Proteomics Analysis of Plasma Membrane Fractions of the Root, Leaf, and Flower of Rice



Abstract
1. Introduction
2. Results
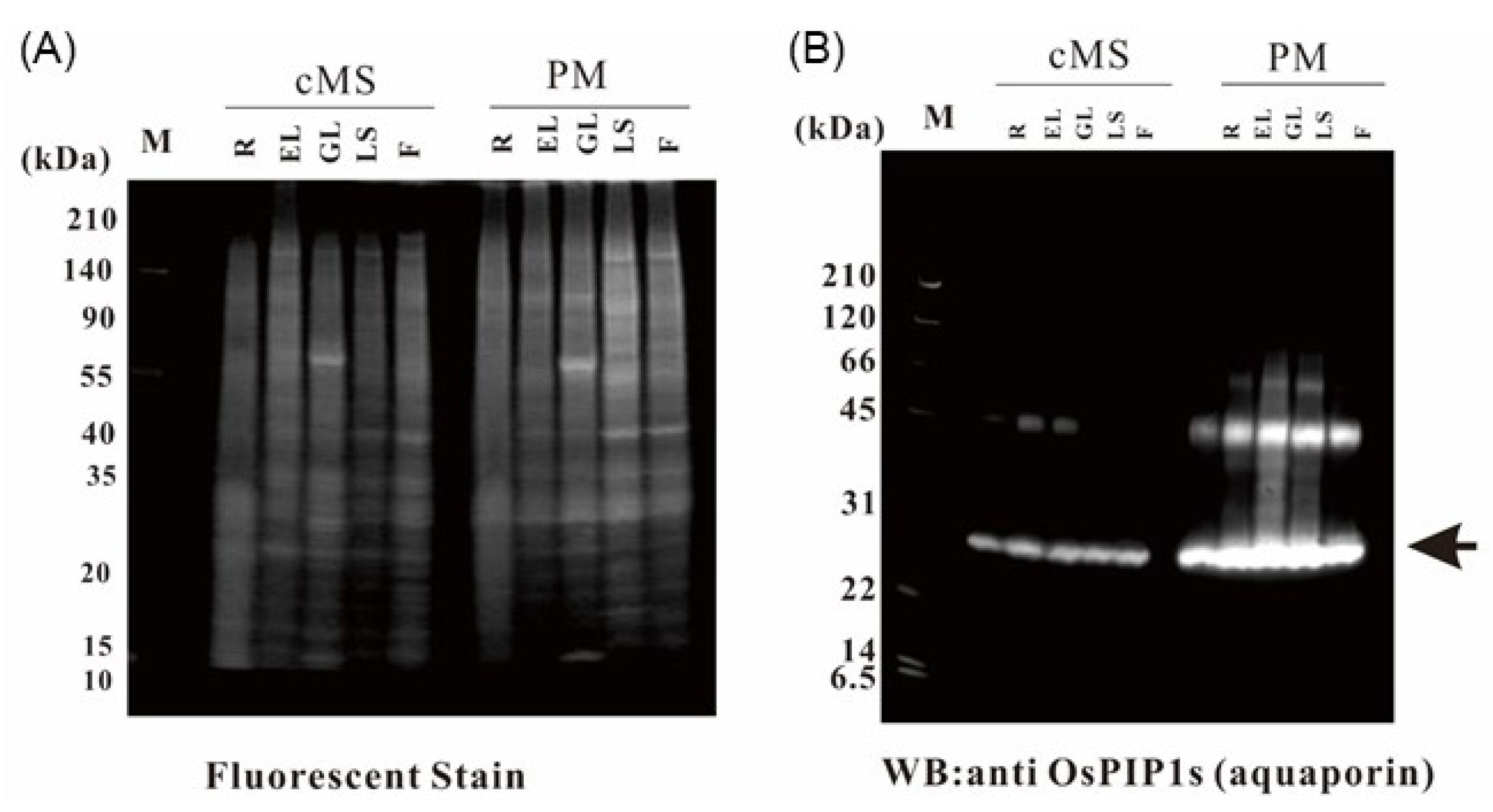
2.1. Purification of Rice Organ Plasma Membrane and Evaluation of Purity
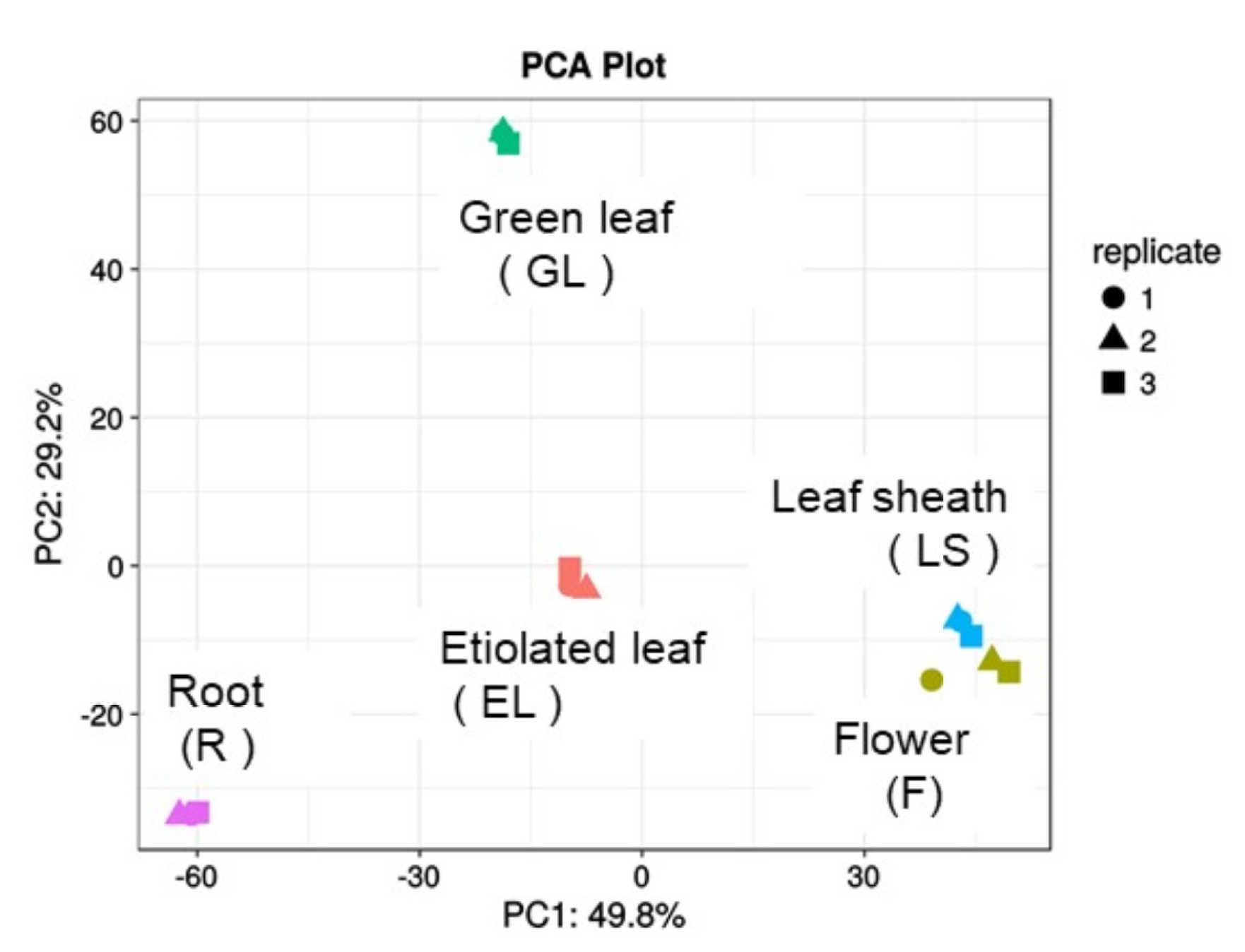
2.2. Principle Component Analysis (PCA) of Plasma Membrane Proteins from Different Organs of Rice
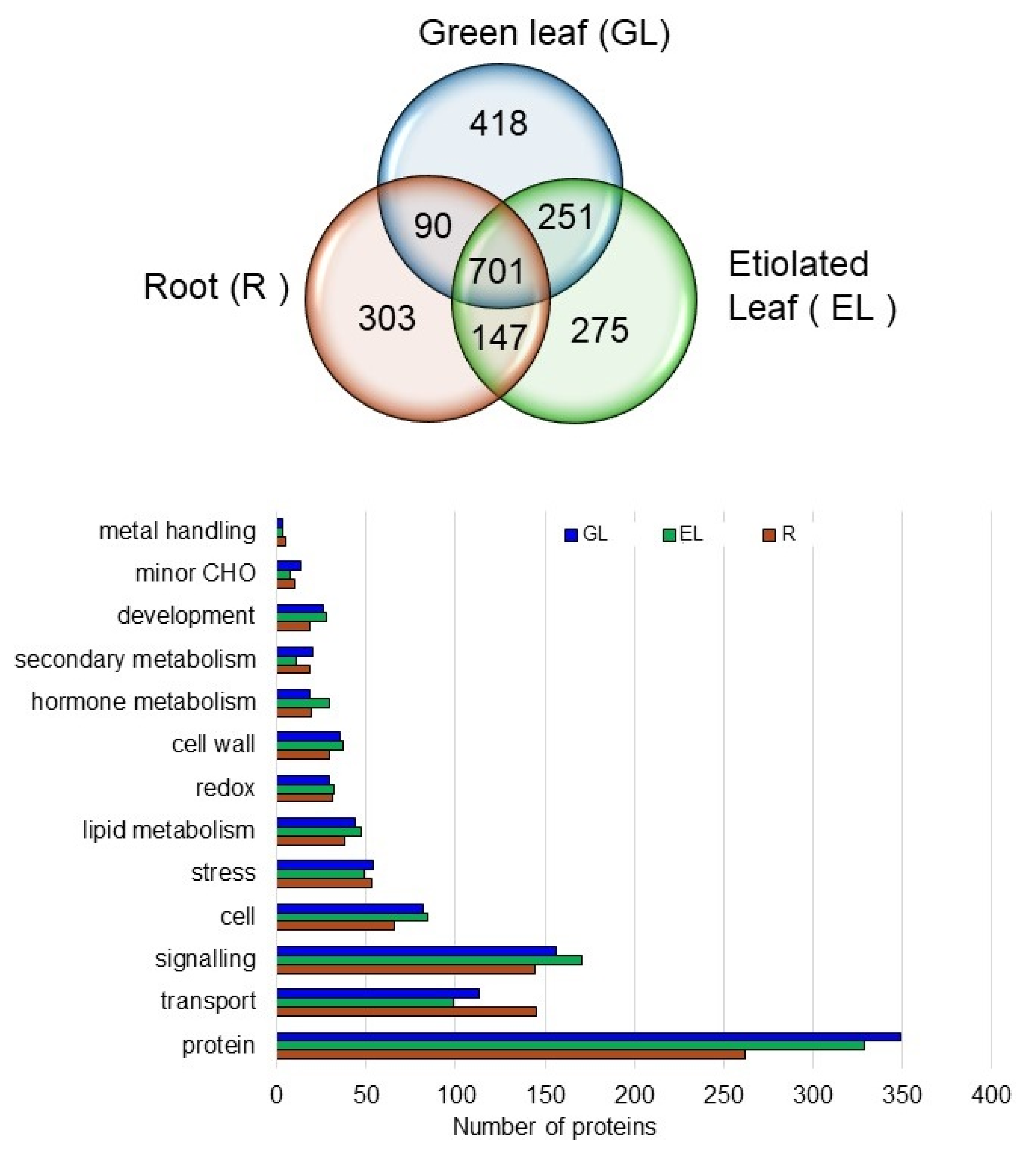
2.3. Overall Aspects of Organ-Specific and Common Accumulated Rice Proteins in the Plasma Membrane Fractions
2.4. Proteins in the Plasma Membrane Fractions in the Root, Etiolated Leaf, and Green Leaf
2.5. Specific Transport-Related Proteins in the Root Plasma Membrane
