New Factors Enhancing the Reactivity of Cysteines in Molten Globule-Like Structures


Abstract
:1. Introduction
2. Results
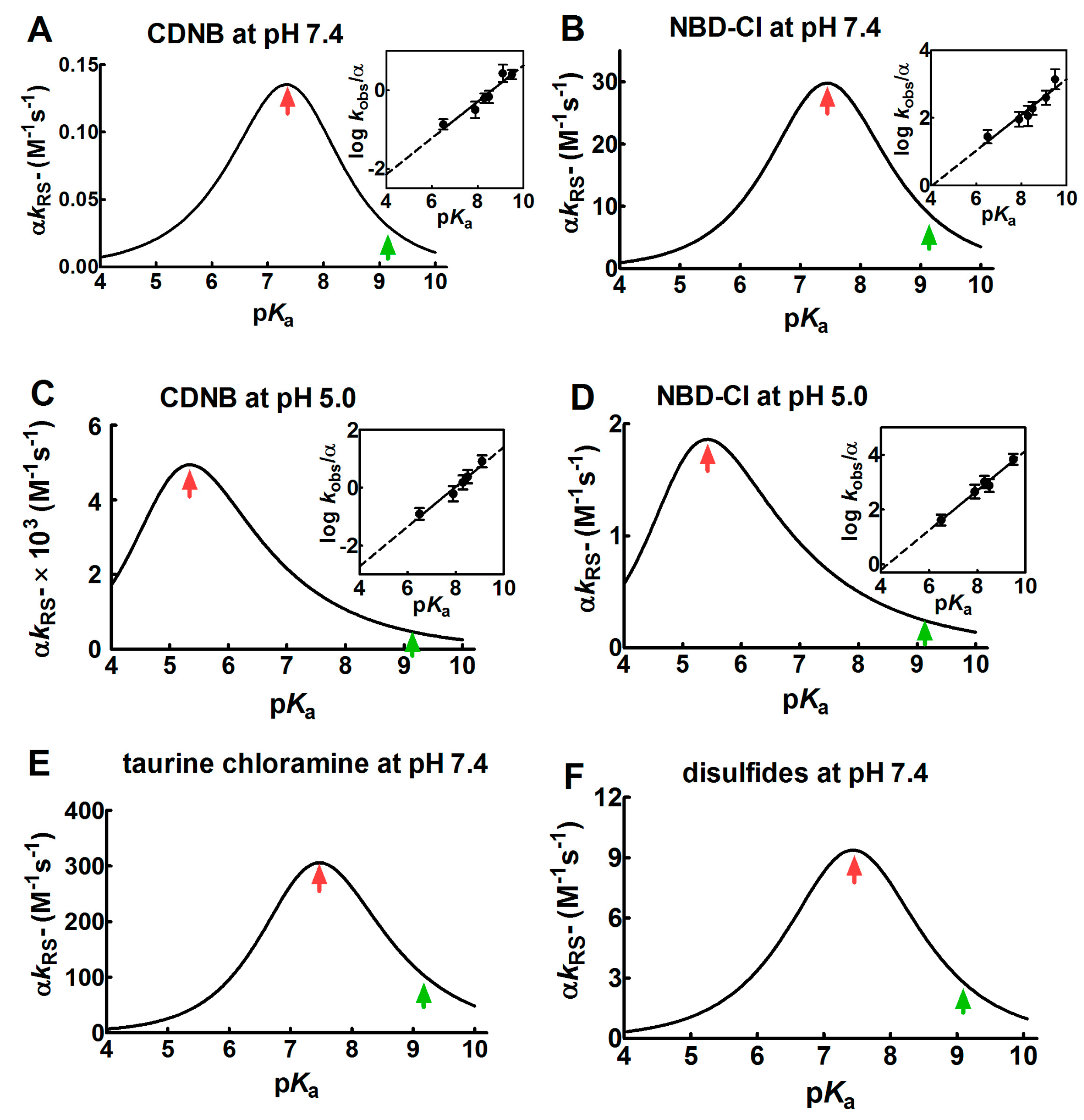
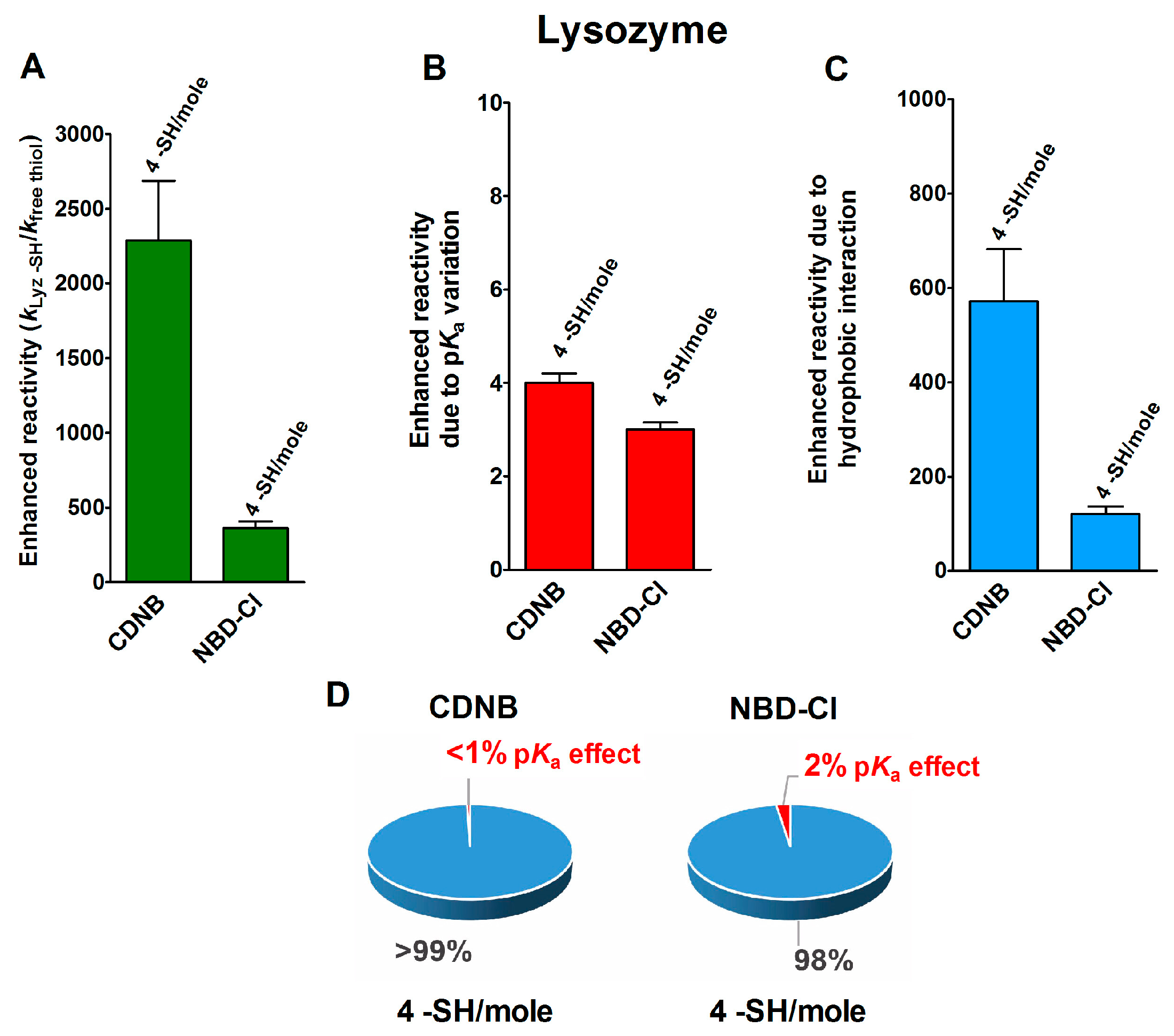
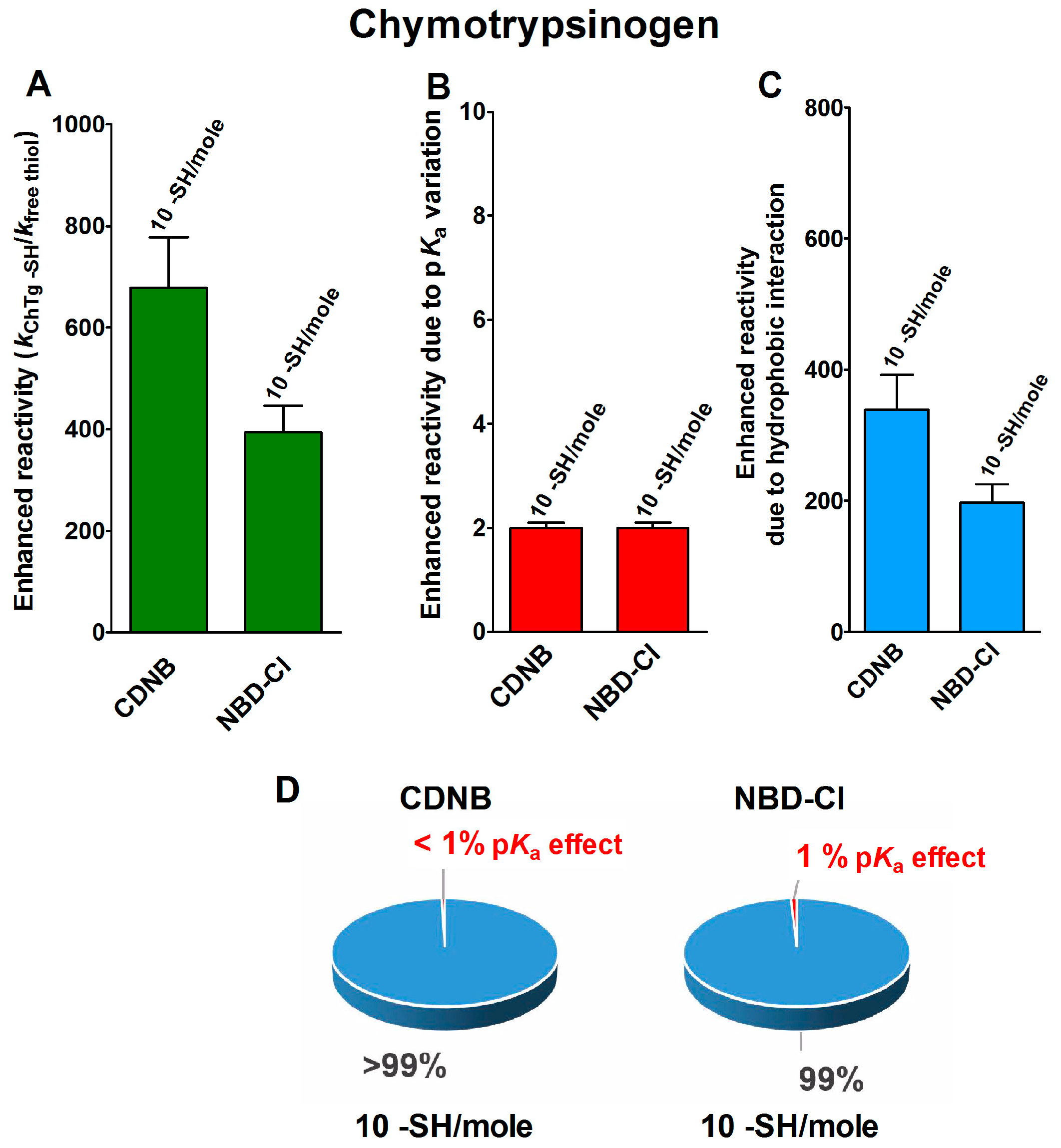
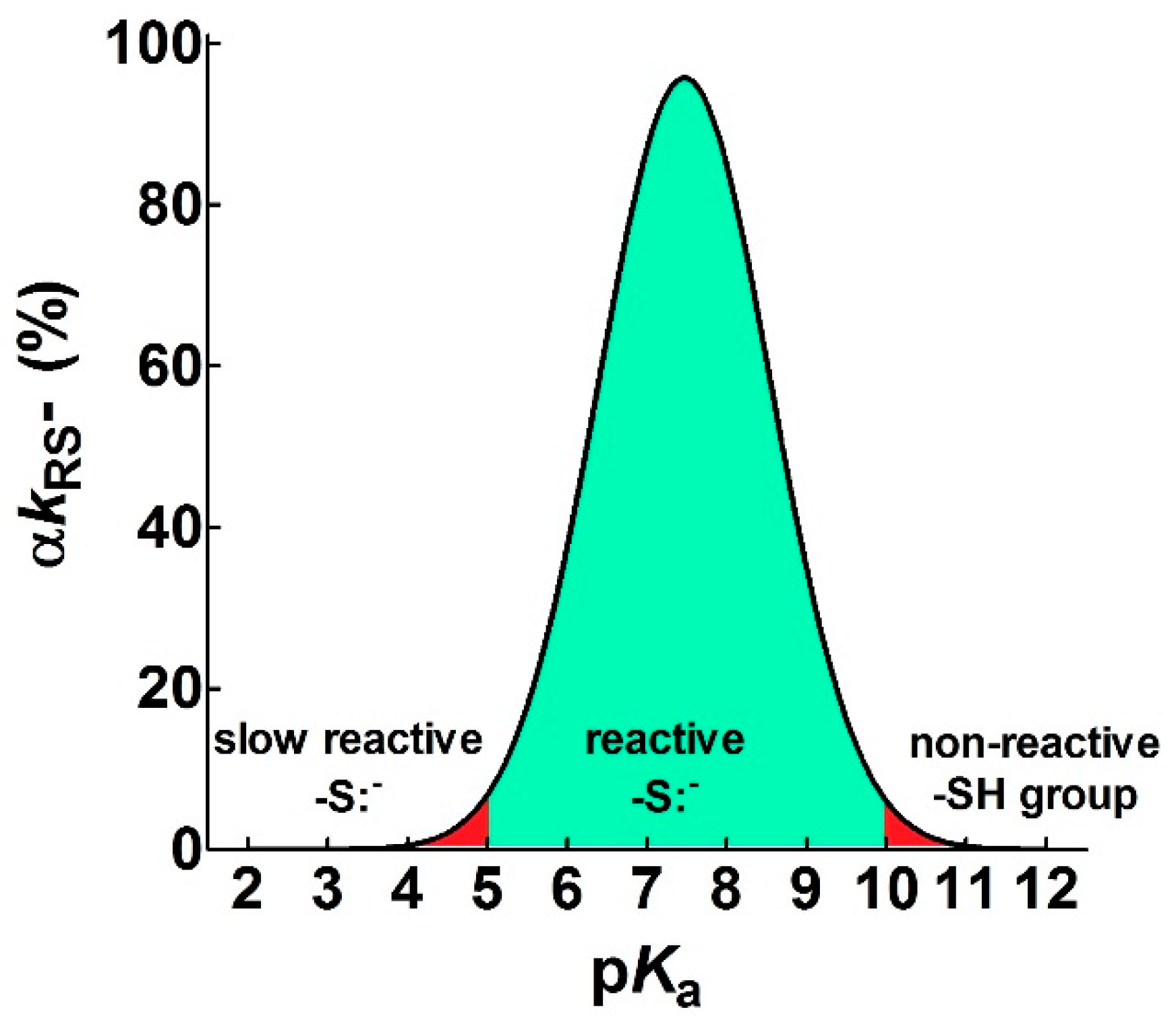
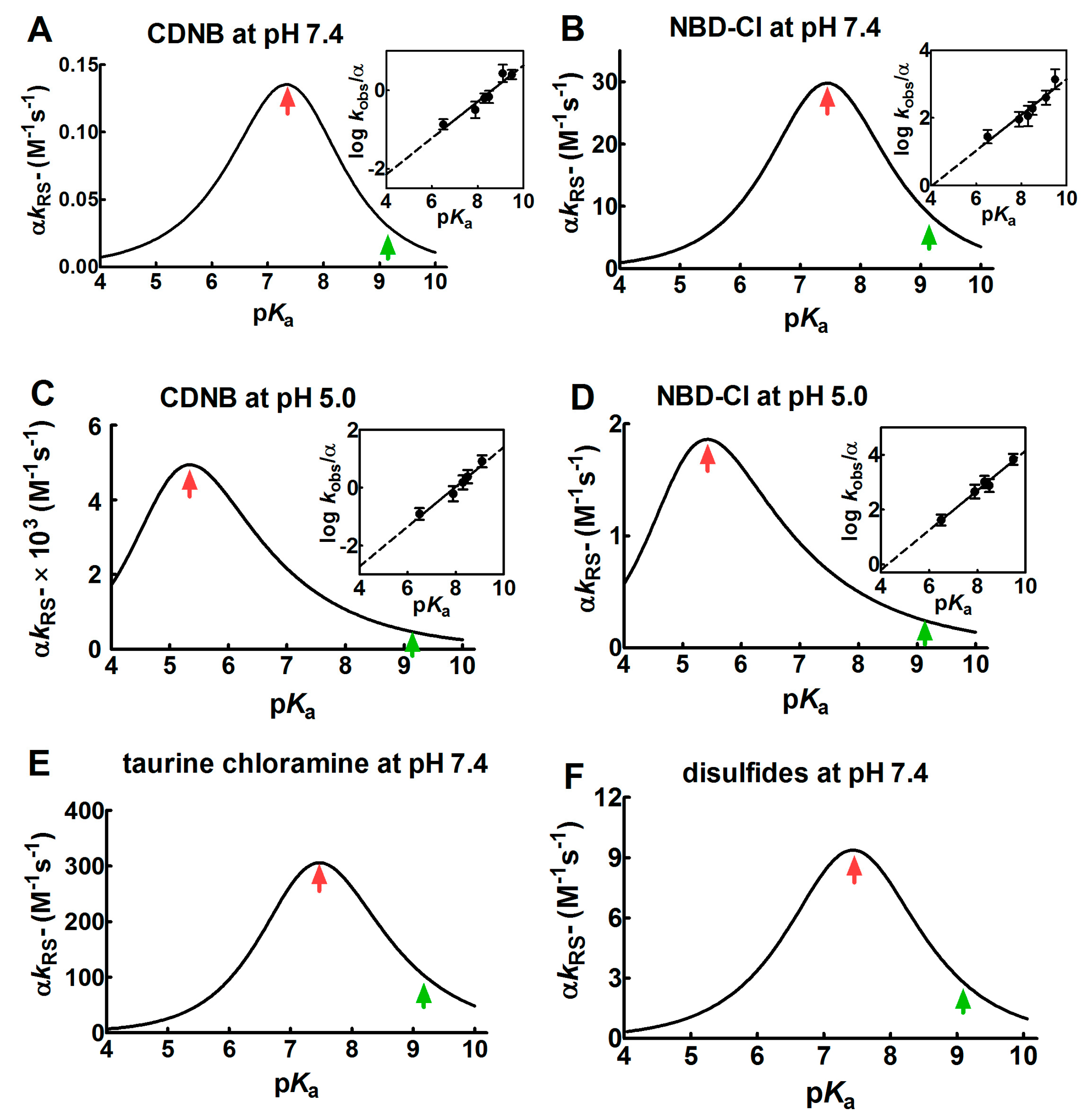
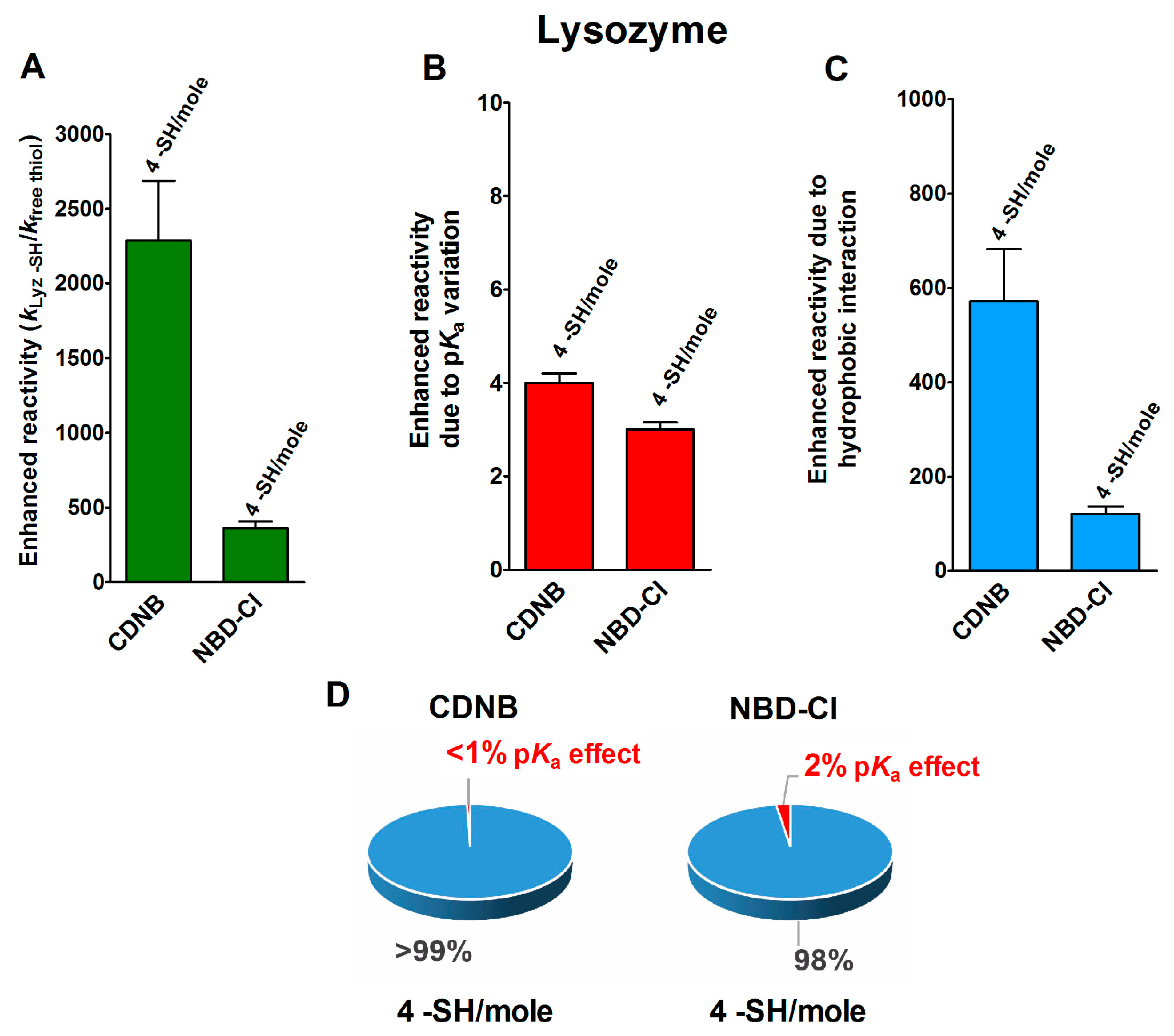
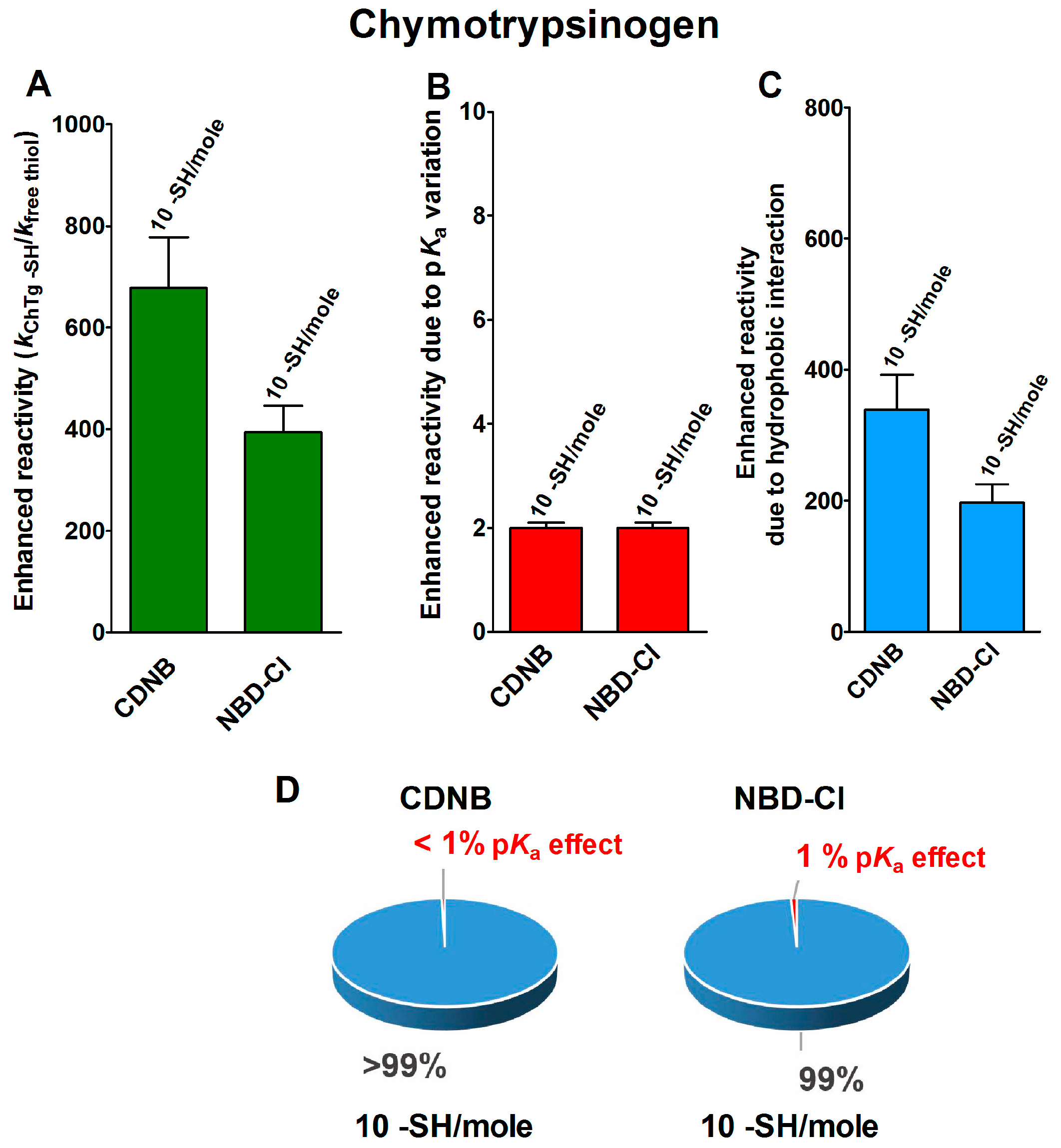
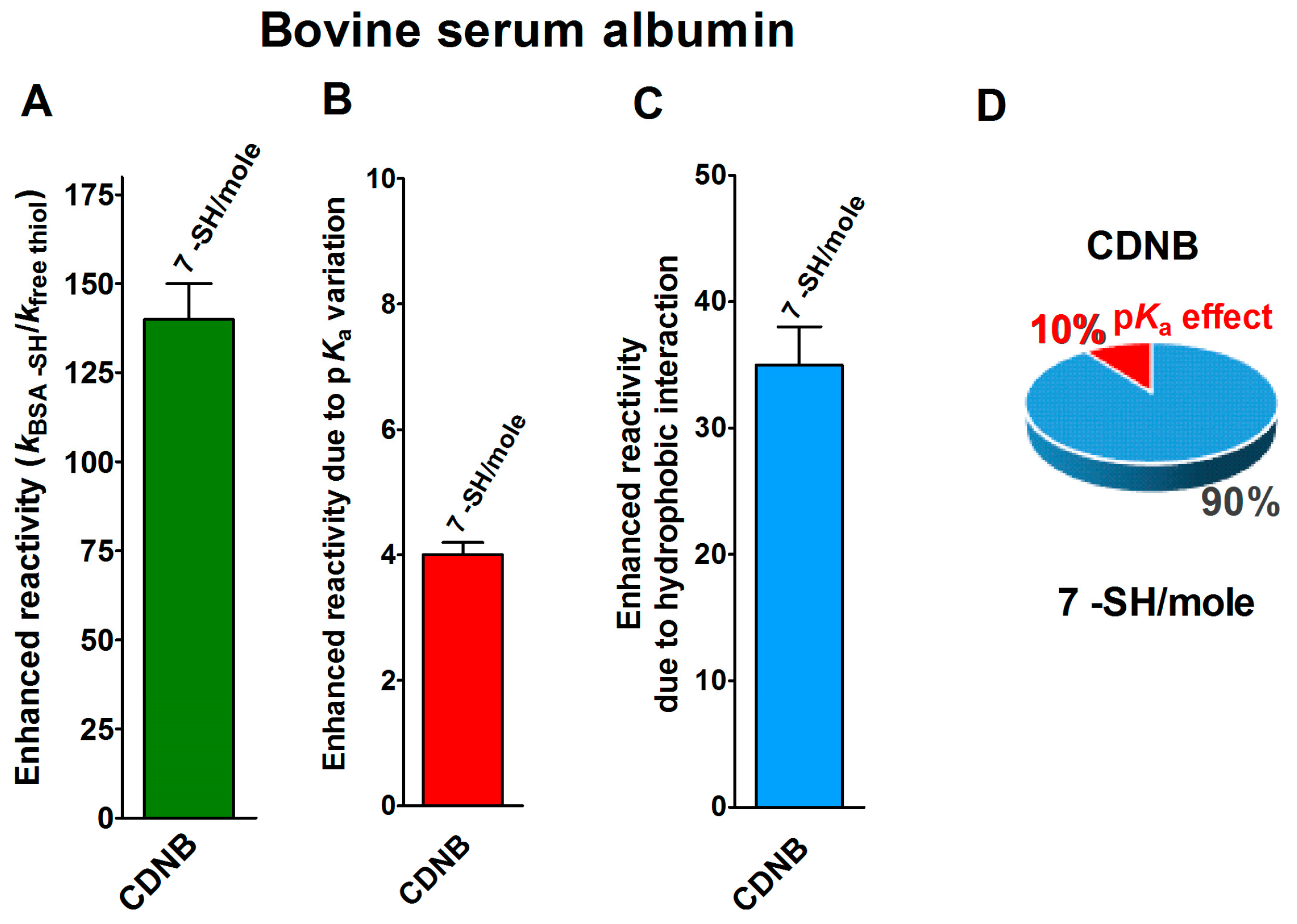
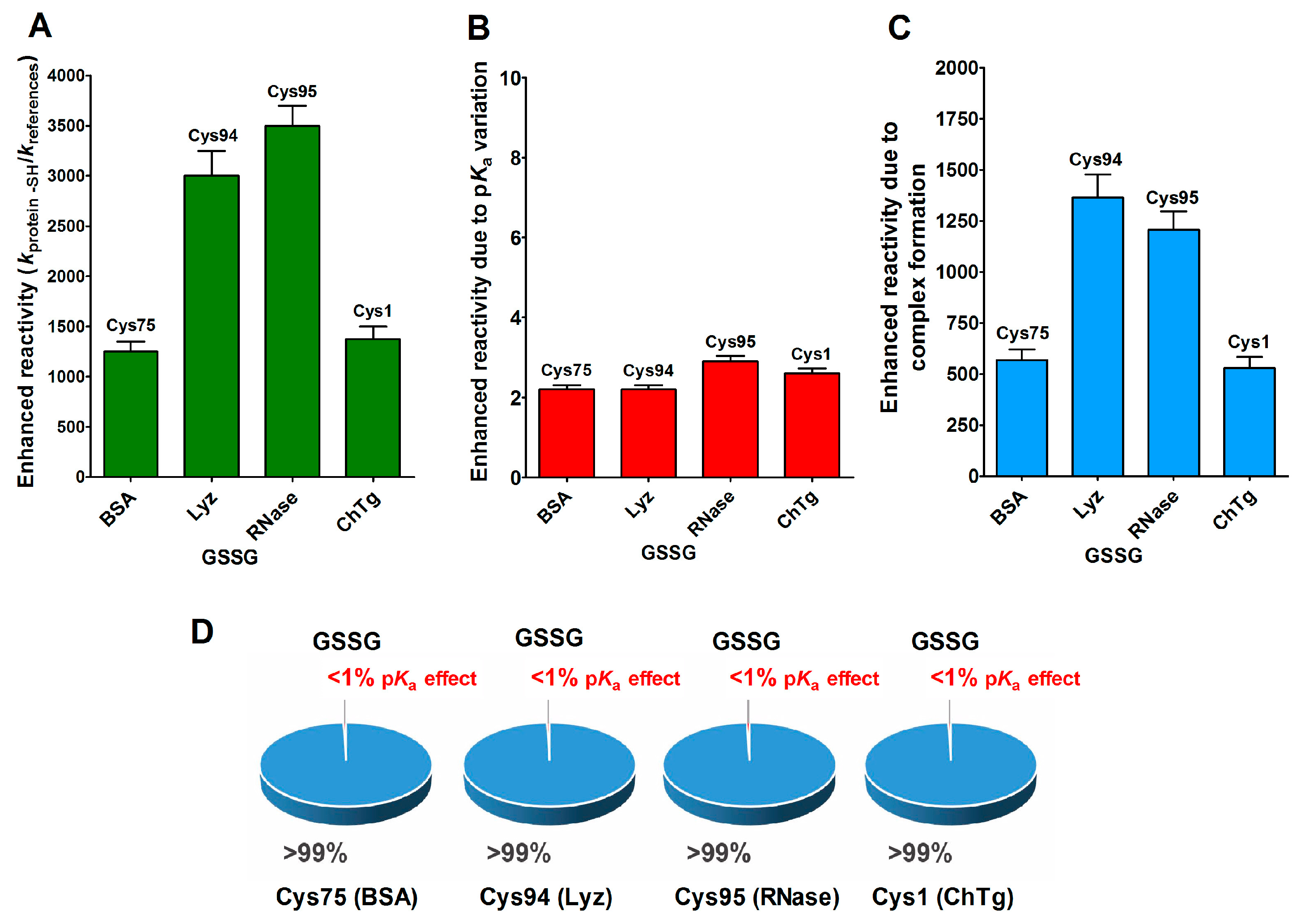
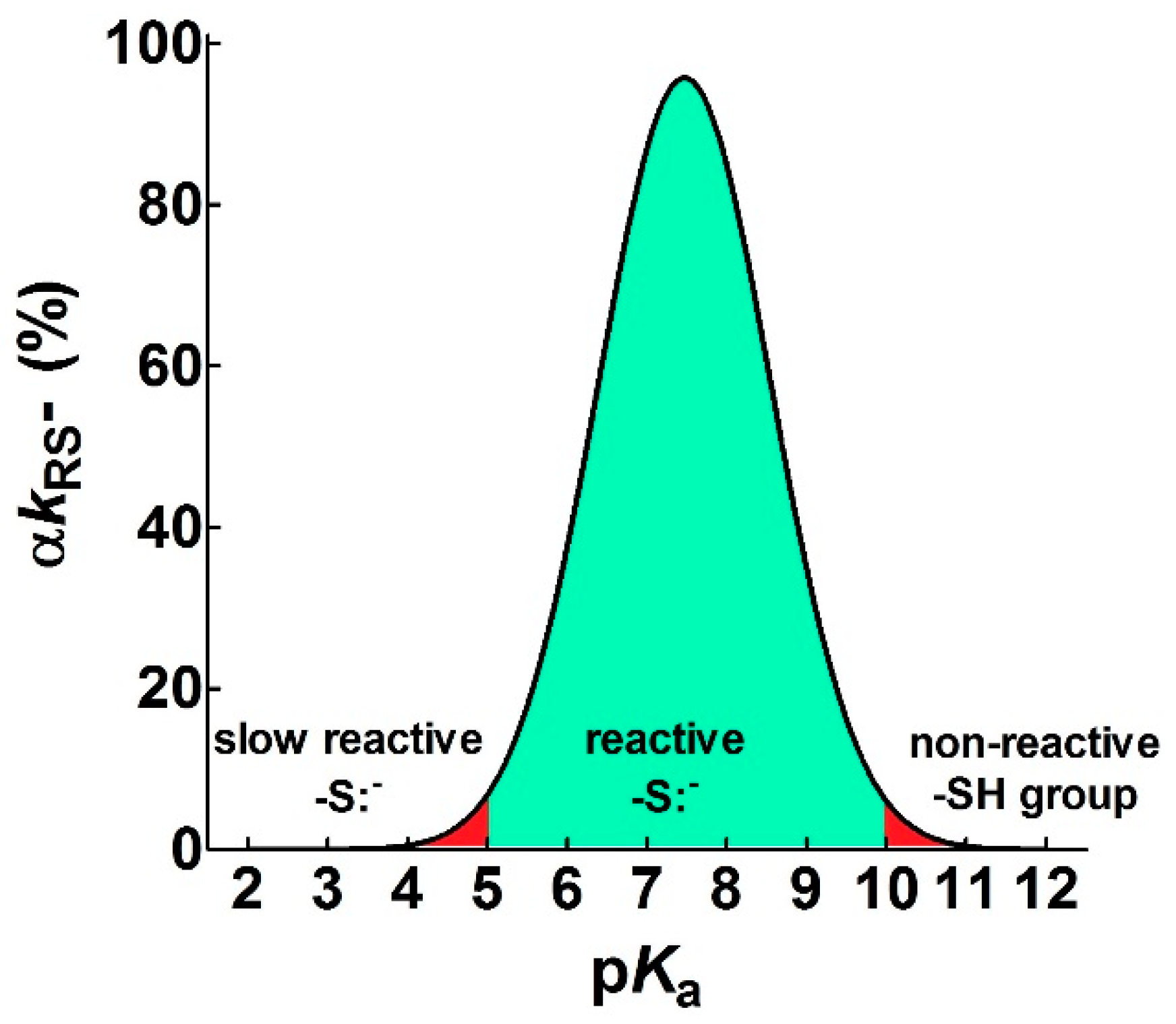
2.1. Poor Enhancement of the Cysteine Reactivity Is Due to pKa Perturbations
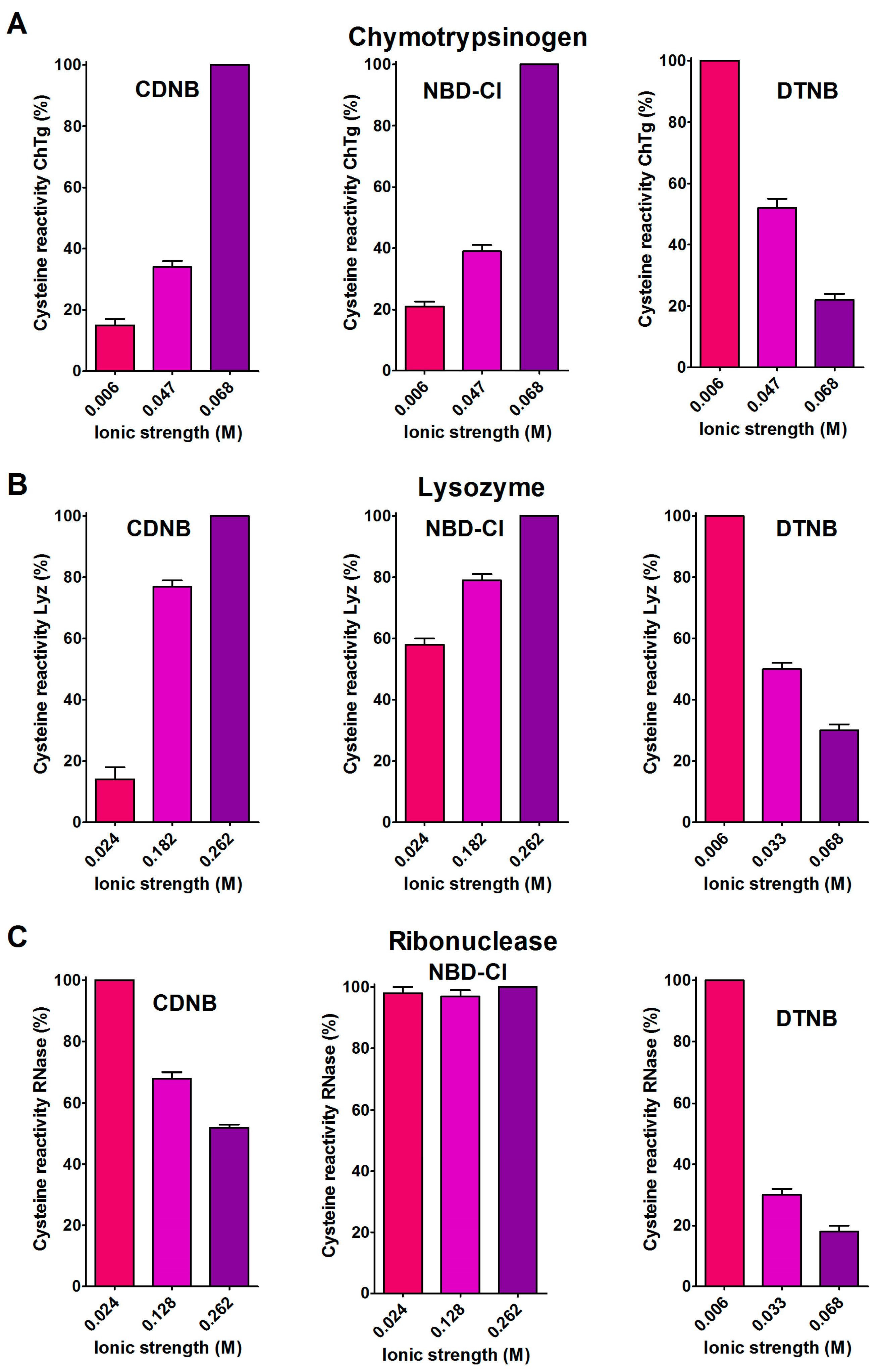
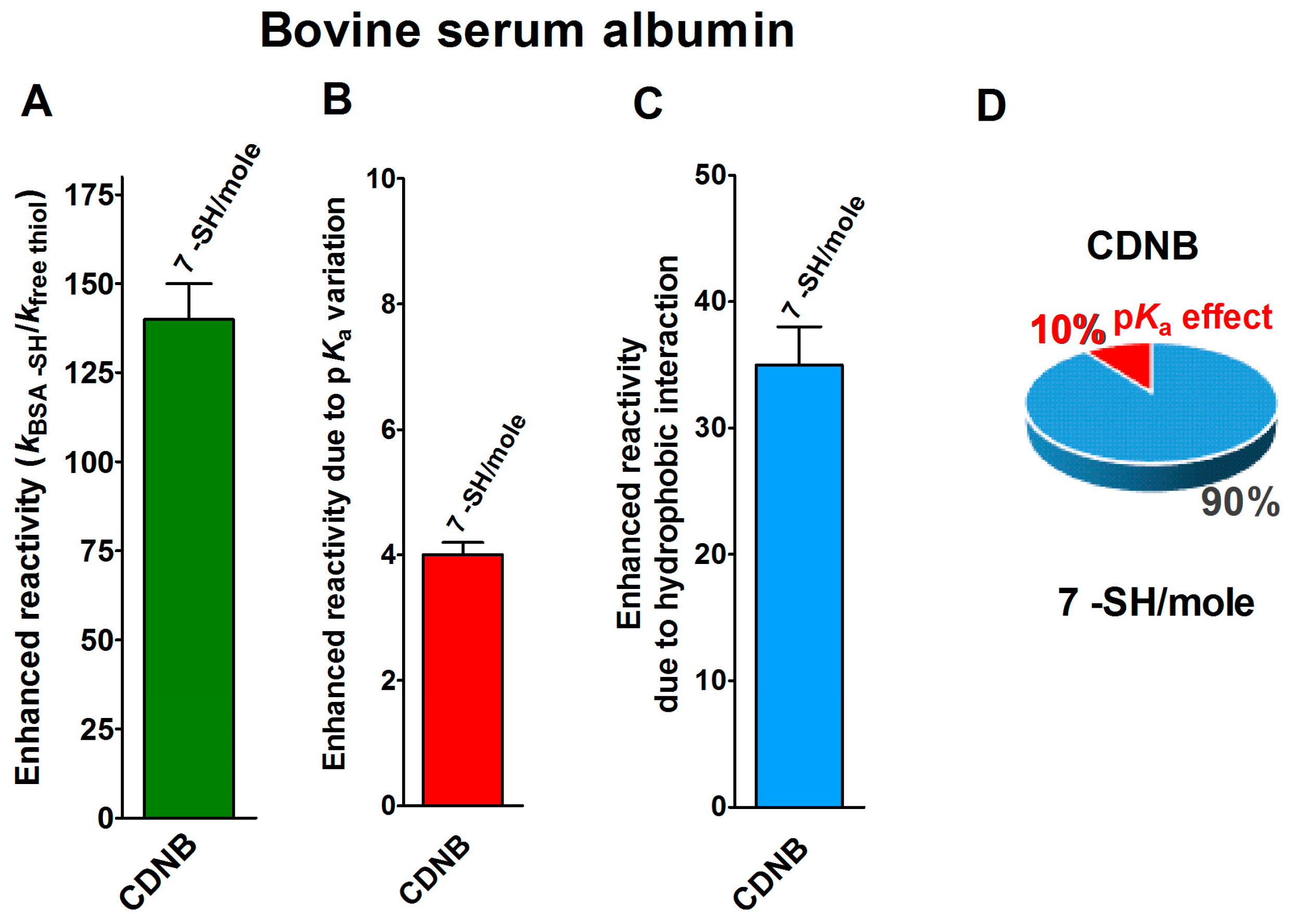
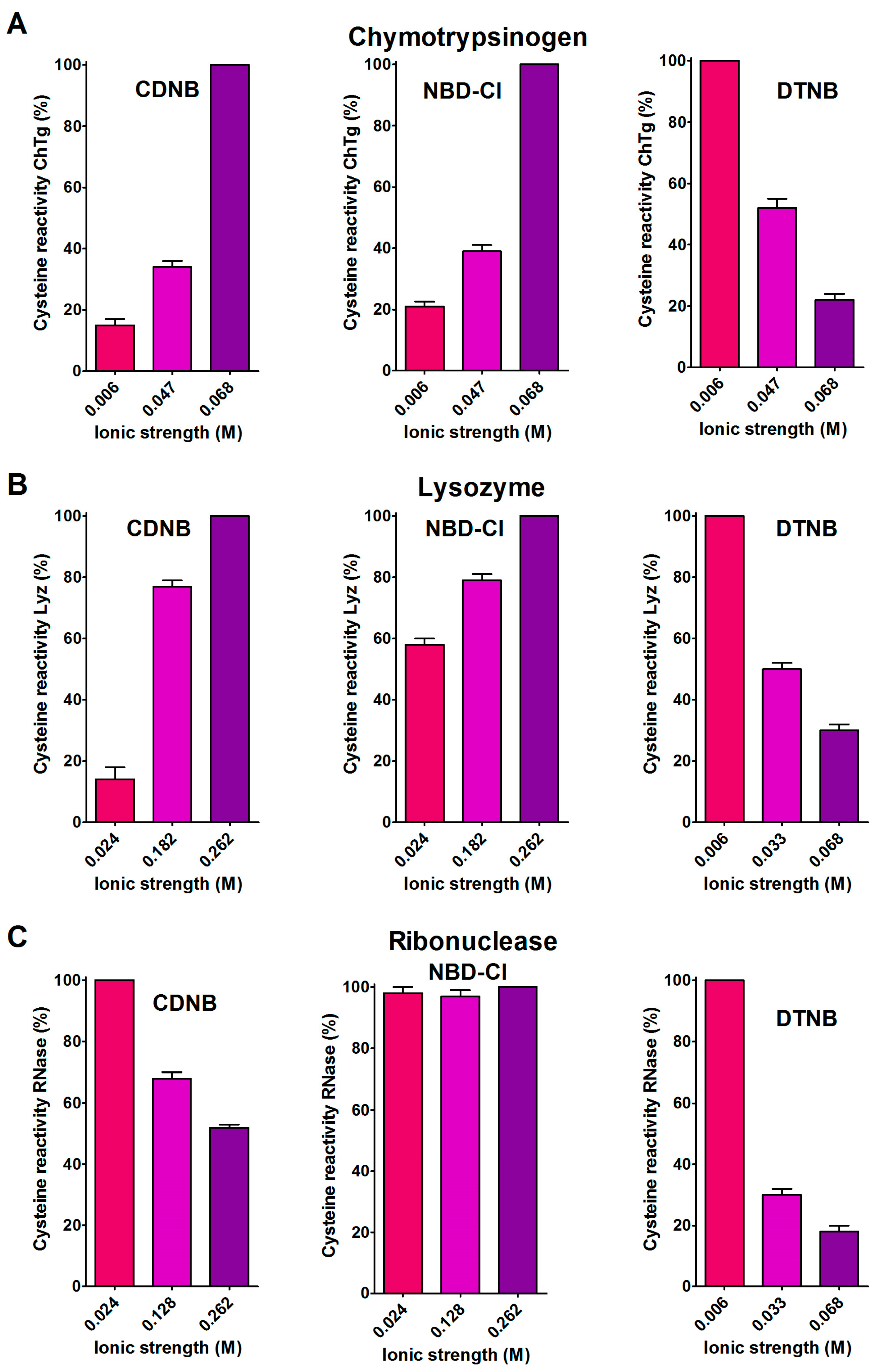
2.2. Hydrophobic Interactions
2.3. The Peculiar Interaction with 5,5′-Dithiobis-(2-Nitrobenzoic Acid)
2.4. CD Analyses and ANS Fluorescence Fulfill Further Insights
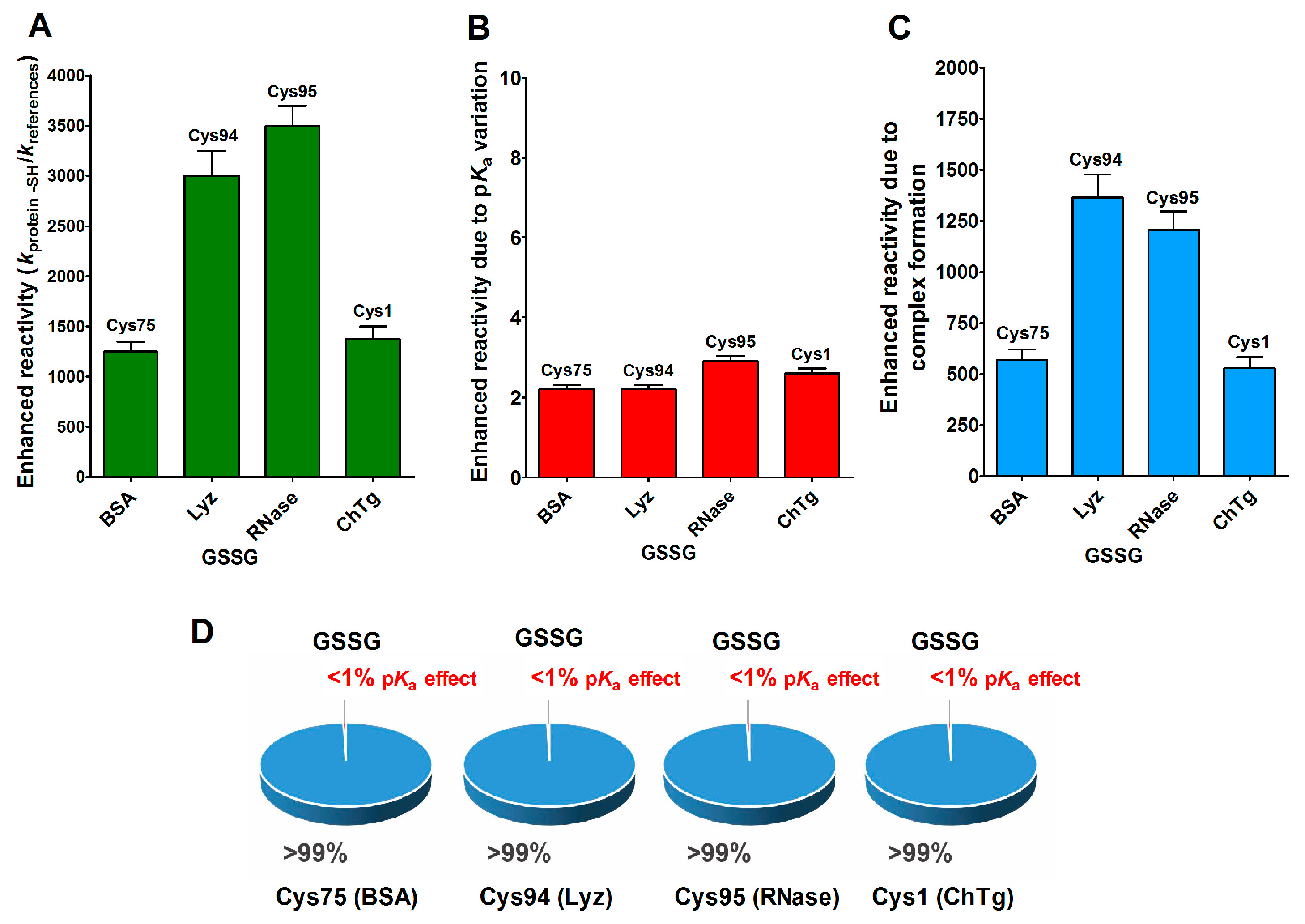
2.5. Productive Transient Complex
3. Discussion
- pKa is not the main determinant in the enhancement of the reactivity of protein cysteines toward various reagents. Conversely, a very low pKa, as well as a very high pKa, may render unreactive these residues (see Figure 1 and Figure 7). What is the utility of some functional cysteine showing very low pKa, such as selected residues in DsbA (pKa = 3.5), DsbC (pKa = 4.1) and Grx1 (pKa = 3.5) [24]? One reasonable explanation is that this property accelerates the reaction of the oxidized form of these enzymes with the thiol substrates stabilizing the products [24]. Another possibility is that a very low pKa that makes the thiolate less reactive and may preserve it against some unproper modifications. This may be the case for GSTP1-1, where the thiolate of Cys47 (pKa = 3.5) is bound to Lys54 in an ion-pair, which is important for the enzyme mechanism and a correct binding of the substrate [25].
