PAI-1, the Plasminogen System, and Skeletal Muscle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Basic Biology of PAI-1 and the Plasminogen System
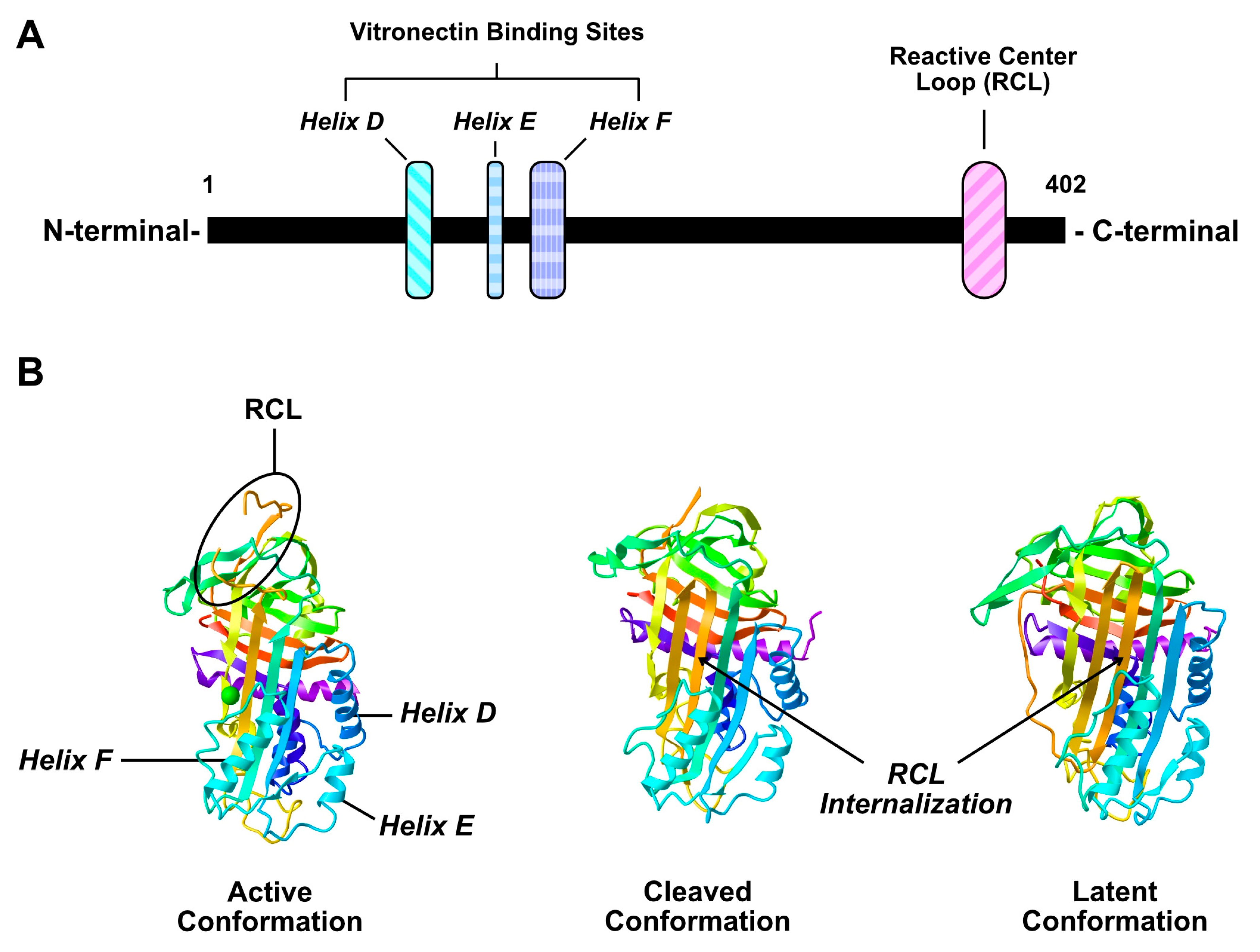
2.1. Structure and Function of PAI-1
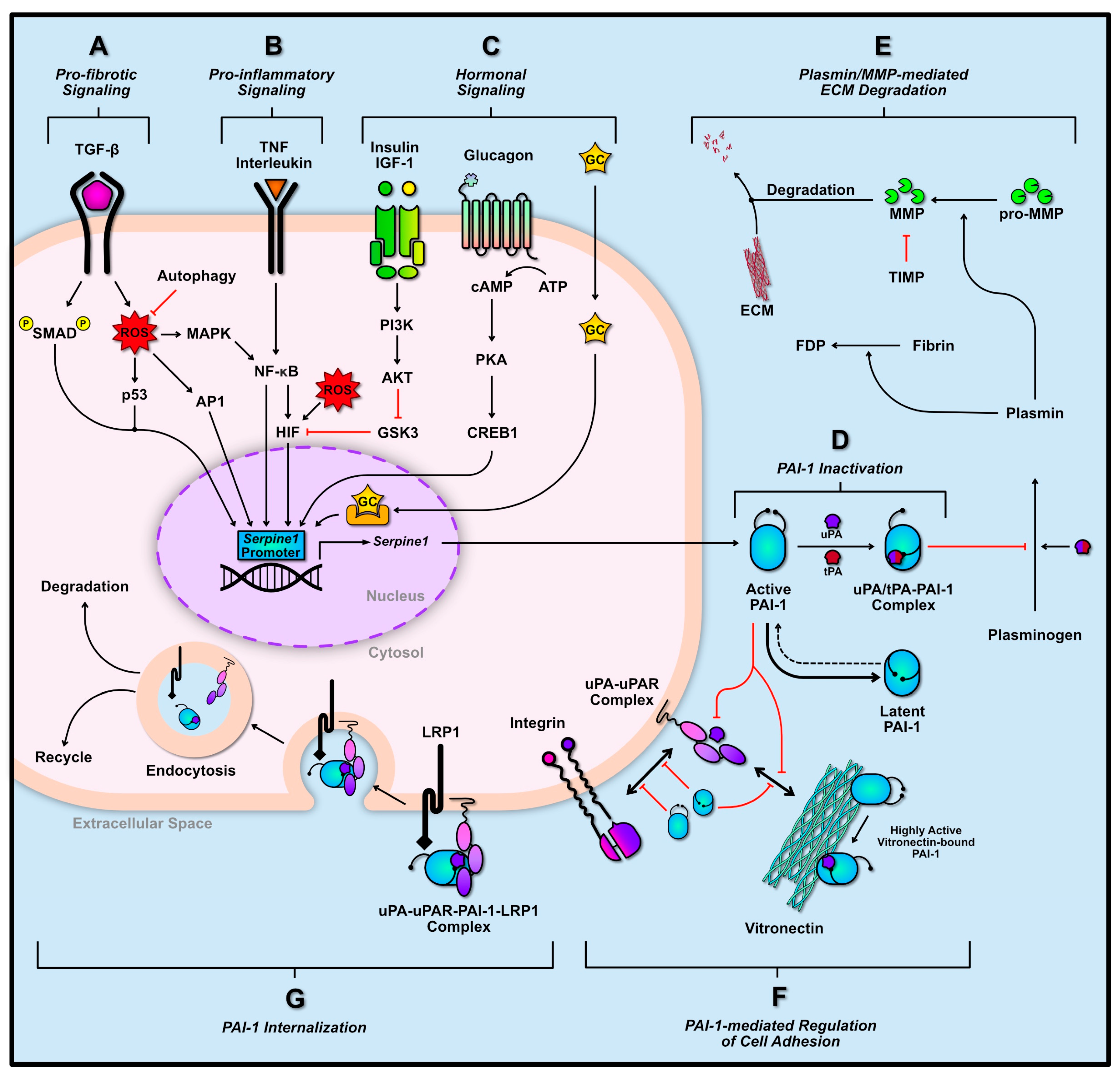
2.2. Transcriptional Regulation of PAI-1
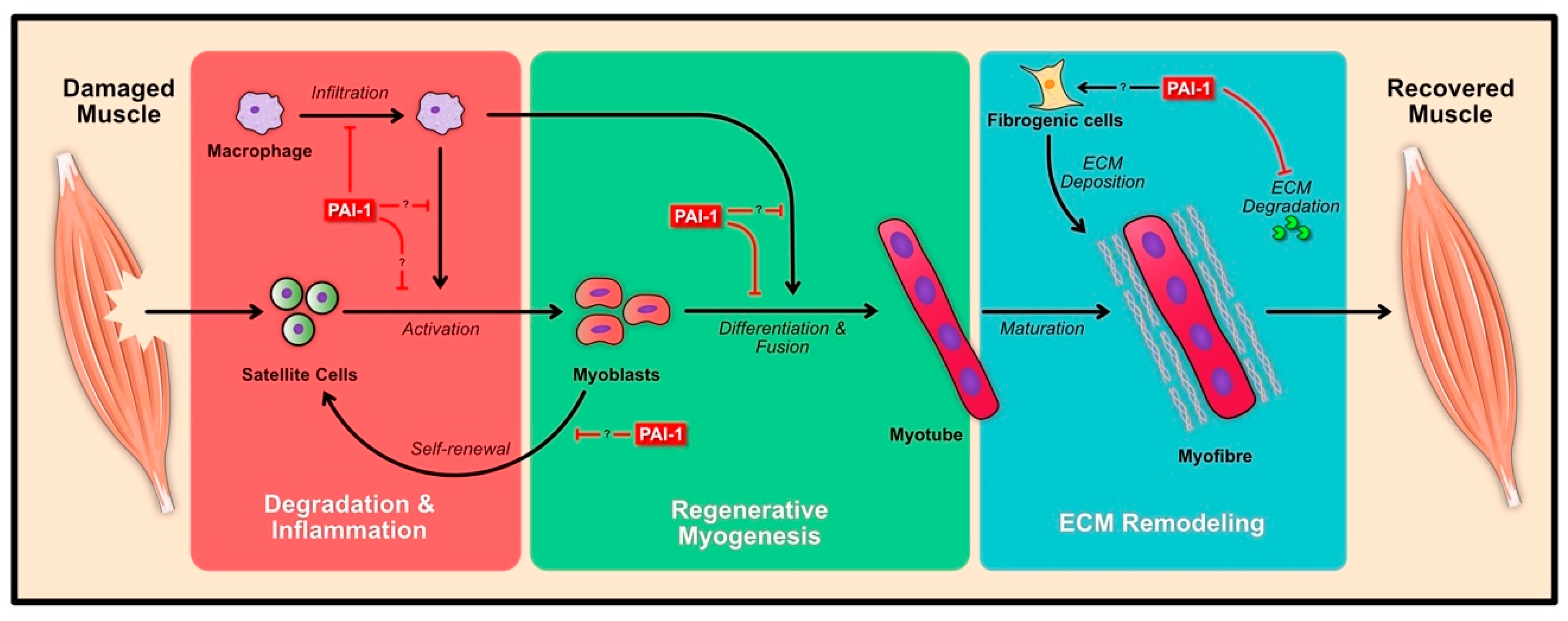
3. Role of PAI-1 Following Skeletal Muscle Damage
3.1. Degeneration and Inflammation
3.2. Regenerative Myogenesis
3.3. ECM Remodeling
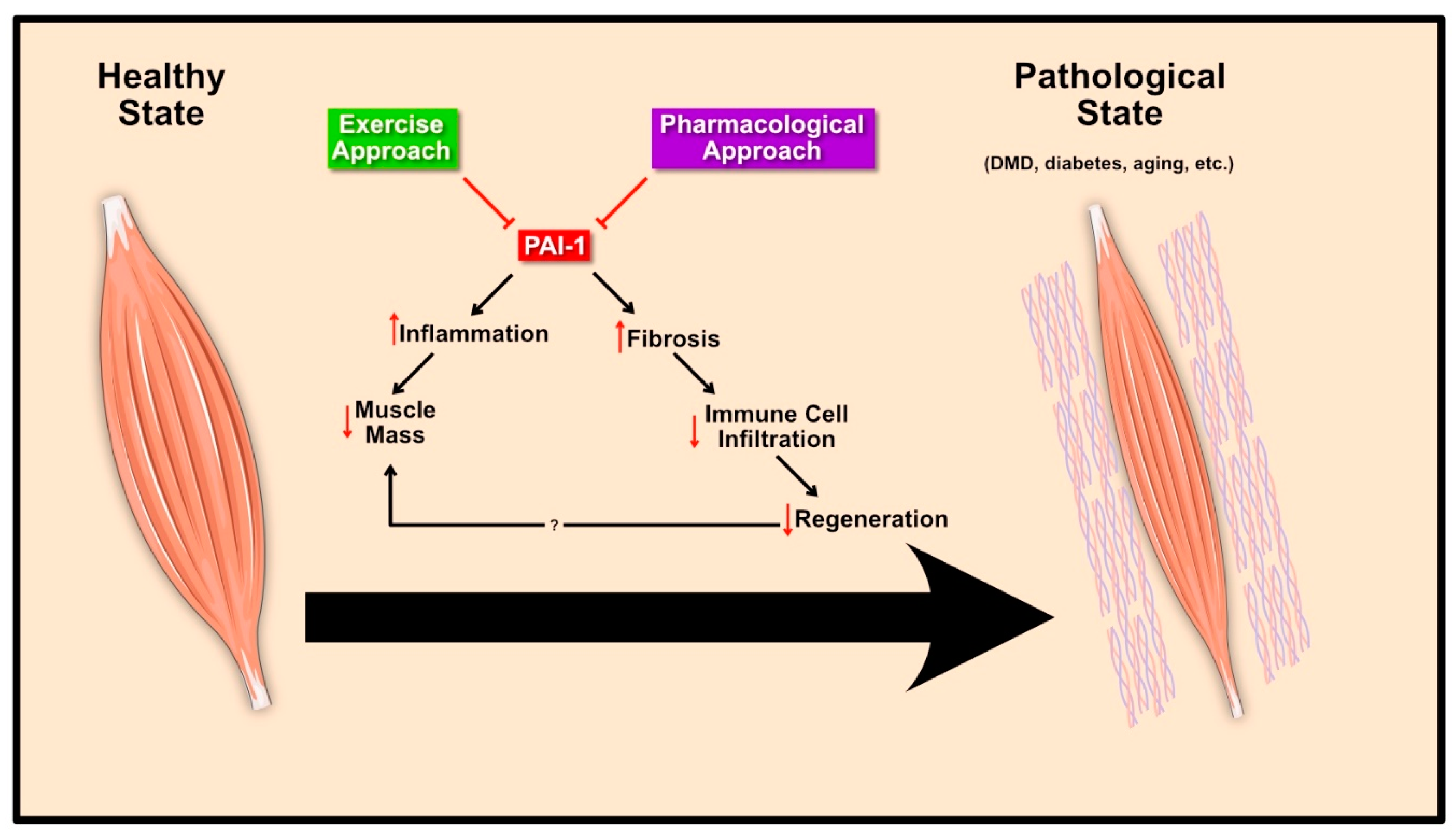
4. Pathological Role of PAI-1 in Skeletal Muscle
4.1. Muscular Dystrophy
4.2. Diabetes Mellitus
4.3. Aging
5. Therapeutic Interventions to Normalize PAI-1
5.1. Exercise Approach
5.2. Pharmacological Approach
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AMPK | Adenosine monophosphate-activated protein kinase |
| AP1 | Activation protein 1 |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| CREB1 | cAMP-responsible element binding protein 1 |
| ECM | Extracellular matrix |
| FDP | Fibrin degradation product |
| GC | Glucocorticoid |
| GSK3 | Glycogen synthase kinase 3 |
| hD/E/F | Helix D/E/F domains |
| HIF | Hypoxia-inducing factor |
| IGF-1 | Insulin-like growth factor-1 |
| IL | Interleukin |
| LRP1 | Low density lipoprotein receptor-related protein 1 |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| NF-κB | Nuclear factor kappa B |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PKA | Protein kinase A |
| RCL | Reactive center loop |
| ROS | Reactive oxygen species |
| TGF-β | Transforming growth factor-β |
| TIMP | Tissue inhibitor of metalloproteinase |
| TNF | Tumor necrosis factor |
| tPA | Tissue-type plasminogen activator |
| uPA | Urokinase-type plasminogen activator |
| uPAR | Urokinase-type plasminogen activator receptor |
References
- Dastre, A. Fibrinolyse dans le sang. Arch. Physiol. Norm. Pathol. 1893, 5, 661–663. [Google Scholar]
- Macfarlane, R.G.; Pilling, J. Observations on fibrinolysis plasminogen, plasmin, and antiplasmin content of human blood. Lancet 1946, 248, 562–565. [Google Scholar] [CrossRef]
- Christensen, L.R. The activation of plasminogen by chloroform. J. Gen. Physiol. 1946, 30, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.R.; Smith, D.H. Plasminogen purification by acid extraction. Proc. Soc. Exp. Biol. Med. 1950, 74, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.H.; Ferguson, J.H.; Howe, A.C.; Rogers, J. Studies on a proteolytic enzyme system of the blood. II. fibrinolysokinase activators for profibrinolysin. J. Clin. Investig. 1950, 29, 1059–1068. [Google Scholar] [CrossRef]
- Kaplan, M. Inhibition of β hemolytic streptococci fibrinolysin by trypsin inhibitor. Science 1944, 100, 198–200. [Google Scholar]
- Kaplan, M. A quantitative study of the fibrinolysin—Antifibrinolysin reaction. Science 1945, 101, 120–122. [Google Scholar]
- Loskutoff, D.J.; van Mourik, J.A.; Erickson, L.A.; Lawrence, D. Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc. Natl. Acad. Sci. USA 1983, 80, 2956–2960. [Google Scholar] [CrossRef]
- Ginsburg, D.; Zeheb, R.; Yang, A.Y.; Rafferty, U.M.; Andreasen, P.A.; Nielsen, L.; Dano, K.; Lebo, R.V.; Gelehrter, T.D. cDNA cloning of human plasminogen activator-inhibitor from endothelial cells. J. Clin. Investig. 1986, 78, 1673–1680. [Google Scholar] [CrossRef]
- Balsara, R.D.; Ploplis, V.A. Plasminogen activator inhibitor-1: The double-edged sword in apoptosis. Thromb. Haemost. 2008, 100, 1029–1036. [Google Scholar] [CrossRef]
- Lademann, U.A.; Rømer, M.U. Regulation of programmed cell death by plasminogen activator inhibitor type 1 (PAI-1). Thromb. Haemost. 2008, 100, 1041–1046. [Google Scholar] [PubMed]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. The TGF-β1/p53/PAI-1 signaling axis in vascular senescence: Role of caveolin-1. Biomolecules 2019, 9, 341. [Google Scholar] [CrossRef]
- Aso, Y. Plasminogen activator inhibitor (PAI)-1 in vascular inflammation and thrombosis. Front. Biosci. 2007, 12, 2957–2966. [Google Scholar] [CrossRef]
- Samad, F.; Ruf, W. Inflammation, obesity, and thrombosis. Blood 2013, 122, 3415–3422. [Google Scholar] [CrossRef] [PubMed]
- Placencio, V.R.; DeClerck, Y.A. Plasminogen activator inhibitor-1 in cancer: Rationale and insight for future therapeutic testing. Cancer Res. 2015, 75, 2969–2974. [Google Scholar] [CrossRef]
- Mashiko, S.; Kitatani, K.; Toyoshima, M.; Ichimura, A.; Dan, T.; Usui, T.; Ishibashi, M.; Shigeta, S.; Nagase, S.; Miyata, T.; et al. Inhibition of plasminogen activator inhibitor-1 is a potential therapeutic strategy in ovarian cancer. Cancer Biol. Ther. 2015, 16, 253–260. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen activator inhibitor-1 in cancer research. Biomed. Pharmacother. 2018, 105, 83–94. [Google Scholar] [CrossRef]
- Peng, Y.; Kajiyama, H.; Yuan, H.; Nakamura, K.; Yoshihara, M.; Yokoi, A.; Fujikake, K.; Yasui, H.; Yoshikawa, N.; Suzuki, S.; et al. PAI-1 secreted from metastatic ovarian cancer cells triggers the tumor-promoting role of the mesothelium in a feedback loop to accelerate peritoneal dissemination. Cancer Lett. 2019, 442, 181–192. [Google Scholar] [CrossRef]
- Stefansson, S.; McMahon, G.A.; Petitclerc, E.; Lawrence, D.A. Plasminogen activator inhibitor-1 in tumor growth, angiogenesis and vascular remodeling. Curr. Pharm. Des. 2003, 9, 1545–1564. [Google Scholar] [CrossRef]
