Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
2. HiPSC-Derived Neurons for AD Modeling and Platform for Screening
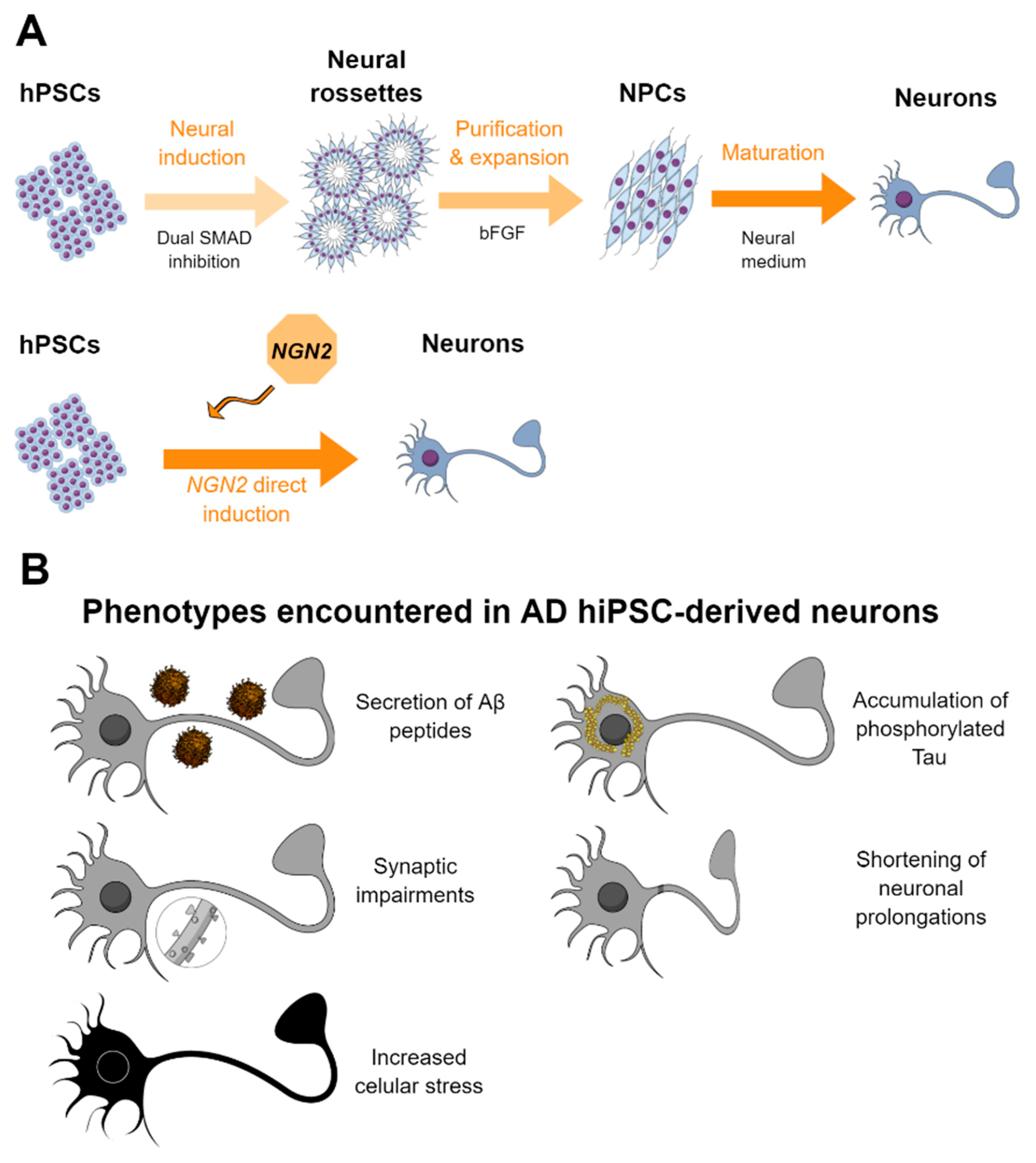
2.1. Methods for the Generation of Neurons from hPSCs
2.2. HPSC-Derived Neurons in AD and Drug Screening
3. HiPSC-Derived Astrocytes for AD Modeling and Platform for Screening
3.1. Definition and Functions of Astrocytes
3.2. Astrocytes in AD
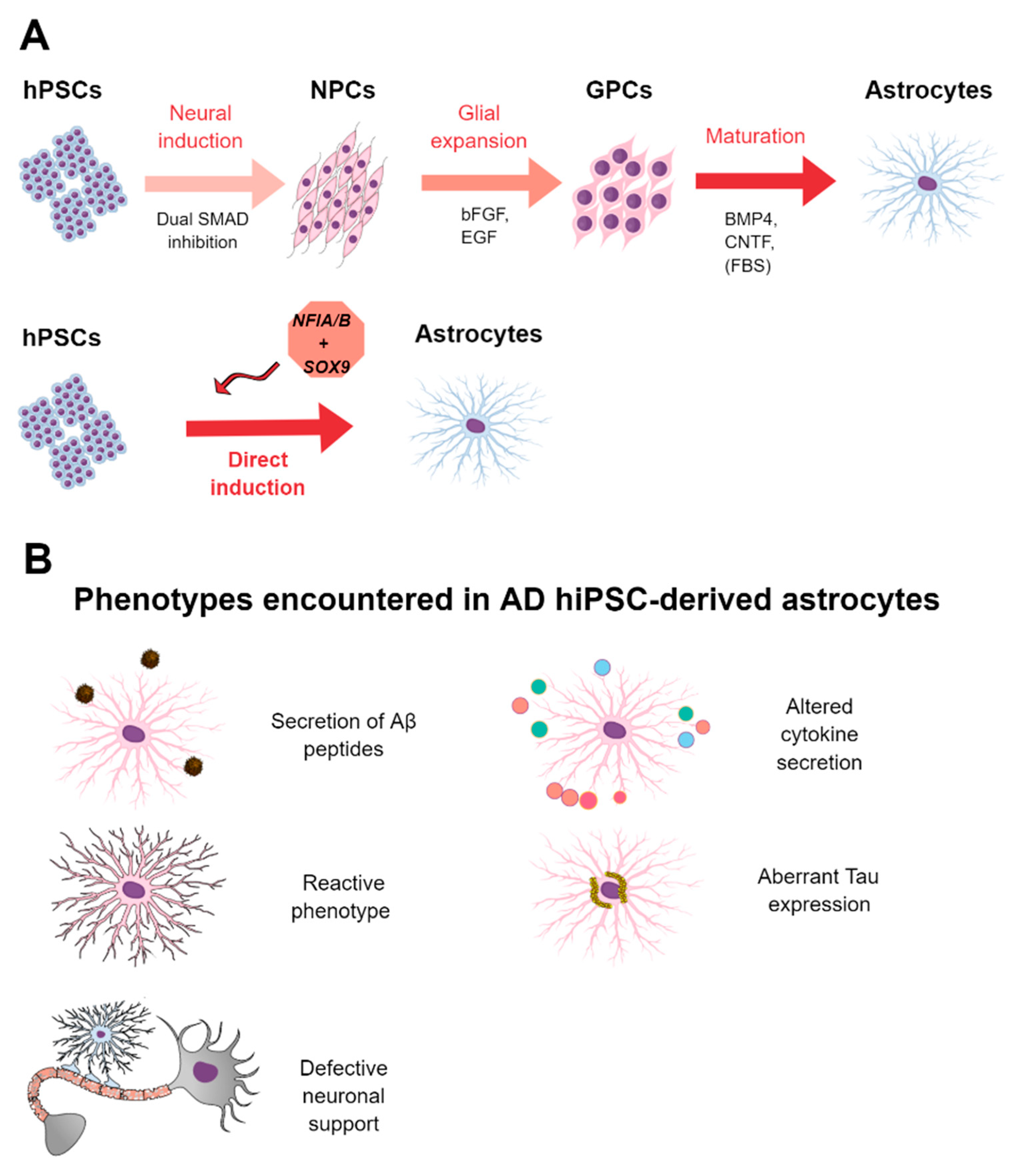
3.3. Methods for the Derivation of Astrocytes from hPSCs
3.4. HPSC-Derived Astrocytes in AD and Drug Screening
4. HiPSC-Derived Oligodendrocytes for AD Modeling and Platform for Screening
4.1. Definition and Functions of Oligodendrocytes
4.2. Oligodendrocytes in AD
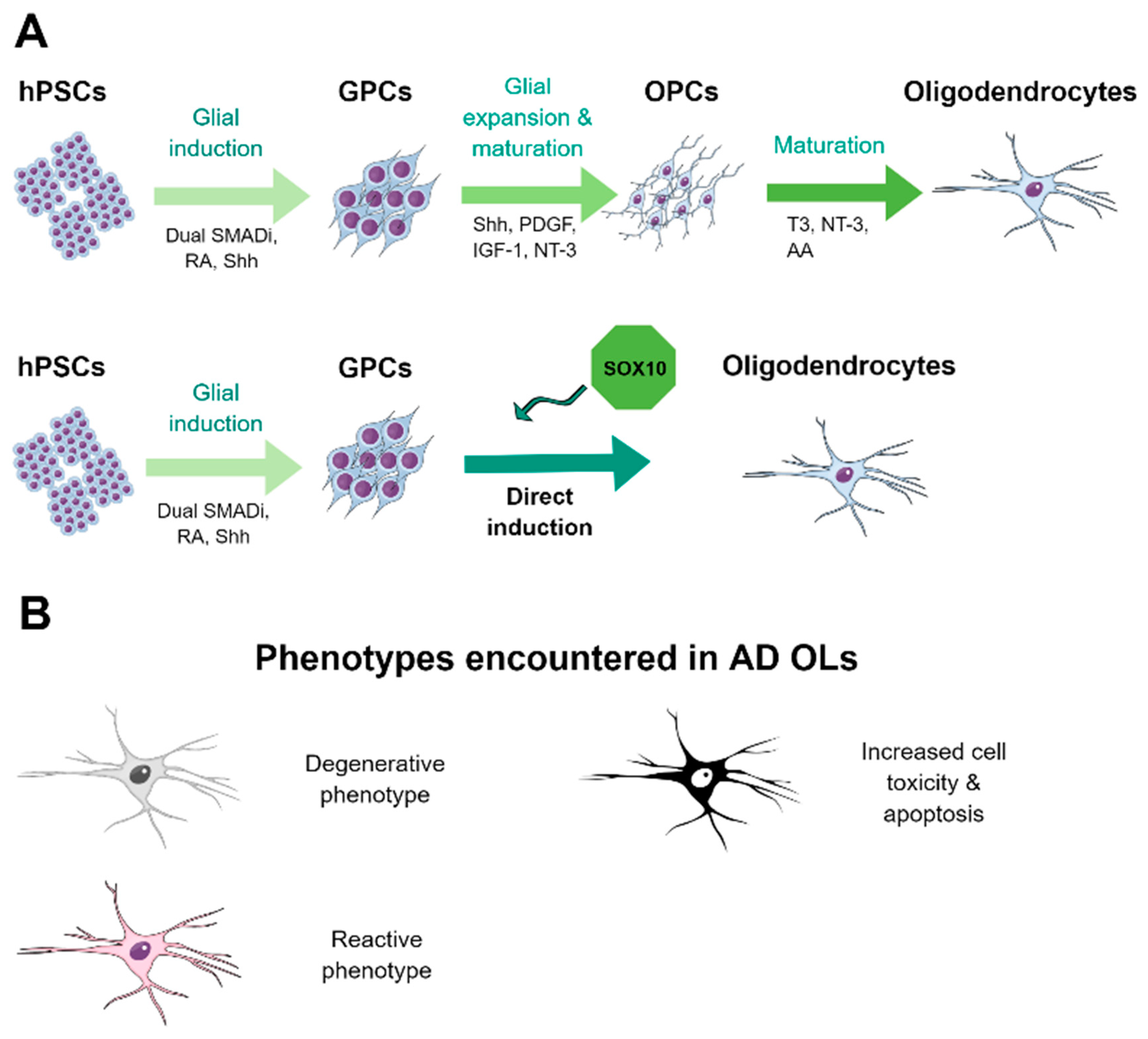
4.3. Methods for the Derivation of Oligodendrocytes from hPSCs
4.4. HPSC-Derived Oligodendrocytes in AD
5. HiPSC-Derived Microglia for AD Modeling and Platform for Screening
5.1. Definition and Functions of Microglia
5.2. Microglia in AD
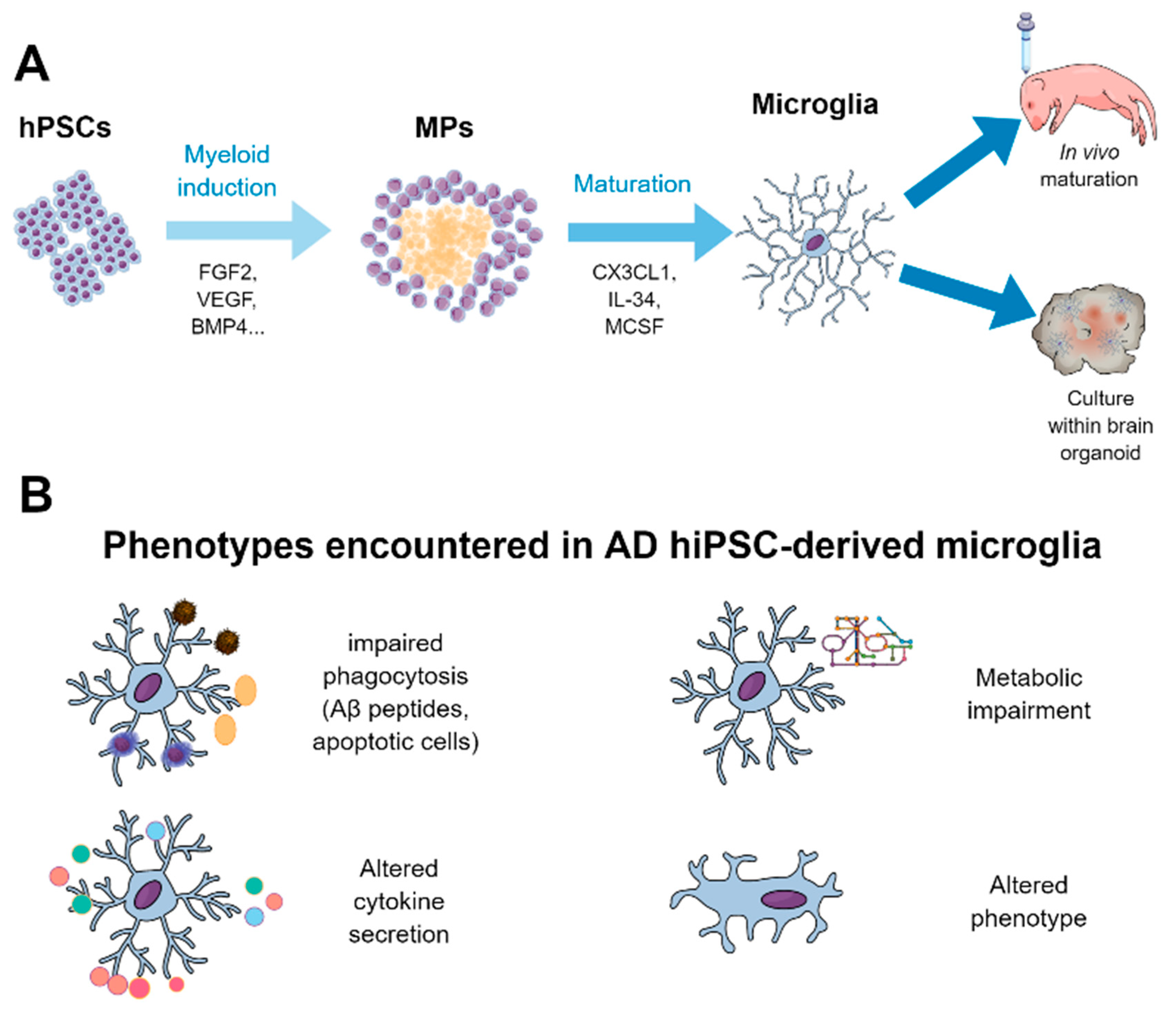
5.3. Derivation of Microglia from hPSCs and their Use for Disease Modeling and Drug Screening
6. HiPSC-Derived 3D Cultures and Brain Organoids for AD Modeling and Platform for Screening
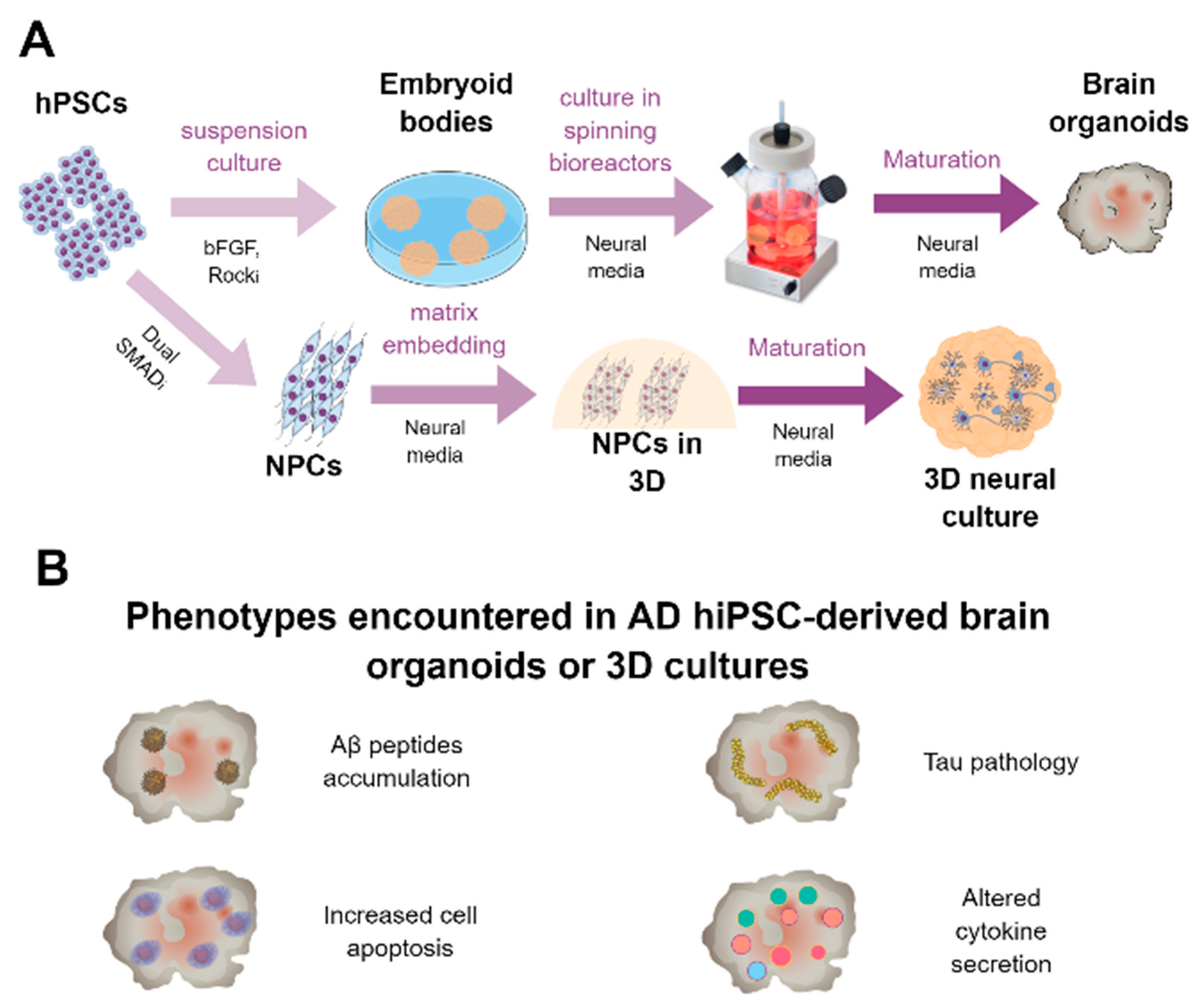
6.1. Generation of Brain Organoids
6.2. Brain Organoids for Modeling AD and to Be Used as a Drug Platform
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013, 12, 92–104. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J. Mol. Biol. 2019, 431, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front. Aging Neurosci. 2018, 10, 140. [Google Scholar] [CrossRef]
- King, A. The search for better animal models of Alzheimer’s disease. Nature 2018, 559, S13–S15. [Google Scholar] [CrossRef]
- Garcia-Leon, J.A.; Vitorica, J.; Gutierrez, A. Use of human pluripotent stem cell-derived cells for neurodegenerative disease modeling and drug screening platform. Future Med. Chem. 2019, 11, 1305–1322. [Google Scholar] [CrossRef]
- Calderon-Garcidueñas, A.L.; Duyckaerts, C. Alzheimer disease. Handb. Clin. Neurol. 2018, 145, 325–337. [Google Scholar]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Gomez-Arboledas, A.; Garcia-Leon, J.A.; Fernandez-Valenzuela, J.J.; Mejias-Ortega, M.; Trujillo-Estrada, L.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; et al. Distinct disease-sensitive GABAergic neurons in the perirhinal cortex of Alzheimer’s mice and patients. Brain Pathol. 2020, 30, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Bouschet, T.; Hourez, R.; Dimidschstein, J.; Naeije, G.; Van Den, J.; Passante, L.; Schiffmann, S.N.; Gaillard, A.; Espuny-camacho, I.; et al. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 2008, 455, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Shi, Y.; Kirwan, P.; Smith, J.; Robinson, H.P.C.; Livesey, F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012, 15, 477–486. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, S.C. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef]
- Ambasudhan, R.; Talantova, M.; Coleman, R.; Yuan, X.; Zhu, S.; Lipton, S.A.; Ding, S. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 2011, 9, 113–118. [Google Scholar] [CrossRef]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011, 476, 224–227. [Google Scholar] [CrossRef]
- Zhang, Y.; Pak, C.H.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Chanda, S.; Ang, C.E.; Lee, Q.Y.; Ghebrial, M.; Haag, D.; Shibuya, Y.; Wernig, M.; Südhof, T.C. Direct Reprogramming of Human Neurons Identifies MARCKSL1 as a Pathogenic Mediator of Valproic Acid-Induced Teratogenicity. Cell Stem Cell 2019, 25, 103–119.e6. [Google Scholar] [CrossRef]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.P.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef]
- Balez, R.; Steiner, N.; Engel, M.; Muñoz, S.S.; Lum, J.S.; Wu, Y.; Wang, D.; Vallotton, P.; Sachdev, P.; O’Connor, M.; et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci. Rep. 2016, 6, 31450. [Google Scholar] [CrossRef]
- Nieweg, K.; Andreyeva, A.; Van Stegen, B.; Tanriöver, G.; Gottmann, K. Alzheimer’s disease-related amyloid-β induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Corbett, G.T.; Moore, S.; Walsh, D.M.; Livesey, F.J.; Rowan, M.J.; Hu, N.; Corbett, G.T.; Moore, S.; Klyubin, I.; et al. Extracellular Forms of A b and Tau from iPSC Models of Alzheimer ’s Disease Disrupt Synaptic Plasticity Report Extracellular Forms of A b and Tau from iPSC Models of Alzheimer’s Disease Disrupt Synaptic Plasticity. Cell Rep. 2018, 23, 1932–1938. [Google Scholar] [CrossRef]
- Chang, K.H.; Lee-Chen, G.J.; Huang, C.C.; Lin, J.L.; Chen, Y.J.; Wei, P.C.; Lo, Y.S.; Yao, C.F.; Kuo, M.W.; Chen, C.M. Modeling Alzheimer’s Disease by Induced Pluripotent Stem Cells Carrying APP D678H Mutation. Mol. Neurobiol. 2019, 56, 3972–3983. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, H.; Ma, Y.; Shi, G.; Song, J.; Tang, Y.; Li, S.; Li, T.; Liu, N.; Tang, F.; et al. Early pathogenic event of Alzheimer’s disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget 2017, 8, 7900–7913. [Google Scholar] [CrossRef]
- Arber, C.; Toombs, J.; Lovejoy, C.C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol. Psychiatry 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S.B. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Sproul, A.A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy failure in fibroblasts and iPSC-derived neurons of alzheimer’s disease-associated presenilin 1 mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Sproul, A.; Martinez, H.; Paquet, D.; Gerges, M.; Noggle, S.; Starkov, A.A. Autophagy Induction by Bexarotene Promotes Mitophagy in Presenilin 1 Familial Alzheimer’s Disease iPSC-Derived Neural Stem Cells. Mol. Neurobiol. 2019, 56, 8220–8236. [Google Scholar] [CrossRef] [PubMed]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron 2019, 104, 256–270.e5. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Virumbrales, M.; Moreno, C.L.; Kruglikov, I.; Marazuela, P.; Sproul, A.; Jacob, S.; Zimmer, M.; Paull, D.; Zhang, B.; Schadt, E.E.; et al. CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer’s PSEN2 N141I neurons. Acta Neuropathol. Commun. 2017, 5, 77. [Google Scholar] [CrossRef]
- Moreno, C.L.; Guardia, L.D.; Shnyder, V.; Ortiz-Virumbrales, M.; Kruglikov, I.; Zhang, B.; Schadt, E.E.; Tanzi, R.E.; Noggle, S.; Buettner, C.; et al. IPSC-derived familial Alzheimer’s PSEN2 N141I cholinergic neurons exhibit mutation-dependent molecular pathology corrected by insulin signaling. Mol. Neurodegener. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-Based Compound Screening and In Vitro Trials Identify a Synergistic Anti-amyloid β Combination for Alzheimer’s Disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef]