- Cysteine hyper-reactivity is not an exclusive property of functional cysteines involved in catalysis and even structural cysteines devoted to the formation of disulfides may display hundred- or thousand-times increased reactivity toward GSSG and various thiol reagents.
- Hydrophobic interactions are the main determinant factors triggering hyper-reactivity toward CDNB and NBD-Cl for rBSA, rChTg and rLyz, while electrostatic interactions are the prominent factors for the reactivity of DTNB toward rRNase, rLyz and rChTg.
- A specific binding site for GSSG is surprisingly present in the reduced molten globule-like conformations of albumin, lysozyme, chymotrypsinogen and ribonuclease. It is the main determinant for the observed hundred- and even thousand-times increased reactivity of one specific cysteine. This phenomenon raises the question of whether a rapid glutathionylation may be the early step of their oxidative pathway.
- Methods for the proteomic identification of cysteines, like the isoTOP-ABPP procedure [1,26], should be used with caution, because they only identify hyper-reactive cysteines toward a specific reagent (i.e., a modified iodacetamide) and this property cannot be referred to an ‘intrinsic reactivity’ because it may be not present in reactions with different thiol reagents. Conversely, some protein cysteines, which are normo-reactive toward the modified iodoacetamide probe, can be hyper-reactive toward some natural intracellular compounds. Our