Patient-Derived Induced Pluripotent Stem Cell-Based Models in Parkinson’s Disease for Drug Identification


Abstract
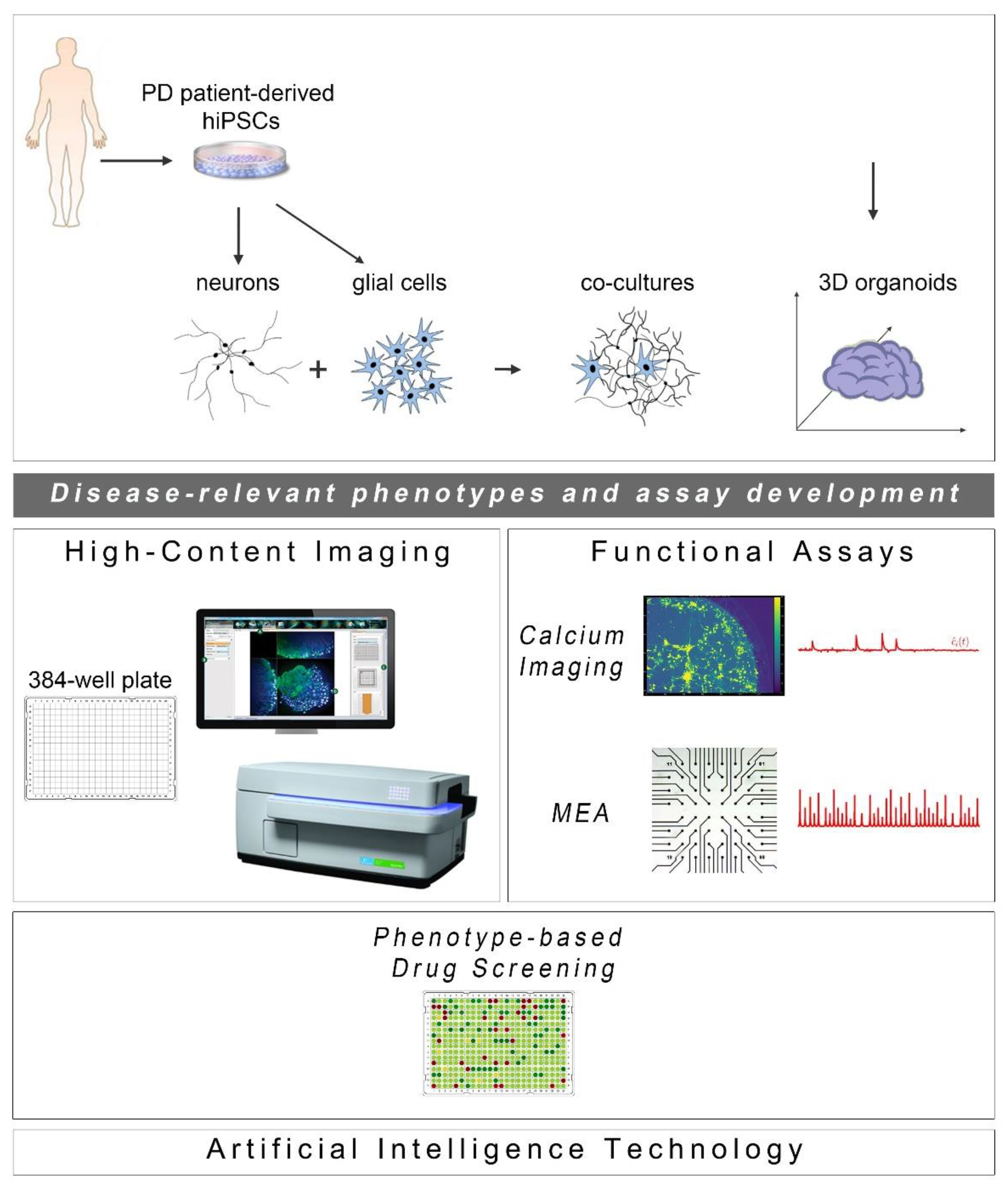
1. Introduction
2. Identification of Disease-Relevant Phenotypes in hiPSC-Derived Models of PD: A Glimpse into Human Pathology
2.1. α-Synuclein Accumulation
2.2. Mitochondrial Defects
2.3. Oxidative Stress
2.4. ER Stress and Autophagy-Related Phenotypes
2.5. Compromised Neuritic Growth, Axonal Degeneration and Decreased Synaptic Connectivity

{kind=link}
| Major PD-Relevant Phenotypes | Patient-Derived iPSC-Based Models in PD | |||||
|---|---|---|---|---|---|---|
| Gene Mutations | ||||||
| SNCA | LRKK2 | PARKIN | PINK1 | GBA | OPA1 | |
| αSyn accumulation and/or aggregation; increased phosphorylated αSyn (Ser 129); presence of oligomeric and fibrillar αSyn forms | G209A [5,36,37,38,39] Duplication [40] Triplication [39,41,42,43,44,45,46] | G2019S [62,67,71,72,73] | Ex2–4 del and Ex6–7 del [49] Ex3del, R42P, Ex3–4del, 1-BP del 255A, R275W, R42P [56] V324A [51] c.255delA [72] | Q456X [51] | L444P [68] N370S [69] | |
| Mitochondrial defects: fragmented mitochondria or mitochondria with abnormal morphology; decreased mitochondrial content; decreased ATP production; reduced membrane potential; dysfunctional mitochondrial mobility | G209A Triplication [39] | G2019S [48,57] | Ex2–4 del and Ex6–7 del [49] Ex3, 5, and 6 del [50] V324A [51] c.1072delT, p.A324fsX110 [52] Ex2–4 del and Ex6–7 del [53] | Q456X [51] G309D; Ex7/del [54] | N370S, L444P, and RecNcil [55] | p.G488R and p.A495V [58] |
| Oxidative stress: increased ROS and carbonylated proteins; upregulation of proteins involved in dopamine oxidation | Triplication [41,43,46] | G2019S [62] I2020T [63] | Ex2–4 del & Ex6–7 del [49] Ex3, 5, and 6 del [50] Ex3 del/Ex5 del and Ex3 del/Ex 3 del [64] | Q456X [60,65] | ||
| ER dysregulation; increased ER stress; autophagy impairment | G209A [37,66] Triplication [44,45] | G2019S [48,67] I2020T [63] | RecNcil, L444P and N370S [68] N370S [69] | |||
| Compromised neurite growth & complexity; neurite swellings; axonal degeneration; decreased synaptic connectivity; impaired axonal transport | G209A [38] Triplication [44] | G2019S [67,71,72,73] | Ex3 del/Ex5 del and Ex3 del/Ex 3 del [75] | |||
3. Rescue of Disease-Related Phenotypes in hiPSC-Derived Models of PD: Setting the Foundations towards Drug Discovery
4. Phenotypic Screens using hiPSC-Derived Models of PD: Empowering Drug Discovery
4.1. Target-Based Versus Phenotype-Based Drug Screening
4.2. Phenotypic-Based Drug Screening in hiPSC-Derived Models of PD
5. Looking into the Future: Optimization of hiPSC-Based Models for Understanding and Treating PD