- Jin, M.; O’Nuallain, B.; Hong, W.; Boyd, J.; Lagomarsino, V.N.; O’Malley, T.T.; Liu, W.; Vanderburg, C.R.; Frosch, M.P.; Young-Pearse, T.; et al. An in vitro paradigm to assess potential anti-Aβ antibodies for Alzheimer’s disease. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector article. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Feldman, H.M.; Lu, T.; Drake, D.; Lim, E.T.; Ling, K.-H.; Bishop, N.A.; Pan, Y.; Seo, J.; Lin, Y.-T.; et al. REST and Neural Gene Network Dysregulation in iPSC Models of Alzheimer’s Disease. Cell Rep. 2019, 26, 1112–1127.e9. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.; Wang, C.; Knoferle, J.; Walker, D.; Balestra, M.E.; Tong, L.M.; Leung, L.; Ring, K.L.; Seeley, W.W.; Karydas, A.; et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 2013, 1, 226–234. [Google Scholar] [CrossRef]
- Wren, M.C.; Zhao, J.; Liu, C.-C.; Murray, M.E.; Atagi, Y.; Davis, M.D.; Fu, Y.; Okano, H.J.; Ogaki, K.; Strongosky, A.J.; et al. Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol. Neurodegener. 2015, 10, 46. [Google Scholar] [CrossRef]
- Iovino, M.; Agathou, S.; González-Rueda, A.; Del Castillo Velasco-Herrera, M.; Borroni, B.; Alberici, A.; Lynch, T.; O’Dowd, S.; Geti, I.; Gaffney, D.; et al. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 2015, 138, 3345–3359. [Google Scholar] [CrossRef]
- Imamura, K.; Sahara, N.; Kanaan, N.M.; Tsukita, K.; Kondo, T.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Kawakami, K.; Hotta, A.; et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 2016, 6, 34904. [Google Scholar] [CrossRef]
- Nakamura, M.; Shiozawa, S.; Tsuboi, D.; Amano, M.; Watanabe, H.; Maeda, S.; Kimura, T.; Yoshimatsu, S.; Kisa, F.; Karch, C.M.; et al. Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived iPSCs. Stem Cell Rep. 2019, 13, 684–699. [Google Scholar] [CrossRef]
- García-León, J.A.; Cabrera-Socorro, A.; Eggermont, K.; Swijsen, A.; Terryn, J.; Fazal, R.; Nami, F.; Ordovás, L.; Quiles, A.; Lluis, F.; et al. Generation of a human induced pluripotent stem cell–based model for tauopathies combining three microtubule-associated protein TAU mutations which displays several phenotypes linked to neurodegeneration. Alzheimer’s Dement. 2018, 14, 1261–1280. [Google Scholar] [CrossRef]
- Medda, X.; Mertens, L.; Versweyveld, S.; Diels, A.; Barnham, L.; Bretteville, A.; Buist, A.; Verheyen, A.; Royaux, I.; Ebneth, A.; et al. Development of a Scalable, High-Throughput-Compatible Assay to Detect Tau Aggregates Using iPSC-Derived Cortical Neurons Maintained in a Three-Dimensional Culture Format. J. Biomol. Screen. 2016, 21, 804–815. [Google Scholar] [CrossRef]
- Wang, C.; Ward, M.E.; Chen, R.; Liu, K.; Tracy, T.E.; Chen, X.; Xie, M.; Sohn, P.D.; Ludwig, C.; Meyer-Franke, A.; et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Rep. 2017, 9, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375.e9. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.-T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.-H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Hallmann, A.L.; Reinhardt, P.; Araúzo-Bravo, M.J.; Korr, S.; Röpke, A.; Psathaki, O.E.; Ehling, P.; Meuth, S.G.; Oblak, A.L.; et al. Distinct Neurodegenerative Changes in an Induced Pluripotent Stem Cell Model of Frontotemporal Dementia Linked to Mutant TAU Protein. Stem Cell Rep. 2015, 5, 83–96. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Ho, M.S.; Parpura, V. Evolution of neuroglia. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1175, pp. 15–44. [Google Scholar]
- Trujillo-Estrada, L.; Gomez-Arboledas, A.; Forner, S.; Martini, A.C.; Gutierrez, A.; Baglietto-Vargas, D.; LaFerla, F.M. Astrocytes: From the Physiology to the Disease. Curr. Alzheimer Res. 2019, 16, 675–698. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Kim, S.K.; Nabekura, J.; Koizumi, S. Astrocyte-mediated synapse remodeling in the pathological brain. Glia 2017, 65, 1719–1727. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.H.; Haydon, P.G. Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 2007, 27, 6473–6477. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond black-and-white. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; Von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martín, A.; Romero-Gómez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275, 305–315. [Google Scholar] [CrossRef]
- Schiweck, J.; Eickholt, B.J.; Murk, K. Important shapeshifter: Mechanisms allowing astrocytes to respond to the changing nervous system during development, injury and disease. Front. Cell. Neurosci. 2018, 12, 261. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Deneen, B. The Emerging Nature of Astrocyte Diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2017, 115, E302–E309. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Li, Z.; Noori, A.; Hyman, B.T.; Serrano-pozo, A. Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflamm. 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2016, 18, 31–41. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef]
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef]
- Bayraktar, O.A.; Bartels, T.; Holmqvist, S.; Kleshchevnikov, V.; Martirosyan, A.; Polioudakis, D.; Ben Haim, L.; Young, A.M.H.; Batiuk, M.Y.; Prakash, K.; et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 2020, 23, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; McGeer, P.L.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef]
- Vehmas, A.K.; Kawas, C.H.; Stewart, W.F.; Troncoso, J.C. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 321–331. [Google Scholar] [CrossRef]
- Simpson, J.E.; Ince, P.G.; Shaw, P.J.; Heath, P.R.; Raman, R.; Garwood, C.J.; Gelsthorpe, C.; Baxter, L.; Forster, G.; Matthews, F.E.; et al. Microarray analysis of the astrocyte transcriptome in the aging brain: Relationship to Alzheimer’s pathology and APOE genotype. Neurobiol. Aging 2011, 32, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013, 27, 187–198. [Google Scholar] [CrossRef]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Sekar, S.; McDonald, J.; Cuyugan, L.; Aldrich, J.; Kurdoglu, A.; Adkins, J.; Serrano, G.; Beach, T.G.; Craig, D.W.; Valla, J.; et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 2015, 36, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation anddysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef]
