HLA-DR11-CLIP | 46.5 ± 0.6 |
| HLA-DR11-RQ13 | 64.1 ± 1.3 |
| HLA-DR11-HA306–318 | 60.6 ± 0.7 |
| pHLA Complex | IC50 (nM) |
|---|---|
| HLA-DR11-RQ13 | 180 ± 50 |
| HLA-DR11-RQ13 + HLA-DM | 35 ± 8 |
| HLA-DR1-RQ13 | 204 ± 30 |
| HLA-DR1-RQ13 + HLA-DM | 307 ± 52 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szeto, C.; Bloom, J.I.; Sloane, H.; Lobos, C.A.; Fodor, J.; Jayasinghe, D.; Chatzileontiadou, D.S.M.; Grant, E.J.; Buckle, A.M.; Gras, S. Impact of HLA-DR Antigen Binding Cleft Rigidity on T Cell Recognition. Int. J. Mol. Sci. 2020, 21, 7081. https://doi.org/10.3390/ijms21197081
Szeto C, Bloom JI, Sloane H, Lobos CA, Fodor J, Jayasinghe D, Chatzileontiadou DSM, Grant EJ, Buckle AM, Gras S. Impact of HLA-DR Antigen Binding Cleft Rigidity on T Cell Recognition. International Journal of Molecular Sciences. 2020; 21(19):7081. https://doi.org/10.3390/ijms21197081
Chicago/Turabian StyleSzeto, Christopher, Joseph I. Bloom, Hannah Sloane, Christian A. Lobos, James Fodor, Dhilshan Jayasinghe, Demetra S. M. Chatzileontiadou, Emma J. Grant, Ashley M. Buckle, and Stephanie Gras. 2020. "Impact of HLA-DR Antigen Binding Cleft Rigidity on T Cell Recognition" International Journal of Molecular Sciences 21, no. 19: 7081. https://doi.org/10.3390/ijms21197081
APA StyleSzeto, C., Bloom, J. I., Sloane, H., Lobos, C. A., Fodor, J., Jayasinghe, D., Chatzileontiadou, D. S. M., Grant, E. J., Buckle, A. M., & Gras, S. (2020). Impact of HLA-DR Antigen Binding Cleft Rigidity on T Cell Recognition. International Journal of Molecular Sciences, 21(19), 7081. https://doi.org/10.3390/ijms21197081