data, in fact, likely indicate that one cysteine may have extraordinary hyper-reactivity toward a specific disulfide (GSSG) and normo-reactivity toward other small disulfides, like cystine and cystamine. Conversely, many cysteines which are present in rBSA and rLyz are hyper-reactive toward hydrophobic reagents like CDNB and NBD-Cl, but (except for one residue) are normo-reactive toward GSSG and other small disulfides. In other words, the “intrinsic” reactivity for a protein cysteine is only determined by its pKa and by the nucleophilicity of its deprotonated form, but it cannot be increased more than three–four-times, as demonstrated in this paper. An evident hyper-reactivity can only be generated by “extrinsic” factors like the protein environment surrounding the cysteine, which may productively and often selectively bind a specific reagent through hydrophobic or electrostatic interactions.
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Reactions of Thiols with Alkylating Reagents
4.3. Reactivity of rChTg Cysteines toward Alkylating Reagents and DTNB Varying the Ionic Strength
4.4. Reactivity of rLyz Cysteines toward Alkylating Reagents and DTNB Varying the Ionic Strength
4.5. Reactivity of rRNase Cysteines toward Alkylating Reagents and DTNB Varying the Ionic Strength
4.6. ANS Fluorescence Assay
4.7. Data Analysis and Graphical Representation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANS | 8-anilinonaphthalene-1-sulfonic acid |
| BSA | bovine serum albumin |
| CD | circular dichroism |
| CDNB | 1-chloro-2,4-dinitrobenzene |
| ChTg | chymotrypsinogen |
| DTNB | 5,5′-dithiobis(2-nitrobenzoic acid) |
| DTT | dithiotreitol |