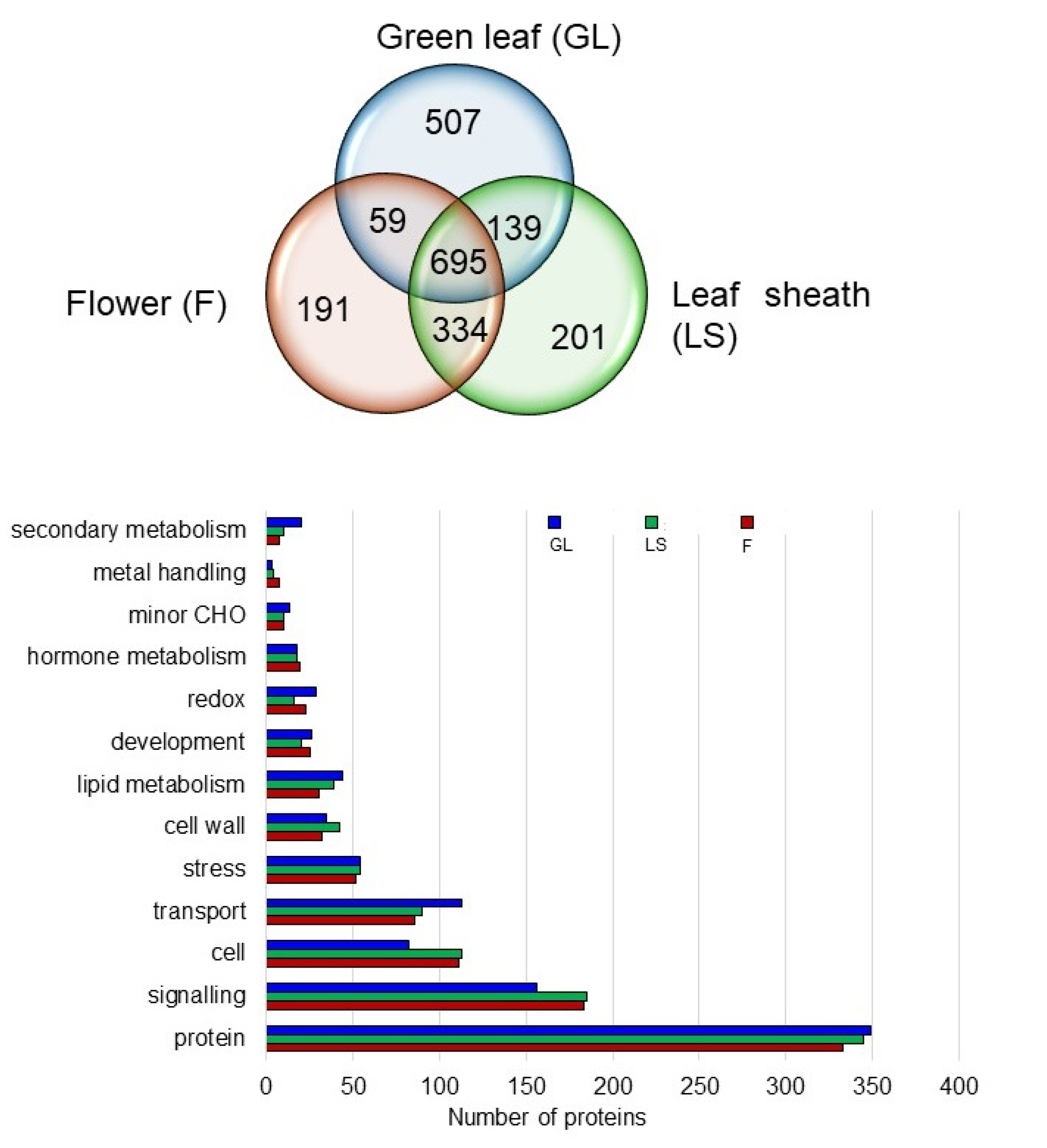
2.6. Proteins in the Plasma Membrane Fractions of the Leaf Sheath and Flower
2.7. Specific Proteins Related to Cell and Signaling in the Plasma Membrane of the Leaf Sheath and Flower
2.8. ATPase and Signal Transduction Proteins in Rice Plasma Membrane
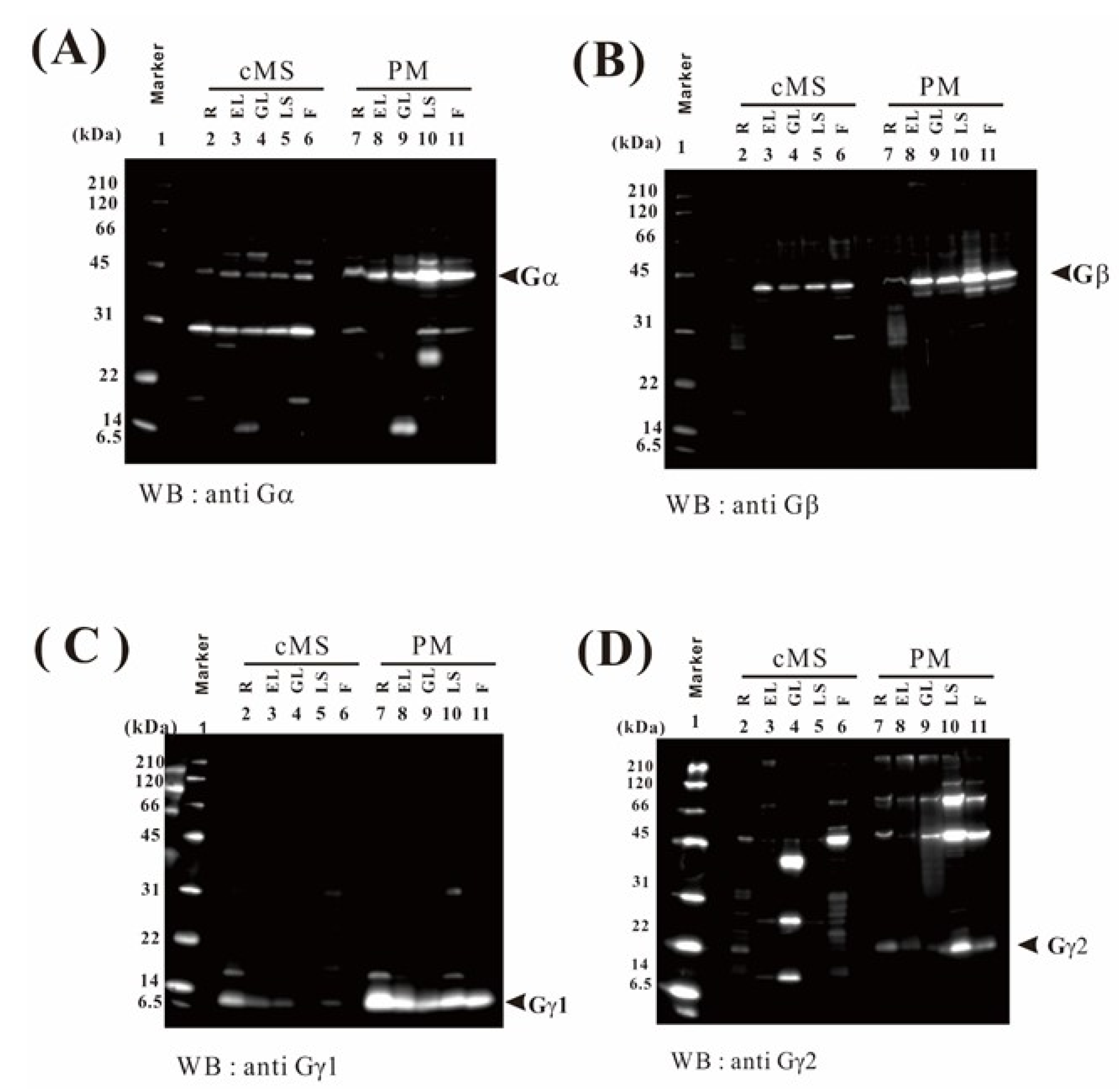
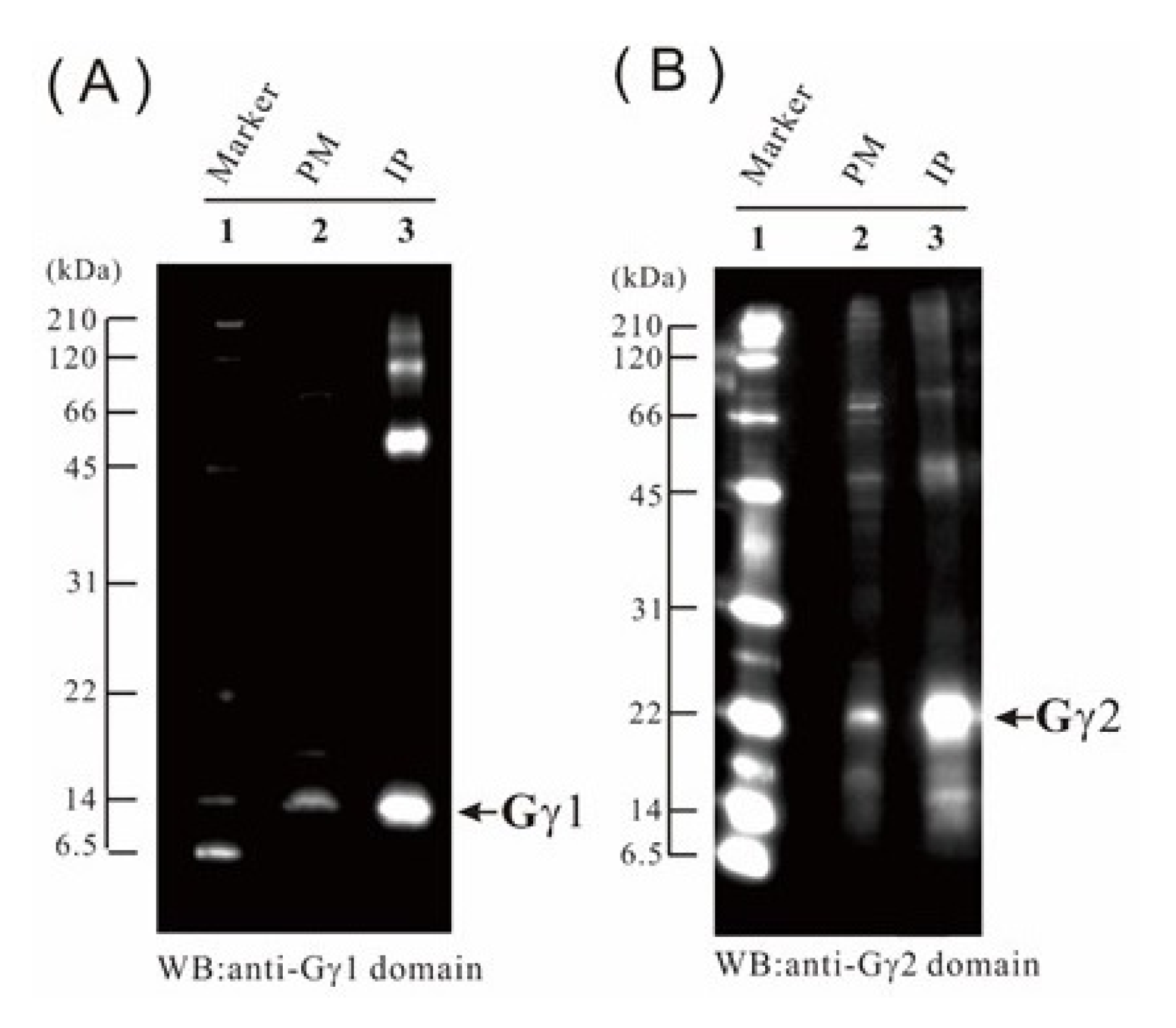
2.9. Verification of the Proteome Data by Immunoblotting
3. Discussion
4. Materials and Methods
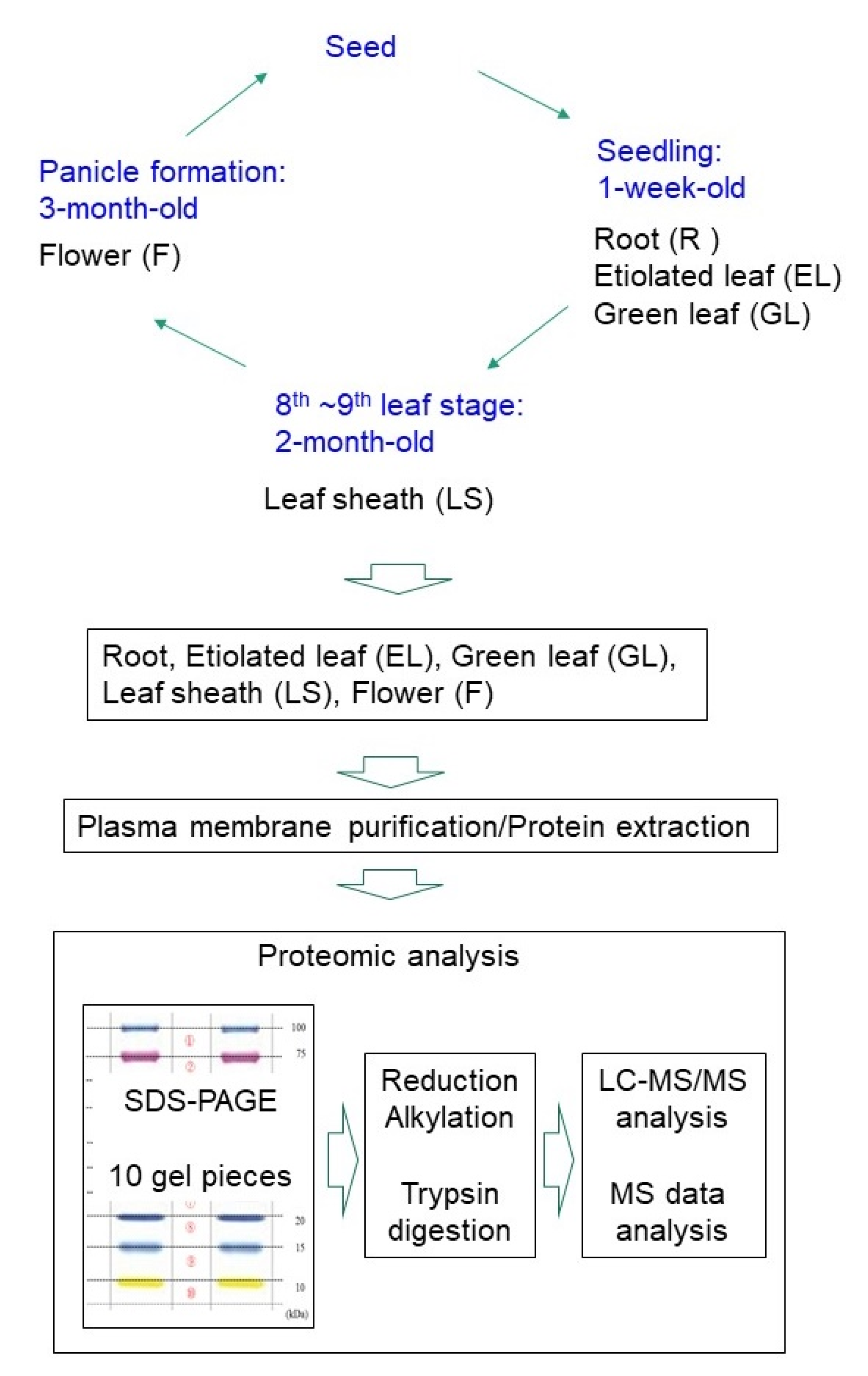
4.1. Plant Materials
4.2. Preparation of Microsomal and Plasma Membrane Fractions
4.3. SDS-PAGE
4.4. Immunoblotting