- Chen, W.-T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Krencik, R.; Weick, J.P.; Liu, Y.; Zhang, Z.-J.; Zhang, S.-C. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat. Biotechnol. 2011, 29, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Shaltouki, A.; Peng, J.; Liu, Q.; Rao, M.S.; Zeng, X. Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells 2013, 31, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Vadodaria, K.C.; Jaeger, B.N.; Mei, A.; Lefcochilos-Fogelquist, S.; Mendes, A.P.D.; Erikson, G.; Shokhirev, M.; Randolph-Moore, L.; Fredlender, C.; et al. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1757–1769. [Google Scholar] [CrossRef]
- TCW, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef]
- Chambers, C.B.; Peng, Y.; Nguyen, H.; Gaiano, N.; Fishell, G.; Nye, J.S. Spatiotemporal selectivity of response to Notch1 signals in mammalian forebrain precursors. Development 2001, 128, 689–702. [Google Scholar] [PubMed]
- Tsai, H.H.; Li, H.; Fuentealba, L.C.; Molofsky, A.V.; Taveira-Marques, R.; Zhuang, H.; Tenney, A.; Murnen, A.T.; Fancy, S.P.J.; Merkle, F.; et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 2012, 337, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Perriot, S.; Mathias, A.; Perriard, G.; Canales, M.; Jonkmans, N.; Merienne, N.; Meunier, C.; El Kassar, L.; Perrier, A.L.; Laplaud, D.A.; et al. Human Induced Pluripotent Stem Cell-Derived Astrocytes Are Differentially Activated by Multiple Sclerosis-Associated Cytokines. Stem Cell Rep. 2018, 11, 1199–1210. [Google Scholar] [CrossRef]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef]
- Li, X.; Tao, Y.; Bradley, R.; Du, Z.; Tao, Y.; Kong, L.; Dong, Y.; Jones, J.; Yan, Y.; Harder, C.R.K.; et al. Fast Generation of Functional Subtype Astrocytes from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 11, 998–1008. [Google Scholar] [CrossRef]
- Tchieu, J.; Calder, E.L.; Guttikonda, S.R.; Gutzwiller, E.M.; Aromolaran, K.A.; Steinbeck, J.A.; Goldstein, P.A.; Studer, L. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat. Biotechnol. 2019, 37, 267–275. [Google Scholar] [CrossRef]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Paşca, S.P. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790.e6. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with iPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Zhao, J.; Davis, M.D.; Martens, Y.A.; Shinohara, M.; Graff-Radford, N.R.; Younkin, S.G.; Wszolek, Z.K.; Kanekiyo, T.; Bu, G. APOE ε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, A.-L.; Araúzo-Bravo, M.J.; Mavrommatis, L.; Ehrlich, M.; Röpke, A.; Brockhaus, J.; Missler, M.; Sterneckert, J.; Schöler, H.R.; Kuhlmann, T.; et al. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci. Rep. 2017, 7, 42991. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C.; Edgar, J.M.; Smith, K.J. Neuroprotection and repair in multiple sclerosis. Nat. Rev. Neurol. 2012, 8, 624–634. [Google Scholar] [CrossRef]
- Li, L.; Tian, E.; Chen, X.; Chao, J.; Klein, J.; Qu, Q.; Sun, G.; Sun, G.; Huang, Y.; Warden, C.D.; et al. GFAP Mutations in Astrocytes Impair Oligodendrocyte Progenitor Proliferation and Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell 2018, 23, 239–251.e6. [Google Scholar] [CrossRef]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Osipovitch, M.; Asenjo Martinez, A.; Mariani, J.N.; Cornwell, A.; Dhaliwal, S.; Zou, L.; Chandler-Militello, D.; Wang, S.; Li, X.; Benraiss, S.J.; et al. Human ESC-Derived Chimeric Mouse Models of Huntington’s Disease Reveal Cell-Intrinsic Defects in Glial Progenitor Cell Differentiation. Cell Stem Cell 2019, 24, 107–122.e7. [Google Scholar] [CrossRef]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H.; et al. Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 2017, 21, 195–208.e6. [Google Scholar] [CrossRef] [PubMed]
- de Vrij, F.M.; Bouwkamp, C.G.; Gunhanlar, N.; Shpak, G.; Lendemeijer, B.; Baghdadi, M.; Gopalakrishna, S.; Ghazvini, M.; Li, T.M.; Quadri, M.; et al. Candidate CSPG4 mutations and induced pluripotent stem cell modeling implicate oligodendrocyte progenitor cell dysfunction in familial schizophrenia. Mol. Psychiatr. 2019, 24, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, R.; Zacharek, A.; Wang, F.; Landschoot-ward, J. ABCA1/ApoE/HDL Signaling Pathway Facilitates Myelination and Oligodendrogenesis after Stroke. Int. J. Mol. Sci. 2020, 21, 4369. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef]
- Falcão, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jäkel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; French-Constant, C.; et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 2018, 24, 1837–1844. [Google Scholar] [CrossRef]
- Chanoumidou, K.; Mozafari, S.; Baron-Van Evercooren, A.; Kuhlmann, T. Stem cell derived oligodendrocytes to study myelin diseases. Glia 2019, 68, 705–720. [Google Scholar] [CrossRef]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef]
- Douvaras, P.; Wang, J.; Zimmer, M.; Hanchuk, S.; O’Bara, M.A.; Sadiq, S.; Sim, F.J.; Goldman, J.; Fossati, V. Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 250–259. [Google Scholar] [CrossRef]
- García-León, J.A.; Kumar, M.; Boon, R.; Chau, D.; One, J.; Wolfs, E.; Eggermont, K.; Berckmans, P.; Gunhanlar, N.; de Vrij, F.; et al. SOX10 Single Transcription Factor-Based Fast and Efficient Generation of Oligodendrocytes from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 655–672. [Google Scholar] [CrossRef]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.-L.; Cui, Q.-L.; Schambach, A.; Kim, K.-P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and early brain development: An intimate journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Primitive Macrophages. Science 2010, 701, 841–845. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1-and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Sierra, A.; Paolicelli, R.C.; Kettenmann, H. Cien Años de Microglía: Milestones in a Century of Microglial Research. Trends Neurosci. 2019, 42, 778–792. [Google Scholar] [CrossRef]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernández-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, Z.; Xiong, S.; Sun, F.; Qin, G.; Hu, G.; Wang, J.; Zhao, L.; Liang, Y.X.; Wu, T.; et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat. Neurosci. 2018, 21, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatr. 2020, 25, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Sanchez-Mico, M.; Sanchez-Varo, R.; Nuñez-Diaz, C.; Trujillo-Estrada, L.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016, 132, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef]
- Cheng-Hathaway, P.J.; Reed-Geaghan, E.G.; Jay, T.R.; Casali, B.T.; Bemiller, S.M.; Puntambekar, S.S.; von Saucken, V.E.; Williams, R.Y.; Karlo, J.C.; Moutinho, M.; et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 29. [Google Scholar] [CrossRef]