5.1. Glial Cell Involvement in PD Pathogenesis: Mimicking the CNS Microenvironment in hiPSC-Based Co-Culture Systems
5.2. Emergence of Three-Dimensional (3D) hiPSC- Based Platforms for PD
6. Functional Assays for PD Studies and Drug Screening in hiPSC-Derived Systems
6.1. Calcium Imaging
6.2. Multi-Electrode Arrays (MEAs)
7. Artificial Intelligence Technologies
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| SNCA (PARK1/4) | α-synuclein gene |
| LRRK2 (PARK8) | leucine-rich repeat kinase 2 gene |
| PINK1 (PARK6) | PTEN-induced putative kinase 1 |
| DJ-1 (PARK7) | protein deglycase |
| ATP13A2 (PARK9) | endo-/lysosomal-associated P5 type transport ATPase |
| GBA | β-glucocerebrosidase gene |
| αSyn | α-synuclein protein |
| hiPSC | human induced pluripotent stem cell |
| GMP | good manufacturing practices |
| ROS | reactive oxygen species |
| ER | endoplasmic reticulum |
| GCase | β-glucocerebrosidase |
| ASO | antisense oligonucleotide |
| 6-OHDA | 6-hydroxydopamine |
| LDH | lactate dehydrogenase |
| OPA1 | mitochondrial dynamin-like GTPase |
| PET | positron emission tomography |
| ECM | extracellular matrix |
| MEA | multi-electrode array |
References
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Non-motor symptoms in Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 22 (Suppl. 1), S119–S122. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Schiffmann, R.; LaMarca, M.E.; Nussbaum, R.L.; McInerney-Leo, A.; Sidransky, E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jeon, B.S.; Jenner, P. Hallmarks of Treatment Aspects: Parkinson’s Disease Throughout Centuries Including l-Dopa. Int. Rev. Neurobiol. 2017, 132, 295–343. [Google Scholar] [CrossRef] [PubMed]
- Kulisevsky, J.; Oliveira, L.; Fox, S.H. Update in therapeutic strategies for Parkinson’s disease. Curr. Opin. Neurol. 2018, 31, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Dijk, J.M.; Espay, A.J.; Katzenschlager, R.; de Bie, R.M.A. The Choice Between Advanced Therapies for Parkinson’s Disease Patients: Why, What, and When? J. Parkinsons Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Petrucci, S.; Ginevrino, M.; Valente, E.M. Phenotypic spectrum of alpha-synuclein mutations: New insights from patients and cellular models. Parkinsonism Relat. Disord. 2016, 22 (Suppl. 1), S16–S20. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Savitt, D.; Jankovic, J. Targeting alpha-Synuclein in Parkinson’s Disease: Progress Towards the Development of Disease-Modifying Therapeutics. Drugs 2019, 79, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci 2019, 42, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Caligiore, D.; Helmich, R.C.; Hallett, M.; Moustafa, A.A.; Timmermann, L.; Toni, I.; Baldassarre, G. Parkinson’s disease as a system-level disorder. NPJ Parkinsons Dis. 2016, 2, 16025. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef]