- Vaughan, D.E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Khoukaz, H.B.; Ji, Y.; Braet, D.J.; Vadali, M.; Abdelhamid, A.A.; Emal, C.D.; Lawrence, D.A.; Fay, W.P. Drug targeting of plasminogen activator inhibitor-1 inhibits metabolic dysfunction and atherosclerosis in a murine model of metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1479–1490. [Google Scholar] [CrossRef]
- Lyon, C.J.; Hsueh, W.A. Effect of plasminogen activator inhibitor-1 in diabetes mellitus and cardiovascular disease. Am. J. Med. 2003, 115 (Suppl. 8A), 62S–68S. [Google Scholar] [CrossRef]
- Yarmolinsky, J.; Bordin Barbieri, N.; Weinmann, T.; Ziegelmann, P.K.; Duncan, B.B.; Inês Schmidt, M. Plasminogen activator inhibitor-1 and type 2 diabetes: A systematic review and meta-analysis of observational studies. Sci. Rep. 2016, 6, 17714. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-J.; Fogo, A.B. PAI-1 and kidney fibrosis. Front. Biosci. 2009, 14, 2028–2041. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Noble, N.A. PAI-1 as a target in kidney disease. Curr. Drug Targets 2007, 8, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.P.; Moradi, J.; Nissar, A.A.; Riddell, M.C.; Hawke, T.J. Inhibition of plasminogen activator inhibitor-1 restores skeletal muscle regeneration in untreated type 1 diabetic mice. Diabetes 2011, 60, 1964–1972. [Google Scholar] [CrossRef]
- Koh, T.J.; Bryer, S.C.; Pucci, A.M.; Sisson, T.H. Mice deficient in plasminogen activator inhibitor-1 have improved skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 2005, 289, C217–C223. [Google Scholar] [CrossRef]
- Rahman, F.A.; Angus, S.A.; Stokes, K.; Karpowicz, P.; Krause, M.P. Impaired ECM remodeling and macrophage activity define necrosis and regeneration following damage in aged skeletal muscle. Int. J. Mol. Sci. 2020, 21, 4575. [Google Scholar] [CrossRef]
- Naderi, J.; Bernreuther, C.; Grabinski, N.; Putman, C.T.; Henkel, B.; Bell, G.; Glatzel, M.; Sultan, K.R. Plasminogen activator inhibitor type 1 up-regulation is associated with skeletal muscle atrophy and associated fibrosis. Am. J. Pathol. 2009, 175, 763–771. [Google Scholar] [CrossRef]
- Flevaris, P.; Vaughan, D. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin. Thromb. Hemost. 2017, 43, 169–177. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen activator inhibitor-1 (pai-1): A key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef]
- Ardite, E.; Perdiguero, E.; Vidal, B.; Gutarra, S.; Serrano, A.L.; Muñoz-Cánoves, P. PAI-1–regulated miR-21 defines a novel age-associated fibrogenic pathway in muscular dystrophy. J. Cell Biol. 2012, 196, 163–175. [Google Scholar] [CrossRef]
- Suelves, M.; Vidal, B.; Serrano, A.L.; Tjwa, M.; Roma, J.; López-Alemany, R.; Luttun, A.; de Lagrán, M.M.; Díaz-Ramos, A.; Díaz, M.A.; et al. uPA deficiency exacerbates muscular dystrophy in MDX mice. J. Cell Biol. 2007, 178, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Suelves, M.; Vidal, B.; Ruiz, V.; Baeza-Raja, B.; Diaz-Ramos, A.; Cuartas, I.; Lluis, F.; Parra, M.; Jardi, M.; Lopez-Alemany, R.; et al. The plasminogen activation system in skeletal muscle regeneration: Antagonistic roles of urokinase-type plasminogen activator (uPA) and its inhibitor (PAI-1). Front. Biosci. 2005, 10, 2978–2985. [Google Scholar] [CrossRef] [PubMed]
- Suelves, M.; López-Alemany, R.; Lluís, F.; Aniorte, G.; Serrano, E.; Parra, M.; Carmeliet, P.; Muñoz-Cánoves, P. Plasmin activity is required for myogenesis in vitro and skeletal muscle regeneration in vivo. Blood 2002, 99, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Lluís, F.; Roma, J.; Suelves, M.; Parra, M.; Aniorte, G.; Gallardo, E.; Illa, I.; Rodríguez, L.; Hughes, S.M.; Carmeliet, P.; et al. Urokinase-dependent plasminogen activation is required for efficient skeletal muscle regeneration in vivo. Blood 2001, 97, 1703–1711. [Google Scholar] [CrossRef]
- Fibbi, G.; Barletta, E.; Dini, G.; Del Rosso, A.; Pucci, M.; Cerletti, M.; Del Rosso, M. Cell invasion is affected by differential expression of the urokinase plasminogen activator/urokinase plasminogen activator receptor system in muscle satellite cells from normal and dystrophic patients. Lab. Investig. 2001, 81, 27–39. [Google Scholar] [CrossRef][Green Version]
- Muñoz-Cánoves, P.; Miralles, F.; Baiget, M.; Félez, J. Inhibition of urokinase-type plasminogen activator (uPA) abrogates myogenesis in vitro. Thromb. Haemost. 1997, 77, 526–534. [Google Scholar] [CrossRef]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.-W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef]
- Zhao, P.; Iezzi, S.; Carver, E.; Dressman, D.; Gridley, T.; Sartorelli, V.; Hoffman, E.P. Slug is a novel downstream target of MyoD. Temporal profiling in muscle regeneration. J. Biol. Chem. 2002, 277, 30091–30101. [Google Scholar] [CrossRef]
- Lira, F.S.; Rosa, J.C.; Lima-Silva, A.E.; Souza, H.A.; Caperuto, E.C.; Seelaender, M.C.; Damaso, A.R.; Oyama, L.M.; Santos, R.V. Sedentary subjects have higher PAI-1 and lipoproteins levels than highly trained athletes. Diabetol. Metab. Syndr. 2010, 2, 7. [Google Scholar] [CrossRef]
- Davis, G.L.; Abildgaard, C.F.; Bernauer, E.M.; Britton, M. Fibrinolytic and hemostatic changes during and after maximal exercise in males. J. Appl. Physiol. 1976, 40, 287–292. [Google Scholar] [CrossRef]
- Fernhall, B.; Szymanski, L.M.; Gorman, P.A.; Milani, J.; Paup, D.C.; Kessler, C.M. Fibrinolytic activity is not dependent upon exercise mode in post-myocardial infarction patients. Eur. J. Appl. Physiol. 1998, 78, 247–252. [Google Scholar] [CrossRef]