- Xiang, X.; Piers, T.M.; Wefers, B.; Zhu, K.; Mallach, A.; Brunner, B.; Kleinberger, G.; Song, W.; Colonna, M.; Herms, J.; et al. The Trem2 R47H Alzheimer’s risk variant impairs splicing and reduces Trem2 mRNA and protein in mice but not in humans. Mol. Neurodegener. 2018, 13, 1–14. [Google Scholar] [CrossRef]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Ohgidani, M.; Kato, T.A.; Setoyama, D.; Sagata, N.; Hashimoto, R.; Shigenobu, K.; Yoshida, T.; Hayakawa, K.; Shimokawa, N.; Miura, D.; et al. Direct induction of ramified microglia-like cells from human monocytes: Dynamic microglial dysfunction in Nasu-Hakola disease. Sci. Rep. 2014, 4, 4957. [Google Scholar] [CrossRef]
- Beutner, C.; Roy, K.; Linnartz, B.; Napoli, I.; Neumann, H. Generation of microglial cells from mouse embryonic stem cells. Nat. Protoc. 2010, 5, 1481–1494. [Google Scholar] [CrossRef]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef]
- McQuade, A.; Coburn, M.; Tu, C.H.; Hasselmann, J.; Davtyan, H.; Blurton-Jones, M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 2018, 13, 1–13. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Espuny-Camacho, I.; Arranz, A.M.; Fiers, M.; Snellinx, A.; Ando, K.; Munck, S.; Bonnefont, J.; Lambot, L.; Corthout, N.; Omodho, L.; et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 2017, 93, 1066–1081.e8. [Google Scholar] [CrossRef]
- Hasselmann, J.; Coburn, M.A.; England, W.; Figueroa Velez, D.X.; Kiani Shabestari, S.; Tu, C.H.; McQuade, A.; Kolahdouzan, M.; Echeverria, K.; Claes, C.; et al. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 2019, 103, 1016–1033.e10. [Google Scholar] [CrossRef]
- Svoboda, D.S.; Barrasa, M.I.; Shu, J.; Rietjens, R.; Zhang, S.; Mitalipova, M.; Berube, P.; Fu, D.; Shultz, L.D.; Bell, G.W.; et al. Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proc. Natl. Acad. Sci. USA 2019, 116, 25293–25303. [Google Scholar] [CrossRef]
- Xu, R.; Li, X.; Boreland, A.J.; Posyton, A.; Kwan, K.; Hart, R.P.; Jiang, P. Human iPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Van Den Daele, J.; Fattorelli, N.; Wolfs, L.; Balusu, S.; Burton, O.; Liston, A.; Sierksma, A.; Fourne, Y.; Poovathingal, S.; et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 2019, 22, 2111–2116. [Google Scholar] [CrossRef] [PubMed]
- Brownjohn, P.W.; Smith, J.; Solanki, R.; Lohmann, E.; Houlden, H.; Hardy, J.; Dietmann, S.; Livesey, F.J. Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem Cell Rep. 2018, 10, 1294–1307. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Reitboeck, P.; Phillips, A.; Piers, T.M.; Villegas-Llerena, C.; Butler, M.; Mallach, A.; Rodrigues, C.; Arber, C.E.; Heslegrave, A.; Zetterberg, H.; et al. Human Induced Pluripotent Stem Cell-Derived Microglia-Like Cells Harboring TREM2 Missense Mutations Show Specific Deficits in Phagocytosis. Cell Rep. 2018, 24, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Claes, C.; Van Den Daele, J.; Boon, R.; Schouteden, S.; Colombo, A.; Monasor, L.S.; Fiers, M.; Ordovás, L.; Nami, F.; Bohrmann, B.; et al. Human stem cell–derived monocytes and microglia-like cells reveal impaired amyloid plaque clearance upon heterozygous or homozygous loss of TREM2. Alzheimer’s Dement. 2018, 15, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Piers, T.M.; Cosker, K.; Mallach, A.; Johnson, G.T.; Guerreiro, R.; Hardy, J.; Pocock, J.M. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J. 2020, 34, 2436–2450. [Google Scholar] [CrossRef]
- Konttinen, H.; Cabral-da-Silva, M.E.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef]
- Hansen, E.; Krautwald, M.; MacZurek, A.E.; Stuchbury, G.; Fromm, P.; Steele, M.; Schulz, O.; Garcia, O.B.; Castillo, J.; Körner, H.; et al. A versatile high throughput screening system for the simultaneous identification of anti-inflammatory and neuroprotective compounds. J. Alzheimer’s Dis. 2010, 19, 451–464. [Google Scholar] [CrossRef]
- Figuera-Losada, M.; Thomas, A.G.; Stathis, M.; Stockwell, B.R.; Rojas, C.; Slusher, B.S. Development of a primary microglia screening assay and its use to characterize inhibition of system xc- by erastin and its analogs. Biochem. Biophys. Rep. 2017, 9, 266–272. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Smith, A.M.; Smyth, L.C.; Jansson, D.; Scotter, E.L.; Swanson, M.E.V.; Aalderink, M.; Coppieters, N.; Narayan, P.; Handley, R.; et al. 1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol. Neurodegener. 2018, 13, 44. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Ormel, P.R.; Vieira de Sá, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Bonfio, C.; Chadwick, J.; Begum, F.; Skehel, M.; Lancaster, M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 2020, 5626, eaaz5626. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; D’Avanzo, C.; Tanzi, R.E.; Kim, D.Y.; Irimia, D. Human Neurospheroid Arrays for In Vitro Studies of Alzheimer’s Disease. Sci. Rep. 2018, 8, 2450. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatr. 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef]
- Labour, M.N.; Vigier, S.; Lerner, D.; Marcilhac, A.; Belamie, E. 3D compartmented model to study the neurite-related toxicity of Aβ aggregates included in collagen gels of adaptable porosity. Acta Biomater. 2016, 37, 38–49. [Google Scholar] [CrossRef]
- Simpson, L.W.; Szeto, G.L.; Boukari, H.; Good, T.A.; Leach, J.B. Collagen hydrogel confinement of Amyloid-β (Aβ) accelerates aggregation and reduces cytotoxic effects. Acta Biomater. 2020, 112, 164–173. [Google Scholar] [CrossRef]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Kwak, S.S.; Washicosky, K.J.; Brand, E.; von Maydell, D.; Aronson, J.; Kim, S.; Capen, D.E.; Cetinbas, M.; Sadreyev, R.; Ning, S.; et al. Amyloid-β42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer’s disease. Nat. Commun. 2020, 11, 1377. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, H.J.; Yang, J.; Chae, S.; Lee, W.; Chung, S.; Kim, J.; Choi, H.; Song, H.; Lee, C.K.; et al. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 2020, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.G.; Schuff, N.; Yaffe, K.; Madison, C.; Miller, B.; Weiner, M.W. Hippocampal atrophy patterns in mild cognitive impairment and alzheimer’s disease. Hum. Brain Mapp. 2010, 31, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Pomeshchik, Y.; Klementieva, O.; Gil, J.; Martinsson, I.; Hansen, M.G.; de Vries, T.; Sancho-Balsells, A.; Russ, K.; Savchenko, E.; Collin, A.; et al. Human iPSC-Derived Hippocampal Spheroids: An Innovative Tool for Stratifying Alzheimer Disease Patient-Specific Cellular Phenotypes and Developing Therapies. Stem Cell Rep. 2020, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neurons | |||||
|---|---|---|---|---|---|