4.5. IP
4.6. Sample Preparation for LC-MS/MS
4.7. Protein Identification by NanoLC-MS/MS
4.8. MS Data Analysis
4.9. PCA
4.10. Functional Predictions
4.11. Accession Codes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IP | Immunoprecipitation |
| LC | Liquid chromatography |
| MS | Mass spectrometry |
| PAGE | Polyacrylamide gel electrophoresis |
| PCA | Principle component analysis |
References
- Alexandersson, E.; Saalbach, G.; Larsson, C.; Kjellbom, P. Arabidopsis plasma membrane proteomics identifies components of transport, signal transduction and membrane trafficking. Plant Cell Physiol. 2004, 45, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Libault, M. Function of plasma membrane microdomain-associated proteins during legume nodulation. Plant Signal Behav. 2017, 12, e1365215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sussman, M. Molecular analysis of proteins in the plant plasma membrane. Annu. Rev. Plant Biol. 1994, 45, 211–234. [Google Scholar] [CrossRef]
- Santoni, V.; Kieffer, S.; Desclaux, D.; Masson, F.; Rabilloud, T. Membrane proteomics: Use of additive main effects with multiplicative interaction model to classify plasma membrane proteins according to their solubility and electrophoretic properties. Electrophoresis 2000, 21, 3329–3344. [Google Scholar] [CrossRef]
- Wijk, K.J.V. Challenges and prospects of plant proteomics. Plant Physiol. 2001, 126, 501–508. [Google Scholar] [CrossRef]
- Reddy, V.S.; Reddy, A.S.N. Review proteomics of calcium-signaling components in plants. Phytochemistry 2004, 65, 1745–1776. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Liu, X.; Li, Z.; Lin, W. The use of comparative quantitative proteomics analysis in rice grain-filling in determining response to moderate soil drying stress. Plant Growth Regul. 2017, 82, 219–232. [Google Scholar] [CrossRef]
- Fischer, R.; Byerlee, D.; Edmeades, G. Crop Yields and Global Food Security; ACIAR: Canberra, Australia, 2014; pp. 8–11.