- Kim, T.W.; Koo, S.Y.; Studer, L. Pluripotent Stem Cell Therapies for Parkinson Disease: Present Challenges and Future Opportunities. Front. Cell Dev. Biol. 2020, 8, 729. [Google Scholar] [CrossRef]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef]
- Marotta, N.; Kim, S.; Krainc, D. Organoid and pluripotent stem cells in Parkinson’s disease modeling: An expert view on their value to drug discovery. Expert Opin. Drug Discov. 2020, 15, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Weykopf, B.; Haupt, S.; Jungverdorben, J.; Flitsch, L.J.; Hebisch, M.; Liu, G.H.; Suzuki, K.; Belmonte, J.C.I.; Peitz, M.; Blaess, S.; et al. Induced pluripotent stem cell-based modeling of mutant LRRK2-associated Parkinson’s disease. Eur. J. Neurosci. 2019, 49, 561–589. [Google Scholar] [CrossRef] [PubMed]
- Sison, S.L.; Vermilyea, S.C.; Emborg, M.E.; Ebert, A.D. Using Patient-Derived Induced Pluripotent Stem Cells to Identify Parkinson’s Disease-Relevant Phenotypes. Curr. Neurol. Neurosci. Rep. 2018, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef]
- Little, D.; Luft, C.; Mosaku, O.; Lorvellec, M.; Yao, Z.; Paillusson, S.; Kriston-Vizi, J.; Gandhi, S.; Abramov, A.Y.; Ketteler, R.; et al. A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 2018, 8, 9033. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schutz, O.; et al. alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schule, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef] [PubMed]
- Flierl, A.; Oliveira, L.M.; Falomir-Lockhart, L.J.; Mak, S.K.; Hesley, J.; Soldner, F.; Arndt-Jovin, D.J.; Jaenisch, R.; Langston, J.W.; Jovin, T.M.; et al. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS ONE 2014, 9, e112413. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.M.; Falomir-Lockhart, L.J.; Botelho, M.G.; Lin, K.H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1994. [Google Scholar] [CrossRef] [PubMed]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology (Basel) 2019, 8, 38. [Google Scholar] [CrossRef]
- Su, Y.C.; Qi, X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 2013, 22, 4545–4561. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef]