| Reference | Cell Type Derived | Subjects | Methodology | Main Findings | Drug Evaluation |
| Yagi et al. 2011 [33] | Neurons | fAD with PS1 A246E (1) and PS2 N141I (1) mutations | Neurosphere-mediated NPC induction followed by adherent terminal differentiation | Neurons presented higher Aβ1-42 production, which was reduced with gamma-secretase inhibitors | Neurons responded to the gamma-secretase inhibitors compounds E and W. |
| Israel et al. 2012 [34] | Neurons | HCs (2), fAD with duplication in APP (2) and sAD (2) | Embryoid bodies followed by FACS purification | Aberrant secretion of Aβ1-40 and increased phospho-Tau and aGSK-3b levels | Neurons responded to beta- but not gamma-secretase inhibitors |
| Muratore et al. 2014 [35] | Neurons | fAD with APP V717I mutation (2), HCs | Embryoid bodies, followed by neural rosette selection, NPC expansion, and terminal differentiation in monoculture | Altered APP processing, secretion of Aβ42 and Aβ38, and increase in total and hyperphosphorylated Tau levels | When neurons were treated with Aβ-specific antibodies, Tau pathology reverted |
| Balez et al. 2016 [36] | Neurons | fAD with PSEN1 P117R mutation (1), sAD with APOE3/4 genotype (1) and HCs | Neurospheres followed by terminal differentiation in monocultures | AD neurons showed hyperexcitable calcium signaling, elevated levels of nitrite, increased cytotoxicity and apoptosis, reduced neurite length, and increased susceptibility to inflammatory stress | Cells responded to short-term treatment with apigenin, reversing most of these phenotypes |
| Nieweg et al. 2015 [37] | Neurons | HCs | Embryoid bodies followed by immunopaning purification | Ab exposure led to a reduction of synapses and electrophysiological activity | - |
| Hu et al. 2018 [38] | Neurons | PS1 Int4 mutation (2), APP Dup (2), trisomy 21 (2) and HCs (2) | Dual SMAD inhibition in adherent cultures plus neuronal maturation | Secretome of cells from mutant lines produced inhibition of hippocampal long-term potentiation when injected into rat brains | Antibody blockade of cellular prion protein ameliorated the synaptic loss. |
| Chang et al. 2019 [39] | Neurons | fAD with APPD678H mutation (3), HCs (including an unaffected sibling) | Dual SMAD inhibition in adherent cultures plus neuronal maturation | Accumulation of Aβ1–42 and Aβ1–40, increased activation of GSK3β, hyperphosphorylation of Tau, and downregulation of synaptophysin | The indole compound NC009-1 partially restored aberrant phenotypes |
| Yang et al. 2017 [40] | NPCs and neurons | fAD with PSEN1 A246E (1) and Ser169del (1) mutations | Generation of neurospheres followed by terminal differentiation in monoculture | Higher levels of Aβ42 and Tau phosphorylation and an accelerated neuronal differentiation in mutant cells | - |
| Arber et al. 2019 [41] | Neurons and brain organoids | fAD PSEN1 mutations (5), APP V717I (2) and HCs (5) | Neurons: neural induction followed by neuronal maturation in monocultures [25]. Organoids: based as Lancaster et al. 2013 [63] | Mutant cells presented an increased secretion of long Aβ peptides (Ab40, Aβ42, and Aβ43) | - |
| Woodruff et al. 2016 [42] | Neurons | Gene-edited iPSC lines with or without PS1 ΔE9, APP V717F, and/or APP Swedish mutations | NPC generation and expansion, FACS purification, and terminal differentiation | Mutant neurons had defects in the recycling state of endocytosis and soma-to-axon transcytosis of APP and lipoproteins | Defects in endocytosis were rescued by beta secretase inhibition |
| Martín-Maestro et al. 2017 [43] | Neurons | fAD with PSEN1 A246E (3) mutations | Dual SMAD inhibition in adherent cultures plus neuronal maturation | Alterations in the autophagic and mitophagy pathways | - |
| Martín-Maestro et al. 2019 [44] | NPCs | Gene-edited WT line to incorporate the PS1 M146L mutation (1) | Neural induction by dual SMAD inhibition | Mitochondrial respiratory chain defects together with aberrant mitophagy | Treating NPCs with autophagy-stimulating drug bexarotene restored autophagy and compensated mitochondrial abnormalities |
| Kwart et al. 2019 [45] | Neurons | Several gene-edited iPSC lines: APP Swe (2), A692G (2), V717G (2), Swe+M146V (1), A673T (1), KO (1), PSEN1 M146V (2), L166P (2), M233L (1), and A246E (2) | Dual SMAD inhibition in adherent cultures plus neuronal maturation | Dysregulation of endosomal pathways in mutant cells, which correlated with accumulation of C-terminal fragments produced by the processing of APP | This could be rescued by pharmacological modulation of beta-secretase (BACE) |
| Birnbaum et al. 2018 [46] | Neurons | sAD (5) and HCs | NGN2-based direct generation of neurons from iPSCs | AD cells: increased production of reactive oxygen species (ROS) and higher levels of DNA damage, which did not correlate with Aβ or Tau phosphorylation | - |
| Ortíz-Virumbrales et al. 2017 [47] | basal forebrain cholinergic neurons | fAD with PSEN2 N141I (2) mutations | Dual SMAD inhibition, FACS selection (CD271), and embryoid body formation followed by neuronal maturation in Brainphys media | Higher production of Aβ42/40 and diminished electrophysiological activity, phenotypes reversed after gene correction | - |
| Moreno et al. 2018 [48] | Basal forebrain cholinergic neurons | fAD PSEN2 N141I (2), HCs (2), and isogenic gene-corrected controls (2) | Dual SMAD inhibition in adherent cultures plus neuronal maturation in Brainphys media | Mutant cells presented increased Aβ40/Ab42 ratio and altered calcium flux | Addition of insulin to the cultures reduced the increase in Aβ40/Aβ42 ratio and the altered calcium flux |
| Kondo et al. 2017 [49] | Neurons | fAD with PSEN1 G384A (2), H163R (1), M146L (1), A246E (1), APP V717L (1) mutations, sAD (4), fAD gene-corrected lines (2), and HC (4) lines | NGN2-based direct generation of neurons from iPSCs | Higher production of Aβ42/40 | Tested >1000 compounds on Aβ production and identified six leading compounds with dose-dependent Aβ42 reduction, with a combination of three of them (bromocriptine, cromolyn, and topiramate) as the most potent anti-Aβ combinations |
| Jin et al. 2018 [50] | Neurons | HC (1) | NGN2-based direct generation of neurons from iPSCs | - | AD brain extracts were toxic to the cells. Addition of Aβ-specific blocking antibodies counteracted Aβ-mediated toxicity |
| Wang et al. 2018 [51] | Neurons | sAD APOE4/4 (3), HCs APOE3/3 (3), isogenic APOE3/3 (1), and APOE−/− (1) | Embryoid bodies, neural induction as monocultures, NPC purification and expansion in neurospheres, and terminal maturation in monocultures | APOE4 neurons had higher levels of Aβ, Tau phosphorylation, and specific degeneration of GABAergic neurons. Converting APOE4 to APOE3 by gene editing rescued these phenotypes | Treating the cells with PH002 changed the conformation of APOE4 to an APOE3-like structure reverted the phenotypes, suggesting a possible treatment for sAD. |