| EDTA | ethylendiamminotetreaacetic acid |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| Lyz | lysozyme |
| NBD-Cl | 4-chloro-7-nitrobenzofurazan |
| rBSA | reduced bovine serum albumin |
| rChTg | reduced chymotrypsinogen |
| rLyz | reduced lysozyme |
| RNase | ribonuclease |
| rRNase | reduced ribonuclease |
| TNBS− | 5-thio-2-nitrobenzoate |
References
- Maurais, A.J.; Weerapana, E. Reactive-cysteine profiling for drug discovery. Curr. Opin. Chem. Biol. 2019, 50, 29–36. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Rossi, R.; Barra, D.; Bellelli, A.; Boumis, G.; Canofeni, S.; Di Simplicio, P.; Lusini, L.; Pascarella, S.; Amiconi, G. Fast-reacting thiols in rat hemoglobins can intercept damaging species in erythrocytes more efficiently than glutathione. J. Biol. Chem. 1998, 273, 19198–19206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, S.M.; Gladyshev, V.N. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 2012, 287, 4419–4425. [Google Scholar] [CrossRef] [Green Version]
- Britto, P.J.; Knipling, L.; Wolff, J. The local electrostatic environment determines cysteine reactivity of tubulin. J. Biol. Chem. 2002, 277, 29018–29027. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P.; Fang, Q.; Liu, L.; Hachey, D.L.; Pegg, A.E. O6-alkylguanine-DNA alkyltransferase: Low pKa and high reactivity of cysteine 145. Biochemistry 2003, 42, 10965–10970. [Google Scholar] [CrossRef]
- Fowler, N.J.; Blanford, C.F.; de Visser, S.P.; Warwicker, J. Features of reactive cysteines discovered through computation: From kinase inhibition to enrichment around protein degrons. Sci. Rep. 2017, 7, 16338. [Google Scholar] [CrossRef]