- Hilberg, T.; Nowacki, P.E.; Müller-Berghaus, G.; Gabriel, H.H. Changes in blood coagulation and fibrinolysis associated with maximal exercise and physical conditioning in women taking low dose oral contraceptives. J. Sci. Med. Sport 2000, 3, 383–390. [Google Scholar] [CrossRef]
- Röcker, L.; Taenzer, M.; Drygas, W.K.; Lill, H.; Heyduck, B.; Altenkirch, H.U. Effect of prolonged physical exercise on the fibrinolytic system. Eur. J. Appl. Physiol. Occup. Physiol. 1990, 60, 478–481. [Google Scholar] [CrossRef]
- Wheeler, M.E.; Davis, G.L.; Gillespie, W.J.; Bern, M.M. Physiological changes in hemostasis associated with acute exercise. J. Appl. Physiol. 1986, 60, 986–990. [Google Scholar] [CrossRef]
- Winther, K.; Hillegass, W.; Tofler, G.H.; Jimenez, A.; Brezinski, D.A.; Schafer, A.I.; Loscalzo, J.; Williams, G.H.; Muller, J.E. Effects on platelet aggregation and fibrinolytic activity during upright posture and exercise in healthy men. Am. J. Cardiol. 1992, 70, 1051–1055. [Google Scholar] [CrossRef]
- Sakata, K.; Kurata, C.; Taguchi, T.; Suzuki, S.; Kobayashi, A.; Yamazaki, N.; Rydzewski, A.; Takada, Y.; Takada, A. Clinical significance of plasminogen activator inhibitor activity in patients with exercise-induced ischemia. Am. Heart J. 1990, 120, 831–838. [Google Scholar] [CrossRef]
- Ivey, F.M.; Womack, C.J.; Kulaputana, O.; Dobrovolny, C.L.; Wiley, L.A.; Macko, R.F. A single bout of walking exercise enhances endogenous fibrinolysis in stroke patients. Med. Sci. Sports Exerc. 2003, 35, 193–198. [Google Scholar] [CrossRef]
- Womack, C.J.; Ivey, F.M.; Gardner, A.W.; Macko, R.F. Fibrinolytic response to acute exercise in patients with peripheral arterial disease. Med. Sci. Sports Exerc. 2001, 33, 214–219. [Google Scholar] [CrossRef]
- Svendsen, O.L.; Hassager, C.; Christiansen, C.; Nielsen, J.D.; Winther, K. Plasminogen activator inhibitor–1, tissue-type plasminogen activator, and fibrinogen. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 381–385. [Google Scholar] [CrossRef]
- Nagelkirk, P.R.; Scalzo, R.; Harber, M.; Kaminsky, L.A. The influence of acute resistance training and body composition on coagulation and fibrinolytic activity in low-risk women. Int. J. Sports Med. 2010, 31, 458–462. [Google Scholar] [CrossRef]
- Kupchak, B.R.; Creighton, B.C.; Aristizabal, J.C.; Dunn-Lewis, C.; Volk, B.M.; Ballard, K.D.; Comstock, B.A.; Maresh, C.M.; Kraemer, W.J.; Volek, J.S. Beneficial effects of habitual resistance exercise training on coagulation and fibrinolytic responses. Thromb. Res. 2013, 131, e227–e234. [Google Scholar] [CrossRef]
- Rossi, F.E.; Gerosa-Neto, J.; Diniz, T.A.; Freitas, I.F.; Lira, F.S.; Cholewa, J.M. Moderate rest intervals are superior to short intervals for improving PAI-1 following exhaustive exercise in recreational weightlifters. J. Exerc. Rehabil. 2016, 12, 559–566. [Google Scholar] [CrossRef]
- Szymanski, L.M.; Pate, R.R. Effects of exercise intensity, duration, and time of day on fibrinolytic activity in physically active men. Med. Sci. Sports Exerc. 1994, 26, 1102–1108. [Google Scholar] [CrossRef]
- Cooper, J.A.; Nagelkirk, P.R.; Coughlin, A.M.; Pivarnik, J.M.; Womack, C.J. Temporal changes in tPA and PAI-1 after maximal exercise. Med. Sci. Sports Exerc. 2004, 36, 1884–1887. [Google Scholar] [CrossRef]
- Booth, N.A.; Walker, E.; Maughan, R.; Bennett, B. Plasminogen activator in normal subjects after exercise and venous occlusion: T-PA circulates as complexes with C1-inhibitor and PAI-1. Blood 1987, 69, 1600–1604. [Google Scholar] [CrossRef]
- Schuit, A.J.; Schouten, E.G.; Kluft, C.; de Maat, M.; Menheere, P.P.; Kok, F.J. Effect of strenuous exercise on fibrinogen and fibrinolysis in healthy elderly men and women. Thromb. Haemost. 1997, 78, 845–851. [Google Scholar] [CrossRef]
- Van Giezen, J.J.J.; Wahlund, G.; Nerme, V.; Abrahamsson, T. The fab-fragment of a PAI-1 inhibiting antibody reduces thrombus size and restores blood flow in a rat model of arterial thrombosis. Thromb. Haemost. 1997, 77, 964–969. [Google Scholar] [CrossRef]
- Akhter, H.; Huang, W.-T.; van Groen, T.; Kuo, H.-C.; Miyata, T.; Liu, R.-M. A small molecule inhibitor of plasminogen activator inhibitor-1 reduces brain amyloid-β load and improves memory in an animal model of alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, 447–457. [Google Scholar] [CrossRef]
- Hennan, J.K.; Elokdah, H.; Leal, M.; Ji, A.; Friedrichs, G.S.; Morgan, G.A.; Swillo, R.E.; Antrilli, T.M.; Hreha, A.; Crandall, D.L. Evaluation of PAI-039 [{1-benzyl-5-[4-(trifluoromethoxy)phenyl]-1H-indol-3-yl}(oxo)acetic acid], a novel plasminogen activator inhibitor-1 inhibitor, in a canine model of coronary artery thrombosis. J. Pharmacol. Exp. Ther. 2005, 314, 710–716. [Google Scholar] [CrossRef]
- Huang, W.-T.; Akhter, H.; Jiang, C.; MacEwen, M.; Ding, Q.; Antony, V.; Thannickal, V.J.; Liu, R.-M. Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp. Gerontol. 2015, 61, 62–75. [Google Scholar] [CrossRef]
- Izuhara, Y.; Yamaoka, N.; Kodama, H.; Dan, T.; Takizawa, S.; Hirayama, N.; Meguro, K.; Strihou, C.V.Y.D.; Miyata, T. A novel inhibitor of plasminogen activator inhibitor-1 provides antithrombotic benefits devoid of bleeding effect in nonhuman primates. J. Cereb. Blood Flow Metab. 2010, 30, 904–912. [Google Scholar] [CrossRef]