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Hong, W.J.; Kim, Y.J.; Chandran, A.K.N.; Jung, K.H. Infrastructures of systems biology that facilitate functional genomic study in rice. Rice (NY) 2019, 12, 15. [Google Scholar] [CrossRef]
- Kudo, T.; Terashima, S.; Takaki, Y.; Tomita, K.; Saito, M.; Kanno, M.; Yokoyama, K.; Yano, K. PlantExpress: A database integrating OryzaExpress and ArthaExpress for single-species and cross-species gene expression network analyses with microarray-based transcriptome data. Plant Cell Physiol. 2017, 58, e1. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Hongo, K.; Suwabe, K.; Shimizu, A.; Nagayama, T.; Abe, R.; Kikuchi, S.; Yamamoto, N.; Fujii, T.; Yokoyama, K.; et al. OryzaExpress: An integrated database of gene expression networks and omics annotations in rice. Plant Cell Physiol. 2011, 52, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, K.B.; Palmgren, M.G. Inventory of the superfamily of P-type ion pumps in Arabidopsis. Plant Physiol. 2001, 126, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Baxter, I.; Tchieu, J.; Sussman, M.R.; Boutry, M.; Palmgren, M.G.; Gribskov, M.; Harper, J.F.; Axelsen, K.B. Genomic comparison of P-type ATPase ion pumps in Arabidopsis and rice. Plant Physiol. 2003, 132, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Komatsu, S. Plant subcellular proteomics: Application for exploring optimal cell function in soybean. J. Proteom. 2016, 143, 4556. [Google Scholar] [CrossRef]
- Komatsu, S.; Wada, T.; Abaléa, Y.; Nouri, M.Z.; Nanjo, Y.; Nakayama, N.; Shimamura, S.; Yamamoto, T.; Nakamura, T.; Furukawa, K. Analysis of plasma membrane proteome in soybean and application to flooding stress response. J. Proteome Res. 2009, 8, 4487–4499. [Google Scholar] [CrossRef]
- Nouri, M.Z.; Komatsu, S. Comparative analysis of soybean plasma membrane proteins under osmotic stress using gel-based and LC MS/MS-based proteomics approaches. Proteomics 2010, 10, 1930–1945. [Google Scholar] [CrossRef]
- Abdallah, C.; Valot, B.; Guillier, C.; Mounier, A.; Balliau, T.; Zivy, M.; van Tuinen, D.; Renaut, J.; Wipf, D.; Dumas-Gaudot, E.; et al. The membrane proteome of Medicago truncatula roots displays qualitative and quantitative changes in response to arbuscular mycorrhizal symbiosis. J. Proteom. 2014, 108, 354–368. [Google Scholar] [CrossRef]
- Cheng, Y.; Qi, Y.; Zhu, Q.; Chen, X.; Wang, N.; Zhao, X.; Chen, H.; Cui, X.; Xu, L.; Zhang, W. New changes in the plasma-membrane-associated proteome of rice roots under salt stress. Proteomics 2009, 9, 3100–3114. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Kim, Y.J.; Kim, S.T. Identification of Msp1-induced signaling components in rice leaves by integrated proteomic and phosphoproteomic analysis. Int. J. Mol. Sci. 2019, 20, 4135. [Google Scholar] [CrossRef]
- Hopff, D.; Wienkoop, S.; Lüthje, S. The plasma membrane proteome of maize roots grown under low and high iron conditions. J. Proteom. 2013, 91, 605–618. [Google Scholar] [CrossRef]
- Takahashi, D.; Kawamura, Y.; Uemura, M. Changes of detergent-resistant plasma membrane proteins in oat and rye during cold acclimation: Association with differential freezing tolerance. J. Proteome Res. 2013, 12, 4998–5011. [Google Scholar] [CrossRef] [PubMed]
- Bernfur, K.; Larsson, O.; Larsson, C.; Gustavsson, N. Relative abundance of integral plasma membrane proteins in Arabidopsis leaf and root tissue determined by metabolic labeling and mass spectrometry. PLoS ONE 2013, 8, e71206. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.; Chiapello, M.; Montero, H.; Gehrig, P.; Grossmann, J.; O’Holleran, K.; Hartken, D.; Walters, F.; Yang, S.Y.; Hillmer, S.; et al. A rice Serine/Threonine receptor-like kinase regulates arbuscular mycorrhizal symbiosis at the peri-arbuscular membrane. Nat. Commun. 2018, 9, 4677. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Yang, N.; Pu, H.; Wang, T. Quantitative proteomics and cytology of rice pollen sterol-rich membrane domains reveals pre-established cell polarity cues in mature pollen. J. Proteome Res. 2018, 17, 1532–1546. [Google Scholar] [CrossRef] [PubMed]
- Falhof, J.; Pedersen, J.T.; Fuglsang, A.T.; Palmgren, M. Plasma membrane H+-ATPase regulation in the center of plant physiology. Mol. Plant 2016, 9, 323–337. [Google Scholar] [CrossRef]