- Harris, T.K.; Turner, G.J. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 2002, 53, 85–98. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 2011, 24, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G.; Kortemme, T.; Goldenberg, D.P. Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry 1998, 37, 8965–8972. [Google Scholar] [CrossRef] [PubMed]
- Szajewski, R.P.; Whitesides, G.M. Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J. Am. Chem. Soc. 1980, 102, 2011–2026. [Google Scholar] [CrossRef]
- Gilbert, H.F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 69–172. [Google Scholar] [PubMed]
- Bocedi, A.; Cattani, G.; Martelli, C.; Cozzolino, F.; Castagnola, M.; Pucci, P.; Ricci, G. The extreme hyper-reactivity of Cys94 in lysozyme avoids its amorphous aggregation. Sci. Rep. 2018, 8, 16050. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Fabrini, R.; Pedersen, J.Z.; Federici, G.; Iavarone, F.; Martelli, C.; Castagnola, M.; Ricci, G. The extreme hyper-reactivity of selected cysteines drives hierarchical disulfide bond formation in serum albumin. FEBS J. 2016, 283, 4113–4127. [Google Scholar] [CrossRef]
- Bocedi, A.; Cattani, G.; Gambardella, G.; Ticconi, S.; Cozzolino, F.; Di Fusco, O.; Pucci, P.; Ricci, G. Ultra-Rapid Glutathionylation of Ribonuclease: Is this the Real Incipit of its Oxidative Folding? Int. J. Mol. Sci. 2019, 20, 5440. [Google Scholar] [CrossRef] [Green Version]
- Bocedi, A.; Gambardella, G.; Cattani, G.; Bartolucci, S.; Limauro, D.; Pedone, E.; Iavarone, F.; Castagnola, M.; Ricci, G. Ultra-rapid glutathionylation of chymotrypsinogen in its molten globule-like conformation. A comparison to archaeal proteins. Sci. Rep. 2020, 10, 8943. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Lilburn, J.E.; Szajewski, R.P. Rates of thiol-disulfide interchange reactions between mono- and dithiols and Ellman’s reagent. J. Org. Chem. 1977, 42, 332–338. [Google Scholar] [CrossRef]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes; Wiley-Interscience Publication: New York City, NY, USA, 1980; pp. 10–13. [Google Scholar]
- Snyder, G.H.; Cennerazzo, M.J.; Karalis, A.J.; Field, D. Electrostatic influence of local cysteine environments on disulfide exchange kinetics. Biochemistry 1981, 20, 6509–6519. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Ptitsyn, O.B. Molten globule and protein folding. Adv. Protein Chem. 1995, 47, 83–229. [Google Scholar]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P. Kinetics an mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antiox. Redox Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef] [Green Version]
- Lo Bello, M.; Parker, M.W.; Desideri, A.; Polticelli, F.; Falconi, M.; Del Boccio, G.; Pennelli, A.; Federici, G.; Ricci, G. Peculiar Spectroscopic and Kinetic Properties of Cys-47 in Human Placental Glutathione Transferase. J. Biol. Chem. 1993, 268, 19033–19038. [Google Scholar]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [Green Version]
- Sastre de Vicente, M.E. The Concept of Ionic Strength Eighty Years after its Introduction in Chemistry. J. Chem. Educ. 2004, 81, 750–753. [Google Scholar] [CrossRef]
- Taylor, J.R. An Introduction to Error Analysis, 2nd ed.; University Science Books: Sausalito, CA, USA, 1997; pp. 45–79. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiol Reagent | Thiol | Exp. pH | Reactive Cysteines | pKa | k (M−1s−1) | Total E.R. a | E.R. Due to pKa | E.R. Due to Other Factors |