| Lin et al. 2018 [52] | Neurons, astrocytes, microglia and 3D organoids | sAD (1) and HC (1) lines and isogenic gene-edited controls | Neurons: NGN2-based induction. Astrocytes: neural induction followed by sorting for GLAST+ cells. Microglia: as described in [64]. Organoids: as described in [65]. | APOE4+ neurons showed a more mature phenotype with higher presence of synapses and higher secretion of Aβ42. APOE4 astrocytes and microglia showed a reduced capacity of Aβ uptake. | - |
| Meyer et al. 2019 [53] | Neurons and organoids | sAD (5), HCs (5), and gene-edited lines | Neurons: (1) neural induction as embryoid bodies, followed by neuronal maturation and (2) NGN2-based direct induction. Organoids: based as Lancaster et al. 2013 [63] | Accelerated neuronal differentiation of APOE4 iPSC-derived neurons and impaired proliferative capacity of NPCs | - |
| Fong et al. 2013 [54] | Neurons | A152T MAPT patients (2) and isogenic lines (2) | Neuronal differentiation by dual SMAD inhibition-based methodology | TAU fragmentation and phosphorylation, leading to neurodegeneration. Genetic correction of the mutation restored those phenotypes and homozygous introduction of the mutation exacerbated the phenotypes. | - |
| Ehrlich et al. 2015 [66] | N279K (1) and V337 M (1) FTDP patients and HCs (3) | Dual SMAD inhibition in embryoid bodies, NPC purification, expansion, and maturation. | Pronounced TAU pathology with increased fragmentation and phosphorylation, decreased neurite extension, and increased oxidative stress. FTD neurons showed an activation of the unfolded protein response and disease-associated gene expression profiles. | ||
| Wren et al. 2015 [55] | NSCs | FTDP-17 N279K MAPT patients (2) and HC (1) | Dual SMAD inhibition in embryoid bodies, NPC purification, and expansion | Increased expression of 4R Tau isoforms, increased cellular stress, and impaired endocytic trafficking, with some of those findings verified using autopsy brains | - |
| Iovino et al. 2015 [56] | Neurons | FTDP-17 MAPT P301L (2), N279K (4), and HCs (3) | Dual SMAD inhibition in adherent cultures plus neuronal maturation | Increase in the 4R/3R ratio of Tau expression, earlier electrophysiological maturation, and an increase in Tau phosphorylation | - |
| Imamura et al. 2016 [57] | Neurons | Patients with I10+14C (1) and R406W (1) mutations and gene-corrected isogenic lines | NGN2-based direct generation of neurons from iPSCs | Diseased neurons showed dysregulation of the augmentation of Ca2+ transients evoked by electrical stimulation which led to the release of misfolded Tau and cell death | Chemogenetic or pharmacological control of Ca2+ influx by the introduction of designer receptors exclusively activated by designer drugs or by treatment with glutamate receptor blockers attenuated misfolded tau accumulation and neuronal death |
| Nakamura et al. 2019 [58] | Neurons | Patients with R406W mutations (2) and gene-corrected isogenic lines | Generation of neural organoids followed by singularization and neuronal culture | Notable Tau fragmentation and mislocation, which led axons to display morphological and functional abnormalities that could be rescued by microtubule stabilization | Morphological and functional abnormalities that could be rescued by a microtubule stabilizer (epothilone D) |
| García-León et al. [59] | Neurons | hiPSCs with the introduction of the N279K, P301L, and E10+16 mutations. | Dual SMAD inhibition in adherent cultures plus neuronal maturation | Mutant neurons expressed higher levels of 4R Tau, higher phosphorylation and aggregation of Tau, an increased electrophysiological activity, deficiencies in neurite outgrowth, aberrant sequence of differentiation to cortical neurons, activation of stress pathways, shift toward GABAergic identity, and an upregulation of neurodegenerative pathways. | - |
| Medda et al. 2016 [60] | Neurons | HCs (2) | NPC generation by dual SMAD inhibition followed by neuronal maturation in 3D | Tau aggregation when pathogenic P301L Tau is overexpressed in the presence of K18 fibrils | Platform compatible with assessing compounds able to reduce Tau aggregation |
| Wang et al. 2017 [61] | Neurons | HCs (2) | NGN2-based direct generation of neurons from iPSCs | Developed a platform to test Ta lowering in a high-throughput manner | Among the 1280 tested compounds, three reduced Tau content without cell toxicity |
| Van der Kant et al. 2019 [62] | NPCs, neurons and astrocytes | fAD, sAD, gene-edited lines, and HCs | NPCs: dual SMAD inhibition followed by FACS sorting. Neurons: neuronal maturation and FACS purification. Astrocytes: sphere-mediated astrocytic induction | Increased Tau phosphorylation and Aβ secretion | Tested >1600 compounds, finding several hits involved in cholesterol pathway. Identified cholesteryl esters (CE), the storage product of excess cholesterol, as upstream regulators of pTau and Aβ secretion, suggesting a therapeutic possibility for AD. |
| Astrocytes | |||||
|---|---|---|---|---|---|
| Reference | Cell Type Derived | Subjects | Methodology | Main Findings | Drug Evaluation |
| Kondo et al. 2013 [123]. | iPSC-derived astrocytes and neurons | HC (1 subject), sAD (1 subject), and fAD (APP E693Δ, 1 subject). | Embryoid bodies, NPC expansion, astrocyte purification by attachment, and terminal differentiation with FBS. | Intracellular Aβ oligomer accumulation. Upregulation of ER and UPR stress. | BSI: reduction of Aβ oligomers and stress. DHA: alleviation of stress. DBM14-26 and NSC23766: no improvement of stress |
| Oksanen et al. 2017 [124] | iPSC-derived astrocytes | HC (3), fAD PSEN1 DE9 (2), pre-symptomatic PSEN1 DE9 (1), and PSEN1 DE9 gene-corrected (2) | Embryoid bodies, NPC expansion, glial induction, and astrocyte maturation with BMP4 and CNTF. | PSEN1-mutants: Increased Aβ1-42 secretion. Reduced capacity of Aβ internalization. Lack of proper support of neurons and induction of toxicity. Altered cytokine secretion. | - |
| Jones et al. 2017 [125] | iPSC-derived astrocytes | HCs, fAD PSEN1 M146L (1), and sAD APOE4+/+ (1) | Dual SMAD inhibition; NPC expansion; glial induction; and astrocyte maturation with FGF2, CNTF, BMP2, EGF, and insulin. | Astrocyte-specific deficiencies in both fAD and sAD cases: Simplified morphology and smaller cell volume and area. Accumulation of astrocytic markers in the nuclei. Altered basal inflammatory cytokine production | - |