- Gevaudant, F.; Duby, G.; von Stedingk, E.; Zhao, R.; Morsomme, P.; Boutry, M. Expression of a constitutively activated plasma membrane H+-ATPase alters plant development and increases salt tolerance. Plant Physiol. 2007, 144, 1763–1776. [Google Scholar] [CrossRef]
- Merlot, S.; Leonhardt, N.; Fenzi, F.; Valon, C.; Costa, M.; Piette, L.; Vavasseur, A.; Genty, B.; Boivin, K.; Müller, A.; et al. Constitutive activation of a plasma membrane H(+)-ATPase prevents abscisic acid-mediated stomatal closure. EMBO J. 2007, 26, 3216–3226. [Google Scholar] [CrossRef]
- Johanson, U.; Gustavsson, S. A new subfamily of major intrinsic proteins in plants. Mol. Biol. Evol. 2002, 19, 456–461. [Google Scholar] [CrossRef]
- Quigley, F.; Rosenberg, J.M.; Shachar-Hill, Y.; Bohnert, H.J. From genome to function: The Arabidopsis aquaporins. Genome Biol. 2002, 3, research0001.1. [Google Scholar]
- Bellati, J.; Champeyroux, C.; Hem, S.; Rofidal, V.; Krouk, G.; Maurel, C.; Santoni, V. Novel aquaporin regulatory mechanisms revealed by interactomics. Mol. Cell. Proteom. 2016, 15, 3473–3487. [Google Scholar] [CrossRef]
- Urano, D.; Chen, J.-G.; Botella, J.R.; Jones, A.M. Heterotrimeric G protein signalling in the plant kingdom. Open Biol. 2013, 3, 120–186. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Tsubouchi, H.; Iwasaki, Y.; Asahi, T. Molecular cloning and characterization of a cDNA for the α subunit of a G protein from rice. Plant Cell Physiol. 1995, 36, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Iwasaki, Y.; Asahi, T. Molecular cloning and characterization of a cDNA for the β subunit of a G protein from rice. Plant Cell Physiol. 1996, 37, 223–228. [Google Scholar] [CrossRef]
- Trusov, Y.; Chakravorty, D.; Botella, J.R. Diversity of heterotrimeric G-protein γ subunits in plants. BMC Res. Notes 2012, 5, 608. [Google Scholar] [CrossRef]
- Sun, S.; Wang, L.; Mao, H.; Shao, L.; Li, X.; Xiao, J.; Ouyang, Y.; Zhang, Q. A G-protein pathway determines grain size in rice. Nat. Commun. 2018, 9, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Qian, Q.; Wu, K.; Lou, J.; Wang, S.; Zhang, C.; Ma, Y.; Lie, Q.; Huang, X.; Yuan, Q.; et al. Heterotrimeric G proteins regulate nitrogen-use efficiency in rice. Nat. Genet. 2014, 46, 652–656. [Google Scholar] [CrossRef]
- Botella, J.R. Can heterotrimeric G proteins help to feed the world? Trends Plant Sci. 2012, 17, 563–568. [Google Scholar] [CrossRef]
- Kato, C.; Mizutani, T.; Tamaki, H.; Kumagai, H.; Kamiya, T.; Hirobe, A.; Fujisawa, Y.; Kato, H.; Iwasaki, Y. Characterization of heterotrimeric G protein complexes in rice plasma membrane. Plant J. 2004, 38, 320–331. [Google Scholar] [CrossRef]
- Abe, Y.; Matsusita, K.; Komatsu, S.; Iwasaki, Y. Identification of heterotrimeric G protein α and β subunits in rice. Protein Pept. Lett. 2012, 19, 277–281. [Google Scholar] [CrossRef]
- Matsuta, S.; Nishiyama, A.; Chaya, G.; Itoh, T.; Miura, K.; Iwasaki, Y. Characterization of heterotrimeric G protein γ4 subunit in rice. Int. J. Mol. Sci. 2018, 19, 3596. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsuta, S.; Chaya, G.; Itoh, T.; Miura, K.; Iwasaki, Y. Identification of heterotrimeric G protein γ3 subunit in rice plasma membrane. Int. J. Mol. Sci. 2018, 19, 3591. [Google Scholar] [CrossRef]
- Yoshida, S.; Uemura, M.; Niki, T.; Sakai, A.; Gusta, L.V. Partition of membrane particles in aqueous two-polymer phase system and its partial use for purification of plasma membranes from plants. Plant Physiol. 1983, 72, 105–114. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Kato, T.; Kaidoh, T.; Ishikawa, A.; Asahi, T. Characterization of the putative α subunit of a heterotrimeric G protein in rice. Plant Mol. Biol. 1997, 34, 563–572. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass, accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Usadel, B.; Nagel, A.; Thimm, O.; Redestig, H.; Blaesing, O.E.; Palacios-Rojas, N.; Selbig, J.; Hannemann, J.; Piques, M.C.; Steinhauser, D.; et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 2005, 138, 1195–1204. [Google Scholar] [CrossRef]
- UniProtKB. Available online: resultshttps://www.uniprot.org/uniprot/ (accessed on 23 September 2020).