- Liu, R.-M.; Eldridge, S.; Watanabe, N.; Deshane, J.; Kuo, H.-C.; Jiang, C.; Wang, Y.; Liu, G.; Schwiebert, L.; Miyata, T.; et al. Therapeutic potential of an orally effective small molecule inhibitor of plasminogen activator inhibitor for asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L328–L336. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Luo, M.; Chen, N.; Deng, X.; He, J.; Zhang, L.; Luo, P.; Wu, J. Inhibition of PAI-1 attenuates perirenal fat inflammation and the associated nephropathy in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E260–E267. [Google Scholar] [CrossRef]
- Tashiro, Y.; Nishida, C.; Sato-Kusubata, K.; Ohki-Koizumi, M.; Ishihara, M.; Sato, A.; Gritli, I.; Komiyama, H.; Sato, Y.; Dan, T.; et al. Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice. Blood 2012, 119, 6382–6393. [Google Scholar] [CrossRef]
- Leik, C.E.; Su, E.J.; Nambi, P.; Crandall, D.L.; Lawrence, D.A. Effect of pharmacologic plasminogen activator inhibitor-1 inhibition on cell motility and tumor angiogenesis. J. Thromb. Haemost. 2006, 4, 2710–2715. [Google Scholar] [CrossRef]
- Gorlatova, N.V.; Cale, J.M.; Elokdah, H.; Li, D.; Fan, K.; Warnock, M.; Crandall, D.L.; Lawrence, D.A. Mechanism of inactivation of plasminogen activator inhibitor-1 by a small molecule inhibitor. J. Biol. Chem. 2007, 282, 9288–9296. [Google Scholar] [CrossRef]
- Cale, J.M.; Lawrence, D.A. Structure-function relationships of plasminogen activator inhibitor-1 and its potential as a therapeutic agent. Curr. Drug Targets 2007, 8, 971–981. [Google Scholar] [CrossRef]
- Boudier, C.; Gils, A.; Declerck, P.J.; Bieth, J.G. The Conversion of Active to Latent Plasminogen Activator Inhibitor-1 Is an Energetically Silent Event. Biophys. J. 2005, 88, 2848–2854. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lawrence, D.A.; Ginsburg, D.; Day, D.E.; Berkenpas, M.B.; Verhamme, I.M.; Kvassman, J.O.; Shore, J.D. Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J. Biol. Chem. 1995, 270, 25309–25312. [Google Scholar] [CrossRef]
- Wilczynska, M.; Fa, M.; Ohlsson, P.I.; Ny, T. The inhibition mechanism of serpins. Evidence that the mobile reactive center loop is cleaved in the native protease-inhibitor complex. J. Biol. Chem. 1995, 270, 29652–29655. [Google Scholar] [CrossRef] [PubMed]
- Dupont, D.M.; Madsen, J.B.; Kristensen, T.; Bodker, J.S.; Blouse, G.E.; Wind, T.; Andreasen, P.A. Biochemical properties of plasminogen activator inhibitor-1. Front. Biosci. 2009, 14, 1337–1361. [Google Scholar] [CrossRef]
- Schroeck, F.R.; De Prada, N.A.; Sperl, S.; Schmitt, M.; Magdolen, V. Interaction of plasminogen activator inhibitor type-1 (PAI-1) with vitronectin (Vn): Mapping the binding sites on PAI-1 and Vn. Biol. Chem. 2002, 383, 1143–1149. [Google Scholar] [CrossRef]
- Wind, T.; Hansen, M.; Jensen, J.K.; Andreasen, P.A. The molecular basis for anti-proteolytic and non-proteolytic functions of plasminogen activator inhibitor type-1: Roles of the reactive centre loop, the shutter region, the flexible joint region and the small serpin fragment. Biol. Chem. 2002, 383, 21–36. [Google Scholar] [CrossRef]
- Gibson, A.; Baburaj, K.; Day, D.E.; Verhamme, I.; Shore, J.D.; Peterson, C.B. The use of fluorescent probes to characterize conformational changes in the interaction between vitronectin and plasminogen activator inhibitor-1. J. Biol. Chem. 1997, 272, 5112–5121. [Google Scholar] [CrossRef]
- Kjøller, L. The urokinase plasminogen activator receptor in the regulation of the actin cytoskeleton and cell motility. Biol. Chem. 2002, 383, 5–19. [Google Scholar] [CrossRef]
- Mondino, A.; Resnati, M.; Blasi, F. Structure and Function of the Urokinase Receptor. Thromb. Haemost. 1999, 82, 19–22. [Google Scholar] [CrossRef]
- Wei, Y.; Eble, J.A.; Wang, Z.; Kreidberg, J.A.; Chapman, H.A. Urokinase receptors promote beta1 integrin function through interactions with integrin alpha3beta1. Mol. Biol. Cell 2001, 12, 2975–2986. [Google Scholar] [CrossRef]
- Chapman, H.A.; Wei, Y. Protease crosstalk with integrins: The urokinase receptor paradigm. Thromb. Haemost. 2001, 86, 124–129. [Google Scholar]
- Stefansson, S.; Lawrence, D.A. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature 1996, 383, 441–443. [Google Scholar] [CrossRef]
- Stefansson, S.; Lawrence, D.A. Old dogs and new tricks: Proteases, inhibitors, and cell migration. Sci. STKE 2003, 2003, pe24. [Google Scholar] [CrossRef]
- Deng, G.; Royle, G.; Wang, S.; Crain, K.; Loskutoff, D.J. Structural and functional analysis of the plasminogen activator inhibitor-1 binding motif in the somatomedin B domain of vitronectin. J. Biol. Chem. 1996, 271, 12716–12723. [Google Scholar] [CrossRef]
- Cubellis, M.V.; Wun, T.C.; Blasi, F. Receptor-mediated internalization and degradation of urokinase is caused by its specific inhibitor PAI-1. EMBO J. 1990, 9, 1079–1085. [Google Scholar] [CrossRef]
- Al-Fakhri, N.; Chavakis, T.; Schmidt-Wöll, T.; Huang, B.; Cherian, S.M.; Bobryshev, Y.V.; Lord, R.S.A.; Katz, N.; Preissner, K.T. Induction of apoptosis in vascular cells by plasminogen activator inhibitor-1 and high molecular weight kininogen correlates with their anti-adhesive properties. Biol. Chem. 2003, 384, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.A.; Olson, S.T.; Palaniappan, S.; Ginsburg, D. Engineering plasminogen activator inhibitor-1 mutants with increased functional stability. Biochemistry 1994, 33, 3643–3648. [Google Scholar] [CrossRef] [PubMed]