|---|---|---|---|---|---|---|---|---|
| CDNB | GSH | 7.4 | 9.0 | 0.07 | 1 | |||
| GSH | 5.0 | 9.0 | 0.00028 | 1 | ||||
| rBSA | 7.4 | 7 | 7.8 b | 9.8 b | 140 b | 4 | 35 | |
| rLyz | 7.4 | 4 | 7.1 c | 160 c | 2286 c | 4 | 572 | |
| rRNase | 7.4 | 8 | 7.9 d | 0.9 d | 13 d | 3 | 4.3 | |
| rChTg | 5 | 10 | 8.1 e | 0.19 e | 678 e | 2 | 339 | |
| NBD-Cl | GSH | 7.4 | 9.0 | 8 | 1 | |||
| GSH | 5.0 | 9.0 | 0.032 | 1 | ||||
| rLyz | 7.4 | 4 | 7.1 c | 2900 c | 362.5 c | 3 | 121 | |
| rRNase | 7.4 | 8 | 7.9 d | 18 d | 2.3 d | 3 | 0.8 | |
| rChTg | 5.0 | 10 | 8.1 e | 12.6 e | 394 e | 2 | 197 | |
| DTNB f | GSH | 6.0 | 9.0 | 150 | 1 | |||
| GSH | 5.0 | 9.0 | 20 | 1 | ||||
| rBSA | 6.0 | 7 | 7.8 b | 30,000 b | 200 b | 4.3 | 46 | |
| rLyz | 5.0 | 7 | 6.6 c | 3200 c | 160 c | 3.3 | 48 | |
| rRNase | 5.0 | 8 | 7.9 d | 2500 d | 125 d | 3.5 | 36 | |
| rChTg | 5.0 | 1 | 8.1 e | 25,000 e | 1250 e | 3.5 | 357 | |
| rChTg | 5.0 | 9 | 8.1 e | 2300 e | 115 e | 3.5 | 33 | |
| GSSG | Cys | 7.4 | 9.1 g | 0.2 g | 1 g | |||
| Cys | 5.0 | 9.1 g | 0.0008 g | 1 g | ||||
| rBSA | 7.4 | Cys75 | 6.6 b | 250 b | 1250 b | 2.2 | 568 | |
| rLyz | 7.4 | Cys94 | 6.6 c | 600 c | 3000 c | 2.2 | 1364 | |
| rRNase | 7.4 | Cys95 | 7.9 d | 700 d | 3500 d | 2.9 | 1207 | |
| rChTg | 5.0 | Cys1 | 8.1 e | 1.1 e | 1375 e | 2.6 | 529 | |
| Cystamine | GSH | 7.4 | 9.0 | 55 | 1 | |||
| GSH | 5.0 | 9.0 | 0.22 | 1 | ||||
| rLyz | 7.4 | 4 | 6.6 c | 43 c | 0.8 c | 2.2 | 0.4 | |
| rRNase | 7.4 | 3 | 7.9 d | 250 d | 4.5 d | 2.9 | 1.5 | |
| rChTg | 5.0 | 5 | 8.1 e | 0.05 e | 0.23 e | 2.6 | 0.09 | |
| Cystine | GSH | 7.4 | 9.0 | 12 | 1 | |||
| GSH | 5.0 | 9.0 | 0.048 | 1 | ||||