- Vizcaíno, J.A.; Côté, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 2013, 41, 1063–1069. [Google Scholar] [CrossRef]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, 1107–1111. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MapMan Bin Code | MapMan Description | R/EL/GL | R/EL | R/GL | EL/GL | R | EL | GL |
|---|---|---|---|---|---|---|---|---|
| 34.1 | p- and v-ATPases | 15 | 0 | 2 | 2 | 5 | 2 | 2 |
| 34.2 | Sugars | 3 | 2 | 2 | 1 | 3 | 1 | 3 |
| 34.3 | H+ transporting pyrophosphatase/amino acids | 3 | 1 | 3 | 0 | 3 | 3 | 3 |
| 34.4 | Nitrate | 0 | 1 | 0 | 0 | 2 | 0 | 2 |
| 34.5 | Ammonium | 1 | 0 | 0 | 0 | 4 | 0 | 0 |
| 34.6 | Sulfate | 0 | 0 | 0 | 0 | 3 | 2 | 2 |
| 34.7 | Phosphate | 0 | 0 | 1 | 1 | 2 | 1 | 1 |
| 34.8 | Transport. Metabolite transporters at the envelope membrane | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| 34.9 | Metabolite transporters at the mitochondrial membrane | 0 | 2 | 0 | 0 | 0 | 0 | 1 |
| 34.12 | Metal | 0 | 2 | 0 | 0 | 6 | 0 | 1 |
| 34.13 | Peptides and oligopeptides | 2 | 1 | 1 | 1 | 6 | 0 | 2 |
| 34.14 | Unspecified cations | 3 | 0 | 0 | 0 | 3 | 0 | 1 |
| 34.15 | Potassium | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| 34.16 | ABC transporters and multidrug resistance systems | 7 | 5 | 5 | 2 | 7 | 5 | 5 |
| 34.18 | Unspecified anions | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| 34.19 | Major intrinsic proteins (PIP etc.). | 7 | 0 | 2 | 3 | 7 | 1 | 1 |
| 34.21 | Calcium | 0 | 2 | 0 | 1 | 0 | 0 | 3 |
| 34.99 | Misc. | 8 | 1 | 1 | 2 | 5 | 4 | 1 |
| Subtotal | □ | 50 | 18 | 18 | 14 | 59 | 19 | 32 |
| MapMan Bin Code | MapMan Description | F/LS/GL | F/LS | F/GL | LS/GL | F | LS | GL |
|---|---|---|---|---|---|---|---|---|
| 31.1 | cell. organization | 24 | 23 | 0 | 2 | 11 | 6 | 15 |
| 31.2 | cell.division | 3 | 1 | 1 | 0 | 0 | 2 | 0 |
| 31.3 | cell.cycle | 2 | 2 | 0 | 1 | 1 | 1 | 1 |
| 31.4 | cell.vesicle transport | 16 | 17 | 2 | 5 | 8 | 8 | 10 |
| Subtotal | □ | 45 | 43 | 3 | 8 | 20 | 17 | 26 |
| MapMan Bin Code | MapMan Description | F/LS/GL | F/LS | F/GL | LS/GL | F | LS | GL |
|---|---|---|---|---|---|---|---|---|
| 30.1 | signaling. in sugar and nutrient physiology | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 30.2 | receptor kinases | 36 | 24 | 2 | 6 | 16 | 11 | 27 |
| 30.3 | calcium | 17 | 9 | 1 | 1 | 2 | 4 | 5 |
| 30.4 | phosphoinositide | 5 | 0 | 0 | 0 | 0 | 3 | 2 |
| 30.5 | G-proteins | 33 | 10 | 1 | 3 | 8 | 6 | 2 |
| 30.6 | MAP kinases | 0 | 2 | 0 | 0 | 3 | 1 | 1 |
| 30.7 | 14-3-3 proteins | 6 | 0 | 0 | 0 | 1 | 0 | 0 |
| 30.9 | lipids | 0 | 0 | 0 | 2 | 1 | 0 | 0 |
| 30.11 | light | 2 | 1 | 0 | 0 | 2 | 3 | 4 |
| 30.99 | unspecified | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Subtotal | □ | 99 | 47 | 4 | 12 | 33 | 28 | 42 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwasaki, Y.; Itoh, T.; Hagi, Y.; Matsuta, S.; Nishiyama, A.; Chaya, G.; Kobayashi, Y.; Miura, K.; Komatsu, S. Proteomics Analysis of Plasma Membrane Fractions of the Root, Leaf, and Flower of Rice. Int. J. Mol. Sci. 2020, 21, 6988. https://doi.org/10.3390/ijms21196988
Iwasaki Y, Itoh T, Hagi Y, Matsuta S, Nishiyama A, Chaya G, Kobayashi Y, Miura K, Komatsu S. Proteomics Analysis of Plasma Membrane Fractions of the Root, Leaf, and Flower of Rice. International Journal of Molecular Sciences. 2020; 21(19):6988. https://doi.org/10.3390/ijms21196988
Chicago/Turabian StyleIwasaki, Yukimoto, Takafumi Itoh, Yusuke Hagi, Sakura Matsuta, Aki Nishiyama, Genki Chaya, Yuki Kobayashi, Kotaro Miura, and Setsuko Komatsu. 2020. "Proteomics Analysis of Plasma Membrane Fractions of the Root, Leaf, and Flower of Rice" International Journal of Molecular Sciences 21, no. 19: 6988. https://doi.org/10.3390/ijms21196988
APA StyleIwasaki, Y., Itoh, T., Hagi, Y., Matsuta, S., Nishiyama, A., Chaya, G., Kobayashi, Y., Miura, K., & Komatsu, S. (2020). Proteomics Analysis of Plasma Membrane Fractions of the Root, Leaf, and Flower of Rice. International Journal of Molecular Sciences, 21(19), 6988. https://doi.org/10.3390/ijms21196988