- Berkenpas, M.B.; Lawrence, D.A.; Ginsburg, D. Molecular evolution of plasminogen activator inhibitor-1 functional stability. EMBO J. 1995, 14, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Fjellström, O.; Deinum, J.; Sjögren, T.; Johansson, C.; Geschwindner, S.; Nerme, V.; Legnehed, A.; McPheat, J.; Olsson, K.; Bodin, C.; et al. Characterization of a small molecule inhibitor of plasminogen activator inhibitor type 1 that accelerates the transition into the latent conformation. J. Biol. Chem. 2013, 288, 873–885. [Google Scholar] [CrossRef]
- Ehnebom, J.; Pusa, S.; Björquist, P.; Deinum, J. Comparison of chromogenic substrates for tissue plasminogen activator and the effects on the stability of plasminogen activator inhibitor type-1. Fibrinolysis Proteolysis 1997, 11, 287–293. [Google Scholar] [CrossRef]
- Hekman, C.M.; Loskutoff, D.J. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J. Biol. Chem. 1985, 260, 11581–11587. [Google Scholar]
- Vaughan, D.E.; Declerck, P.J.; Van Houtte, E.; De Mol, M.; Collen, D. Studies of recombinant plasminogen activator inhibitor-1 in rabbits. Pharmacokinetics and evidence for reactivation of latent plasminogen activator inhibitor-1 in vivo. Circ. Res. 1990, 67, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.F.; Hutzelmann, J.E. Plasminogen activator inhibitor-1 binds to fibrin and inhibits tissue-type plasminogen activator-mediated fibrin dissolution. J. Biol. Chem. 1992, 267, 17128–17135. [Google Scholar]
- Lee, E.; Vaughan, U.E.; Parikh, S.H.; Grodzinsky, A.J.; Libby, P.; Lark, M.W.; Lee, R.T. Regulation of Matrix Metalloproteinases and Plasminogen Activator Inhibitor-1 Synthesis by Plasminogen in Cultured Human Vascular Smooth Muscle Cells. Circ. Res. 1996, 78, 44–49. [Google Scholar] [CrossRef]
- Duymelinck, C.; Dauwe, S.E.; De Greef, K.E.; Ysebaert, D.K.; Verpooten, G.A.; De Broe, M.E. TIMP-1 gene expression and PAI-1 antigen after unilateral ureteral obstruction in the adult male rat. Kidney Int. 2000, 58, 1186–1201. [Google Scholar] [CrossRef][Green Version]
- Ahmed, M.M.; King, K.C.; Pearce, S.M.; Ramsey, M.A.; Miranpuri, G.S.; Resnick, D.K. Novel target for Spinal Cord Injury related neuropathic pain. Ann. Neurosci. 2011, 18, 162–167. [Google Scholar] [CrossRef]
- Leivonen, S.-K.; Lazaridis, K.; Decock, J.; Chantry, A.; Edwards, D.R.; Kähäri, V.-M. TGF-β-Elicited Induction of Tissue Inhibitor of Metalloproteinases (TIMP)-3 Expression in Fibroblasts Involves Complex Interplay between Smad3, p38α, and ERK1/2. PLoS ONE 2013, 8, e57474. [Google Scholar] [CrossRef]
- Clark, I.M.; Rowan, A.D.; Edwards, D.R.; Bech-Hansen, T.; Mann, D.A.; Bahr, M.J.; Cawston, T.E. Transcriptional activity of the human tissue inhibitor of metalloproteinases 1 (TIMP-1) gene in fibroblasts involves elements in the promoter, exon 1 and intron 1. Biochem. J. 1997, 324 Pt 2, 611–617. [Google Scholar] [CrossRef]
- Qureshi, H.Y.; Sylvester, J.; El Mabrouk, M.; Zafarullah, M. TGF-beta-induced expression of tissue inhibitor of metalloproteinases-3 gene in chondrocytes is mediated by extracellular signal-regulated kinase pathway and Sp1 transcription factor. J. Cell. Physiol. 2005, 203, 345–352. [Google Scholar] [CrossRef]
- Stout, T.J.; Graham, H.; Buckley, D.I.; Matthews, D.J. Structures of active and latent PAI-1: A possible stabilizing role for chloride ions. Biochemistry 2000, 39, 8460–8469. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.K.; Gettins, P.G.W. High-resolution structure of the stable plasminogen activator inhibitor type-1 variant 14-1B in its proteinase-cleaved form: A new tool for detailed interaction studies and modeling. Protein Sci. 2008, 17, 1844–1849. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K. Plasminogen activator inhibitor-1 and the circadian clock in metabolic disorders. Clin. Exp. Hypertens. 2009, 31, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.R.; Riccio, A.; Andreasen, P.A.; Nielsen, L.S.; Kristensen, P.; Laiho, M.; Saksela, O.; Blasi, F.; Danø, K. Transforming growth factor-beta is a strong and fast acting positive regulator of the level of type-1 plasminogen activator inhibitor mRNA in WI-38 human lung fibroblasts. EMBO J. 1987, 6, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Keski-Oja, J.; Raghow, R.; Sawdey, M.; Loskutoff, D.J.; Postlethwaite, A.E.; Kang, A.H.; Moses, H.L. Regulation of mRNAs for type-1 plasminogen activator inhibitor, fibronectin, and type I procollagen by transforming growth factor-beta. Divergent responses in lung fibroblasts and carcinoma cells. J. Biol. Chem. 1988, 263, 3111–3115. [Google Scholar]
- Kwon, I.-S.; Kim, J.; Rhee, D.-K.; Kim, B.-O.; Pyo, S. Pneumolysin induces cellular senescence by increasing ROS production and activation of MAPK/NF-κB signal pathway in glial cells. Toxicon 2017, 129, 100–112. [Google Scholar] [CrossRef]
- You, W.; Hong, Y.; He, H.; Huang, X.; Tao, W.; Liang, X.; Zhang, Y.; Li, X. TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging 2019, 11, 3574–3584. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Jaffer, O.A.; Carter, A.B.; Sanders, P.N.; Dibbern, M.E.; Winters, C.J.; Murthy, S.; Ryan, A.J.; Rokita, A.G.; Prasad, A.M.; Zabner, J.; et al. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-β–mediated collagen production in a murine asthma model. Am. J. Respir. Cell Mol. Biol. 2015, 52, 106–115. [Google Scholar] [CrossRef]
- Jiang, Z.; Seo, J.Y.; Ha, H.; Lee, E.A.; Kim, Y.S.; Han, D.C.; Uh, S.T.; Park, C.S.; Lee, H.B. Reactive oxygen species mediate TGF-beta1-induced plasminogen activator inhibitor-1 upregulation in mesangial cells. Biochem. Biophys. Res. Commun. 2003, 309, 961–966. [Google Scholar] [CrossRef]