| rLyz | 7.4 | 1 | 6.6 c | 770 c | 64 c | 2.2 | 29 | |
| rRNase | 7.4 | 6 | 7.9 d | 53 d | 4.4 d | 2.9 | 1.5 | |
| rChTg | 5 | 8 | 8.1 e | 0.56 e | 12 e | 2.6 | 4.6 |
| Secondary Structure | Oxidized BSA | Reduced BSA | Oxidized Lyz | Reduced Lyz | Oxidized RNase | Reduced RNase | Oxidized ChTg | Reduced ChTg |
|---|---|---|---|---|---|---|---|---|
| Helix | 70% | 54% | 50% | 6% | 22% | 27% | 12% | 10% |
| Strand | 0% | 16% | 24% | 27% | 20% | 24% | 25% | 39% |
| Turn | 9% | 2% | 11% | 14% | 15% | 12% | 14% | 11% |
| Others | 21% | 28% | 15% | 53% | 43% | 37% | 49% | 40% |
| Proteins | Fcred/Fcox |
|---|---|
| rLyz | 4.4 ± 0.1 |
| rRNase | 1.0 ± 0.1 |
| rChTg | 11.3 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambardella, G.; Cattani, G.; Bocedi, A.; Ricci, G. New Factors Enhancing the Reactivity of Cysteines in Molten Globule-Like Structures. Int. J. Mol. Sci. 2020, 21, 6949. https://doi.org/10.3390/ijms21186949
Gambardella G, Cattani G, Bocedi A, Ricci G. New Factors Enhancing the Reactivity of Cysteines in Molten Globule-Like Structures. International Journal of Molecular Sciences. 2020; 21(18):6949. https://doi.org/10.3390/ijms21186949
Chicago/Turabian StyleGambardella, Giorgia, Giada Cattani, Alessio Bocedi, and Giorgio Ricci. 2020. "New Factors Enhancing the Reactivity of Cysteines in Molten Globule-Like Structures" International Journal of Molecular Sciences 21, no. 18: 6949. https://doi.org/10.3390/ijms21186949
APA StyleGambardella, G., Cattani, G., Bocedi, A., & Ricci, G. (2020). New Factors Enhancing the Reactivity of Cysteines in Molten Globule-Like Structures. International Journal of Molecular Sciences, 21(18), 6949. https://doi.org/10.3390/ijms21186949