| Zhao et al. 2017 ([126] | iPSC-derived astrocytes and neurons | Cognitively normal APOE3+/+ and APOE4+/+ individuals | Embryoid bodies; NPC expansion; glial induction; and astrocyte maturation with BMP4, CNTF, heregulin-b, and FBS. | In APOE4+/+-derived astrocytes, APOE with a less lipidation status. Defective neuronal support | - |
| Hallmann et al. 2017 [127] | iPSC-derived astrocytes | HCs, FTD TAU N279K (2), and N279K gene-corrected (2) | SOX10-induction of astrocytes | Aberrant expression of 4R TAU. Larger cell volume and reactivity. Upregulation in ROS and UPR stress. Toxic to neurons | - |
| Microglia | |||||
|---|---|---|---|---|---|
| Reference | Cell Type Derived | Subjects | Methodology | Main Findings | Drug Evaluation |
| Muffat et al. 2016 [64] | Microglia | HCs (8), adrenoleukodystrophy (4), adrenomyeloneuropathy (3), Rett syndrome (1), or AD (3) | Embryoid body-based using a defined neural medium supplemented with CSF1 and IL-34 to generate myeloid progenitors and later microglia | Resembles primary microglia at the transcriptome and functional (phagocytosis, cytokine secretion, and response to injury) levels and interacted with neurons and glial cells | - |
| Pandya et al. 2017 [162] | Microglia | HCs (2) | Induction towards myeloid progenitors, FACS purification, and maturation in the presence of hPSC-derived astrocytes | Murine and human microglia resembled primary microglia at the transcriptome level as well as functionally (phagocytosis, ROS production, and improved outcomes of mice bearing brain gliomas). | - |
| Abud et al. 2017 [163] | Microglia | HCs (10) | Induction towards hematopoietic progenitors, CD43+ enrichment, and maturation in the presence of MCSF, IL34, TGFb1, CD200, and CX3CL1 | Generated microglial cells resembling human fetal and adult microglia at the transcriptome level and responded to inflammatory stimuli, phagocytose Aβ, and phosphorylated Tau | - |
| Douvaras et al. 2017 [164] | Microglia | HCs (11), Parkinson’s (3), multiple sclerosis (1), and AD (1) | Induction towards hematopoietic progenitors, CD14+/CX3CR1+ enrichment, and maturation in the presence of GM-CSF and IL34. | Similar to primary microglia at the transcriptome level and cytokine expression profile, able to phagocytose and responded to ADP. | - |
| Haenseler et al. 2017 [165] | Microglia | HCs (6) | Induction towards hematopoietic progenitors in embryoid bodies, collection of hematopoietic progenitors, and maturation in the presence of GM-CSF and IL34 and in co-culture with neurons | Similar to primary microglia at the transcriptome level and cytokine expression profile, able to phagocytose, activated with LPS, and surveyed the environment as the primary microglia. | - |
| Takata et al. 2017 [166] | Murine and human microglia and other macrophages | Parkinson’s (1) and familial Mediterranean fever (2) | Differentiation towards early macrophages following molecular cues present during yolk sac hematopoiesis and microglial maturation in co-culture with neurons | Generated cells resembled fetal microglia, interacted with neurons, responded to injury, were able to phagocytose, and synthesized cytokines | - |
| McQuade et al. 2018 [167] | Microglia | HCs (5) | Induction towards hematopoietic CD43+ progenitors and maturation in the presence of MCSF, IL34, TGFb1, CD200, and CX3CL1 | This paper describes a simplified and more efficient protocol compared to the previously developed by the group [163], which results in microglia with the same features as the previous report. | - |
| Hasselmann et al. 2019 [170] | Microglia precursors | HC (1) and gene-edited TREM2 R47H (1) | Differentiation towards microglial progenitors as previously described [167] and transplanted in immunodeficient mice | Transplanted cells better resembled primary human microglia that cultured cells, perform homeostatic functions, responded to injury, and reacted to Aβ plaques with a differential transcriptome signature compared to murine microglia | - |
| Svoboda et al. 2019 [171] | Microglial precursors and mature microglia | HC (1) | Differentiation towards microglial progenitors and matured cells according to Douvaras et al. 2017 [164] and transplanted in immunodeficient mice | Transplanted cells colonized the whole brain of hCSF1-expressing mice. Injected precursors acquired a mature microglial phenotype after in vivo maturation resembling human primary microglia in homeostatic state. | - |
| Xu et al. 2020 [172] | Primitive macrophage progenitors | HCs (2) | Differentiation towards macrophage progenitors as previously described [165] and transplanted into immunodeficient mice | Transplanted cells colonized the whole brain of hCSF1-expressing mice. Injected precursors acquired a mature microglial homeostatic phenotype as adult human microglia, expressed neurodegenerative disease-associated genes, and responded to acute demyelination. | - |
| Mancuso et al. 2019 [173] | Monocyte-derived microglia from hPSCs | HC (1) | First, monocytes were differentiated from hPSCs as described [176], and then, monocytes were induced towards neural lineage in neural medium. | Embryonic stem cell-derived microglia survive and integrate in mouse brain and mimic primary human cells at the transcriptome level. Human ESC-derived and host mouse microglia display a divergent response to oligomeric amyloid-β. | - |
| Brownjohn et al. 2018 [174] | Microglia | HCs (3), Nasu-Hakola disease patients (2), or unaffected family members (2) | Derivation of primitive macrophage precursors, and microglia and microglia maturation in the presence of GM-CSF and IL34. | Lower TREM2 levels were expressed by mutant cell-derived microglia, but this did not affect considerably their phagocytic capacity or response to inflammatory stimuli | - |
| Garcia-Reitboeck et al. 2018 [175] | Microglia | HCs (4), Nasu-Hakola disease patients (2), or unaffected family members (2) | Derivation of primitive macrophage precursors through an embryoid body-based method and microglia maturation. | TREM2 mutant cells presented impaired survival and reduced phagocytic capacity of apoptotic bodies. | - |
| Claes et al. 2018 [176] | Monocyte-derived microglia from hPSCs | HC (1) and gene-edited lines (3) | First, monocytes were differentiated from hPSCs and, then, monocytes were induced towards neural lineage in neural medium. | Mutant cells presented a reduced phagocytic capacity of E. coli fragments as well as of Aβ plaques using co-cultures of derived microglial-like cells with brain slices from APP/PS1 mice. | - |
| Piers et al. 2020 [177] | Microglia | HCs and diseased or gene-edited lines with R47H, T66M, or W50C mutations | As previously described [158] | The AD-related TREM2 R47H mutation produced impaired Aβ phagocytosis and led to metabolic impairments | - |