- Samarakoon, R.; Chitnis, S.S.; Higgins, S.P.; Higgins, C.E.; Krepinsky, J.C.; Higgins, P.J. Redox-Induced Src Kinase and Caveolin-1 Signaling in TGF-β1-Initiated SMAD2/3 Activation and PAI-1 Expression. PLoS ONE 2011, 6, e22896. [Google Scholar] [CrossRef]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.-P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell. Signal. 2018, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Hyman, M.C.; Lawrence, D.A.; Pinsky, D.J. Molecular regulation of the PAI-1 gene by hypoxia: Contributions of Egr-1, HIF-1 α, and C/EBPα. FASEB J. 2007, 21, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef]
- Fink, T.; Kazlauskas, A.; Poellinger, L.; Ebbesen, P.; Zachar, V. Identification of a tightly regulated hypoxia-response element in the promoter of human plasminogen activator inhibitor-1. Blood 2002, 99, 2077–2083. [Google Scholar] [CrossRef]
- Westerhausen, D.R.; Hopkins, W.E.; Billadello, J.J. Multiple transforming growth factor-beta-inducible elements regulate expression of the plasminogen activator inhibitor type-1 gene in Hep G2 cells. J. Biol. Chem. 1991, 266, 1092–1100. [Google Scholar]
- Dimova, E.Y.; Kietzmann, T. Metabolic, hormonal and environmental regulation of plasminogen activator inhibitor-1 (PAI-1) expression: Lessons from the liver. Thromb. Haemost. 2008, 100, 992–1006. [Google Scholar] [CrossRef]
- Sakamoto, T.; Woodcock-Mitchell, J.; Marutsuka, K.; Mitchell, J.J.; Sobel, B.E.; Fujii, S. TNF-alpha and insulin, alone and synergistically, induce plasminogen activator inhibitor-1 expression in adipocytes. Am. J. Physiol. 1999, 276, C1391–C1397. [Google Scholar] [CrossRef]
- Hou, B.; Eren, M.; Painter, C.A.; Covington, J.W.; Dixon, J.D.; Schoenhard, J.A.; Vaughan, D.E. Tumor necrosis factor alpha activates the human plasminogen activator inhibitor-1 gene through a distal nuclear factor kappaB site. J. Biol. Chem. 2004, 279, 18127–18136. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Y.; Liu, L.; Lin, C.; Liao, C.; Xin, L.; Zhong, S.; Cheng, Q.; Zhang, L. Adiponectin Inhibits TNF-α-Activated PAI-1 Expression Via the cAMP-PKA-AMPK-NF-κB Axis in Human Umbilical Vein Endothelial Cells. Cell. Physiol. Biochem. 2017, 42, 2342–2352. [Google Scholar] [CrossRef]
- Kocić, J.; Santibañez, J.F.; Krstić, A.; Mojsilović, S.; Ilić, V.; Bugarski, D. Interleukin-17 modulates myoblast cell migration by inhibiting urokinase type plasminogen activator expression through p38 mitogen-activated protein kinase. Int. J. Biochem. Cell Biol. 2013, 45, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Wiman, B.; Hamsten, A.; Green, F.; Humphries, S.; Henney, A.M. The two allele sequences of a common polymorphism in the promoter of the plasminogen activator inhibitor-1 (PAI-1) gene respond differently to interleukin-1 in HepG2 cells. J. Biol. Chem. 1993, 268, 10739–10745. [Google Scholar] [PubMed]
- Nordt, T.K.; Bode, C.; Sobel, B.E. Stimulation in vivo of expression of intra-abdominal adipose tissue plasminogen activator inhibitor Type I by proinsulin. Diabetologia 2001, 44, 1121–1124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heaton, J.H.; Nebes, V.L.; O’Dell, L.G.; Morris, S.M.; Gelehrter, T.D. Glucocorticoid and cyclic nucleotide regulation of plasminogen activator and plasminogen activator-inhibitor gene expression in primary cultures of rat hepatocytes. Mol. Endocrinol. 1989, 3, 185–192. [Google Scholar] [CrossRef]
- Tidball, J.G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef]
- Bryer, S.C.; Koh, T.J. The urokinase-type plasminogen activator receptor is not required for skeletal muscle inflammation or regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1152–R1158. [Google Scholar] [CrossRef]
- Novak, M.L.; Bryer, S.C.; Cheng, M.; Nguyen, M.-H.; Conley, K.L.; Cunningham, A.K.; Xue, B.; Sisson, T.H.; You, J.-S.; Hornberger, T.A.; et al. Macrophage-specific expression of urokinase-type plasminogen activator promotes skeletal muscle regeneration. J. Immunol. 2011, 187, 1448–1457. [Google Scholar] [CrossRef]
- Krause, M.P.; Al-Sajee, D.; D’Souza, D.M.; Rebalka, I.A.; Moradi, J.; Riddell, M.C.; Hawke, T.J. Impaired Macrophage and Satellite Cell Infiltration Occurs in a Muscle-Specific Fashion Following Injury in Diabetic Skeletal Muscle. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Hawke, T.J.; Garry, D.J. Myogenic satellite cells: Physiology to molecular biology. J. Appl. Physiol. 2001, 91, 534–551. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Kherif, S.; Dehaupas, M.; Lafuma, C.; Fardeau, M.; Alameddine, H.S. Matrix metalloproteinases MMP-2 and MMP-9 in denervated muscle and injured nerve. Neuropathol. Appl. Neurobiol. 1998, 24, 309–319. [Google Scholar] [CrossRef]
- Tamura, Y.; Kawao, N.; Shimoide, T.; Okada, K.; Matsuo, O.; Kaji, H. Role of plasminogen activator inhibitor-1 in glucocorticoid-induced muscle change in mice. J. Bone Miner. Metab. 2018, 36, 148–156. [Google Scholar] [CrossRef]
- Elfahime, E.; Mills, P.; Lafreniere, J.F.; Torrente, Y.; Tremblay, J.P. The urokinase plasminogen activator: An interesting way to improve myoblast migration following their transplantation. Exp. Cell Res. 2002, 280, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Kherif, S.; Lafuma, C.; Dehaupas, M.; Lachkar, S.; Fournier, J.G.; Verdière-Sahuqué, M.; Fardeau, M.; Alameddine, H.S. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: A study in experimentally injured and mdx muscles. Dev. Biol. 1999, 205, 158–170. [Google Scholar] [CrossRef]
- Lei, H.; Leong, D.; Smith, L.R.; Barton, E.R. Matrix metalloproteinase 13 is a new contributor to skeletal muscle regeneration and critical for myoblast migration. Am. J. Physiol. Cell Physiol. 2013, 305, C529–C538. [Google Scholar] [CrossRef]
- Lewis, M.P.; Tippett, H.L.; Sinanan, A.C.M.; Morgan, M.J.; Hunt, N.P. Gelatinase-B (Matrix Metalloproteinase-9; MMP-9) secretion is involved in the migratory phase of human and murine muscle cell cultures. J. Muscle Res. Cell Motil. 2000, 21, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Zimowska, M.; Brzoska, E.; Swierczynska, M.; Streminska, W.; Moraczewski, J. Distinct patterns of MMP-9 and MMP-2 activity in slow and fast twitch skeletal muscle regeneration in vivo. Int. J. Dev. Biol. 2008, 52, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Elfahime, E.; Torrente, Y.; Caron, N.J.; Bresolin, M.D.; Tremblay, J.P. In Vivo Migration of Transplanted Myoblasts Requires Matrix Metalloproteinase Activity. Exp. Cell Res. 2000, 258, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Lluri, G.; Jaworski, D.M. Regulation of TIMP-2, MT1-MMP, and MMP-2 expression during C2C12 differentiation. Muscle Nerve 2005, 32, 492–499. [Google Scholar] [CrossRef]
- Bedair, H.; Liu, T.T.; Kaar, J.L.; Badlani, S.; Russell, A.J.; Li, Y.; Huard, J. Matrix metalloproteinase-1 therapy improves muscle healing. J. Appl. Physiol. 2007, 102, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Kaar, J.L.; Li, Y.; Blair, H.C.; Asche, G.; Koepsel, R.R.; Huard, J.; Russell, A.J. Matrix metalloproteinase-1 treatment of muscle fibrosis. Acta Biomater. 2008, 4, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, H.; Murray, K.; Jefferson, B.S.; Li, Y. Matrix metalloproteinase-1 promotes muscle cell migration and differentiation. Am. J. Pathol. 2009, 174, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Kok, H.J.; Zhang, B.; Chung, D.; Spradlin, R.A.; Rakoczy, K.D.; Lei, H.; Boesze-Battaglia, K.; Barton, E.R. Matrix Metalloproteinase 13 from Satellite Cells is Required for Efficient Muscle Growth and Regeneration. Cell. Physiol. Biochem. 2020, 54, 333–353. [Google Scholar] [CrossRef]
- Zellweger, H.; Antonik, A. Newborn screening for Duchenne muscular dystrophy. Pediatrics 1975, 55, 30–34. [Google Scholar] [CrossRef][Green Version]
- Fibbi, G.; D’Alessio, S.; Pucci, M.; Cerletti, M.; Del Rosso, M. Growth factor-dependent proliferation and invasion of muscle satellite cells require the cell-associated fibrinolytic system. Biol. Chem. 2002, 383, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.P.; Riddell, M.C.; Hawke, T.J. Effects of type 1 diabetes mellitus on skeletal muscle: Clinical observations and physiological mechanisms. Pediatr. Diabetes 2011, 12, 345–364. [Google Scholar] [CrossRef]
- Krause, M.P.; Riddell, M.C.; Gordon, C.S.; Imam, S.A.; Cafarelli, E.; Hawke, T.J. Diabetic myopathy differs between Ins2Akita+/− and streptozotocin-induced Type 1 diabetic models. J. Appl. Physiol. 2009, 106, 1650–1659. [Google Scholar] [CrossRef]
- Monaco, C.M.F.; Hughes, M.C.; Ramos, S.V.; Varah, N.E.; Lamberz, C.; Rahman, F.A.; McGlory, C.; Tarnopolsky, M.A.; Krause, M.P.; Laham, R.; et al. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia 2018, 61, 1411–1423. [Google Scholar] [CrossRef]
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: Impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 4. [Google Scholar] [CrossRef]
- Zeitler, P.; Thiede, A.; Müller, H.L. Prospective study on plasma clotting parameters in diabetic children—No evidence for specific changes in coagulation system. Exp. Clin. Endocrinol. Diabetes 2001, 109, 146–150. [Google Scholar] [CrossRef]
- Brazionis, L.; Rowley, K.; Jenkins, A.J.; Itsiopoulos, C.; O’Dea, K. Plasminogen Activator Inhibitor-1 Activity in Type 2 Diabetes. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 786–791. [Google Scholar] [CrossRef]
- Rebalka, I.A.; Cao, A.W.; Raleigh, M.J.; Henriksbo, B.D.; Coleman, S.K.; Schertzer, J.D.; Hawke, T.J. Statin Therapy Negatively Impacts Skeletal Muscle Regeneration and Cutaneous Wound Repair in Type 1 Diabetic Mice. Front. Physiol. 2017, 8, 8. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeshita, K.; Kojima, T.; Takamatsu, J.; Saito, H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: Implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc. Res. 2005, 66, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Eren, M.; Boe, A.E.; Klyachko, E.A.; Vaughan, D.E. Role of plasminogen activator inhibitor-1 in senescence and aging. Semin. Thromb. Hemost. 2014, 40, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Bogue, M.A.; Brind, J.; Fernandez, E.; Flurkey, K.; Javors, M.; Ladiges, W.; Leeuwenburgh, C.; et al. Glycine supplementation extends lifespan of male and female mice. Aging Cell 2019, 18, e12953. [Google Scholar] [CrossRef] [PubMed]
- Diéval, J.; Nguyen, G.; Gross, S.; Delobel, J.; Kruithof, E.K. A lifelong bleeding disorder associated with a deficiency of plasminogen activator inhibitor type 1. Blood 1991, 77, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Fay, W.P.; Parker, A.C.; Condrey, L.R.; Shapiro, A.D. Human Plasminogen Activator Inhibitor-1 (PAI-1) Deficiency: Characterization of a Large Kindred with a Null Mutation in the PAI-1 Gene. Blood 1997, 90, 204–208. [Google Scholar] [CrossRef]