| Lin et al. 2018 [52] | Neurons, astrocytes, microglia and organoids | HC (1), sAD (1), and gene-edited lines with APOE3/3 and APOE4/4 genotypes | Microglia was generated as in Muffat et al. [64]. | Microglia derived from APOE4/4 hiPSC lines showed upregulation of inflammatory genes, an activated phenotype, and a reduced capacity to phagocytose Aβ in comparison with isogenic APOE3/3 lines. | - |
| Konttinen et al. 2019 [178] | Microglia | HCs (5), APOE4 sAD (3), PSEN1ΔE9 (4), and APP Swedish (2). | Small molecule generation of myeloid progenitors and maturation in the presence of MSCF and IL-34. | APOE4 microglia had impairment in key microglial functions such as phagocytosis, migration, and metabolic activity but exacerbated cytokine secretion to inflammatory stimuli. APP or PSEN1 mutations had little impact on microglial functionality. | - |
| Organoids | |||||
|---|---|---|---|---|---|
| Reference | Cell Type Derived | Subjects | Methodology | Main Findings | Drug Evaluation |
| Choi et al. 2014 [185] | An immortalized human neural stem cell line (ReNcell) | Lentiviral-mediated overexpression of the fAD mutations APP Swe/Lon and/or PSEN1 DE9 | Neural differentiation in a defined medium with cells embedded in a gel matrix (Matrigel) | Able to model AD in vitro, as this system led to Aβ accumulation, which subsequently induced Tau pathology | Beta-secretase inhibitors (DAPT, compound E and b-secretase inhibitor IV) or gamma-secretase inhibition (SGSM41) reverted Aβ and Tau pathology |
| Jorfi et al. 2018 [186] | An immortalized human neural stem cell line (ReNcell) | An immortalized human neural stem cell line (ReNcell) | Neural differentiation in a defined medium with cells embedded in a gel matrix (Matrigel) and these neurospheroids allocated within defined arrays | Cells presented extensive neurite outgrowth, and Aβ and Tau pathologies | Treatment of the spheroids with five tested compounds (gamma–secretase inhibitor (compound E), beta–secretase inhibitor (LY2886721), methotrexate, and imatinib) resulted in impaired neurite outgrowth and/or spheroid size. |
| Raja et al. 2016 [65] | iPSC-derived neural cells | HCs (2), fAD APPdup (2), and fAD PSEN1 M146I (1) PSEN1 A264E (1). | Embryoid bodies formation, neural induction, and neural maturation in neural medium with Matrigel | higher accumulation of Aβ40 and Aβ 42, an increased expression of phosphorylated Tau, and higher expression of the early endosome antigen 1 (EEA1). | Treatment of the organoids with gamma-secretase (compound E) and beta-secretase (beta-secretase inhibitor IV) inhibitors attenuated Aβ and Tau pathology |
| Gonzalez et al. 2018 [187] | iPSC-derived neural cells | HC (1), fAD PSEN1 A246E (1), Down syndrome (1), Creutzfeldt–Jakob disease (2), mouse ESCs, and iPSCs. | Brain organoid generation according to Lancaster et al. 2013 [63]. | Organoids derived from both fAD and Down syndrome patients presented accumulation of amyloid-like plaques and neurofibrillary tangles, which led to higher cell death | - |
| Zhang et al. 2014 [188] | iPSC-derived neural cells | HCs (1) | Neural cells were cultured in self-assembling peptide hydrogels or 2D and allowed to mature by growth factor withdrawal. | Cytoskeleton abnormalities linked to AD could be best modeled in 3D, especially when Aβ peptides were added to the culture | - |
| Labour et al. 2016 [189] | Immortalized human cell lines (PC12 and SH-SY6Y) | Non-demented subjects | Neural cell cultured in 3D collagen-based scaffolds | Physical contacts between cells and Aβ aggregates resulted in cell toxicity and neurite dystrophy | - |
| Park et al. 2018 [191] | Immortalized human neural stem cell line (ReNcell), human immortalized microglia cell line, and iPSC-derived neural cells | Immortalized human neural stem cell line (ReNcell), human immortalized microglia cell line, and iPSC-derived neural cells | Neural differentiation in defined medium with cells embedded in a gel matrix (Matrigel) with these neurospheroids allocated into a microfluidic device | Aβ40 and Aβ42 secretion, Tau phosphorylation, and expression of inflammatory cytokines and chemokines. Introduction of microglia led to neurotoxicity and astrogliosis | - |
| Kwak et al. 2020 [192] | Immortalized human neural stem cell line (ReNcell) | Immortalized human neural stem cell line (ReNcell) overexpressing APPSL and/or PS1ΔE9 isoforms | Neural differentiation in a defined medium with cells embedded in a gel matrix (Matrigel) | Tau pathology was demonstrated to be induced in cells with a high Aβ42/Aβ40 ratio | Treatment with BPN-15606, a γ-secretase modulator, which reduced Aβ42/40, effectively reducing p-Tau accumulation. |
| Choi et al. 2020 [193] | iPSC-derived neural cells | HC (1) and AD (1) | Embryoid body formation, neural induction, and neural maturation in a neural medium | Increased Tau phosphorylation | A HDAC6 inhibitor, CKD-504, led to reduced Tau phosphorylation by increasing the degradation of pathological Tau |
| Pomeshchik et al. 2020 [195] | Hippocampal spheroids formed by iPSC-derived neural cells | HCs, fAD with APP p.V717I (1) and PS1 p.R278K genes (1) | Embryoid body formation, neural induction, and neural maturation in neural medium with specific morphogens | increased Aβ42/40 peptide ratios and decreased levels of synaptic proteins | NeuroD1 overexpression partially restored aberrant phenotypes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Leon, J.A.; Caceres-Palomo, L.; Sanchez-Mejias, E.; Mejias-Ortega, M.; Nuñez-Diaz, C.; Fernandez-Valenzuela, J.J.; Sanchez-Varo, R.; Davila, J.C.; Vitorica, J.; Gutierrez, A. Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6867. https://doi.org/10.3390/ijms21186867
Garcia-Leon JA, Caceres-Palomo L, Sanchez-Mejias E, Mejias-Ortega M, Nuñez-Diaz C, Fernandez-Valenzuela JJ, Sanchez-Varo R, Davila JC, Vitorica J, Gutierrez A. Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(18):6867. https://doi.org/10.3390/ijms21186867
Chicago/Turabian StyleGarcia-Leon, Juan Antonio, Laura Caceres-Palomo, Elisabeth Sanchez-Mejias, Marina Mejias-Ortega, Cristina Nuñez-Diaz, Juan Jose Fernandez-Valenzuela, Raquel Sanchez-Varo, Jose Carlos Davila, Javier Vitorica, and Antonia Gutierrez. 2020. "Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 18: 6867. https://doi.org/10.3390/ijms21186867
APA StyleGarcia-Leon, J. A., Caceres-Palomo, L., Sanchez-Mejias, E., Mejias-Ortega, M., Nuñez-Diaz, C., Fernandez-Valenzuela, J. J., Sanchez-Varo, R., Davila, J. C., Vitorica, J., & Gutierrez, A. (2020). Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(18), 6867. https://doi.org/10.3390/ijms21186867