- Khan, S.S.; Shah, S.J.; Klyachko, E.; Baldridge, A.S.; Eren, M.; Place, A.T.; Aviv, A.; Puterman, E.; Lloyd-Jones, D.M.; Heiman, M.; et al. A null mutation in SERPINE1 protects against biological aging in humans. Sci. Adv. 2017, 3, eaao1617. [Google Scholar] [CrossRef]
- Akhmedov, D.; Berdeaux, R. The effects of obesity on skeletal muscle regeneration. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef]
- Fu, X.; Zhu, M.; Zhang, S.; Foretz, M.; Viollet, B.; Du, M. Obesity Impairs Skeletal Muscle Regeneration through Inhibition of AMPK. Diabetes 2016, 65, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Lee, J.D.; Mula, J.; Kirby, T.J.; Jackson, J.R.; Liu, F.; Yang, L.; Mendias, C.L.; Dupont-Versteegden, E.E.; McCarthy, J.J.; et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 2015, 21, 76–80. [Google Scholar] [CrossRef]
- Richard-Bulteau, H.; Serrurier, B.; Crassous, B.; Banzet, S.; Peinnequin, A.; Bigard, X.; Koulmann, N. Recovery of skeletal muscle mass after extensive injury: Positive effects of increased contractile activity. Am. J. Physiol. Cell Physiol. 2008, 294, C467–C476. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, E.M.; Mendoza, S.G.; Wang, P.; Glueck, C.J. Metformin therapy is associated with a decrease in plasma plasminogen activator inhibitor-1, lipoprotein(a), and immunoreactive insulin levels in patients with the polycystic ovary syndrome. Metabolism 1997, 46, 454–457. [Google Scholar] [CrossRef]
- Anfosso, F.; Chomiki, N.; Alessi, M.C.; Vague, P.; Juhan-Vague, I. Plasminogen activator inhibitor-1 synthesis in the human hepatoma cell line Hep G2. Metformin inhibits the stimulating effect of insulin. J. Clin. Investig. 1993, 91, 2185–2193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tan, B.K.; Adya, R.; Chen, J.; Farhatullah, S.; Heutling, D.; Mitchell, D.; Lehnert, H.; Randeva, H.S. Metformin decreases angiogenesis via NF-κB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovasc. Res. 2009, 83, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Langone, F.; Cannata, S.; Fuoco, C.; Lettieri Barbato, D.; Testa, S.; Nardozza, A.P.; Ciriolo, M.R.; Castagnoli, L.; Gargioli, C.; Cesareni, G. Metformin Protects Skeletal Muscle from Cardiotoxin Induced Degeneration. PLoS ONE 2014, 9, e114018. [Google Scholar] [CrossRef] [PubMed]
- Zagotta, I.; Dimova, E.Y.; Funcke, J.-B.; Wabitsch, M.; Kietzmann, T.; Fischer-Posovszky, P. Resveratrol suppresses PAI-1 gene expression in a human in vitro model of inflamed adipose tissue. Oxidative Med. Cell. Longev. 2013, 2013, 793525. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, H.; Kim, S.; Ha, T. Resveratrol inhibits TNF-alpha-induced changes of adipokines in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2007, 364, 972–977. [Google Scholar] [CrossRef]
- Suenaga, F.; Hatsushika, K.; Takano, S.; Ando, T.; Ohnuma, Y.; Ogawa, H.; Nakao, A. A possible link between resveratrol and TGF-β: Resveratrol induction of TGF-β expression and signaling. FEBS Lett. 2008, 582, 586–590. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Ren, W.-Y.; Zhu, L.; Hu, L.-J. Plasminogen activator inhibitor-1 regulates LPS induced inflammation in rat macrophages through autophagy activation. Sci. World J. 2014, 2014, 189168. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.M.; Prabhu, A.; Bhandary, Y.P. Curcumin alleviates IL-17A-mediated p53-PAI-1 expression in bleomycin-induced alveolar basal epithelial cells. J. Cell. Biochem. 2018, 119, 2222–2230. [Google Scholar] [CrossRef]
- Hu, Y.; Liang, H.; Du, Y.; Zhu, Y.; Wang, X. Curcumin inhibits transforming growth factor-beta activity via inhibition of Smad signaling in HK-2 cells. Am. J. Nephrol. 2010, 31, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.G.; Zhou, D.Y. Efficiency of Ginkgo biloba extract (EGb 761) in antioxidant protection against myocardial ischemia and reperfusion injury. Biochem. Mol. Biol. Int. 1995, 35, 125–134. [Google Scholar] [PubMed]
- Leiherer, A.; Stoemmer, K.; Muendlein, A.; Saely, C.H.; Kinz, E.; Brandtner, E.M.; Fraunberger, P.; Drexel, H. Quercetin Impacts Expression of Metabolism- and Obesity-Associated Genes in SGBS Adipocytes. Nutrients 2016, 8, 282. [Google Scholar] [CrossRef]
- Gaedeke, J.; Noble, N.A.; Border, W.A. Curcumin blocks multiple sites of the TGF-beta signaling cascade in renal cells. Kidney Int. 2004, 66, 112–120. [Google Scholar] [CrossRef]
- Han, T.; Zhang, G.; Yan, D.; Yang, H.; Ma, T.; Ye, Z. Modulation of plasminogen activator inhibitor-1 (PAI-1) by the naphthoquinone shikonin. Fitoterapia 2016, 113, 117–122. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, F.A.; Krause, M.P. PAI-1, the Plasminogen System, and Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 7066. https://doi.org/10.3390/ijms21197066
Rahman FA, Krause MP. PAI-1, the Plasminogen System, and Skeletal Muscle. International Journal of Molecular Sciences. 2020; 21(19):7066. https://doi.org/10.3390/ijms21197066
Chicago/Turabian StyleRahman, Fasih Ahmad, and Matthew Paul Krause. 2020. "PAI-1, the Plasminogen System, and Skeletal Muscle" International Journal of Molecular Sciences 21, no. 19: 7066. https://doi.org/10.3390/ijms21197066
APA StyleRahman, F. A., & Krause, M. P. (2020). PAI-1, the Plasminogen System, and Skeletal Muscle. International Journal of Molecular Sciences, 21(19), 7066. https://doi.org/10.3390/ijms21197066