- Aboud, A.A.; Tidball, A.M.; Kumar, K.K.; Neely, M.D.; Han, B.; Ess, K.C.; Hong, C.C.; Erikson, K.M.; Hedera, P.; Bowman, A.B. PARK2 patient neuroprogenitors show increased mitochondrial sensitivity to copper. Neurobiol. Dis. 2015, 73, 204–212. [Google Scholar] [CrossRef]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and alpha-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Zanon, A.; Kalvakuri, S.; Rakovic, A.; Foco, L.; Guida, M.; Schwienbacher, C.; Serafin, A.; Rudolph, F.; Trilck, M.; Grunewald, A.; et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2017, 26, 2412–2425. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Amadeo, A.; Calogero, A.M.; Casagrande, F.V.M.; De Gregorio, C.; Gioria, M.; Kuzumaki, N.; Costa, I.; Sassone, J.; Ciammola, A.; et al. Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol. Aging 2018, 61, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Azkona, G.; Lopez de Maturana, R.; Del Rio, P.; Sousa, A.; Vazquez, N.; Zubiarrain, A.; Jimenez-Blasco, D.; Bolanos, J.P.; Morales, B.; Auburger, G.; et al. LRRK2 Expression Is Deregulated in Fibroblasts and Neurons from Parkinson Patients with Mutations in PINK1. Mol. Neurobiol. 2018, 55, 506–516. [Google Scholar] [CrossRef]
- Schondorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Shaltouki, A.; Sivapatham, R.; Pei, Y.; Gerencser, A.A.; Momcilovic, O.; Rao, M.S.; Zeng, X. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep. 2015, 4, 847–859. [Google Scholar] [CrossRef]
- Schwab, A.J.; Sison, S.L.; Meade, M.R.; Broniowska, K.A.; Corbett, J.A.; Ebert, A.D. Decreased Sirtuin Deacetylase Activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 2017, 9, 1839–1852. [Google Scholar] [CrossRef]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates alpha-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef]
- Toulorge, D.; Schapira, A.H.; Hajj, R. Molecular changes in the postmortem parkinsonian brain. J. Neurochem. 2016, 139 (Suppl. 1), 27–58. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schule, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Ohta, E.; Nihira, T.; Uchino, A.; Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Takahashi, K.; Hayakawa, H.; Nagai, M.; Ohyama, M.; et al. I2020T mutant LRRK2 iPSC-derived neurons in the Sagamihara family exhibit increased Tau phosphorylation through the AKT/GSK-3beta signaling pathway. Hum. Mol. Genet. 2015, 24, 4879–4900. [Google Scholar] [CrossRef]
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3, 668. [Google Scholar] [CrossRef]
- Vera, E.; Bosco, N.; Studer, L. Generating Late-Onset Human iPSC-Based Disease Models by Inducing Neuronal Age-Related Phenotypes through Telomerase Manipulation. Cell Rep. 2016, 17, 1184–1192. [Google Scholar] [CrossRef]
- Khurana, V.; Peng, J.; Chung, C.Y.; Auluck, P.K.; Fanning, S.; Tardiff, D.F.; Bartels, T.; Koeva, M.; Eichhorn, S.W.; Benyamini, H.; et al. Genome-Scale Networks Link Neurodegenerative Disease Genes to alpha-Synuclein through Specific Molecular Pathways. Cell Syst. 2017, 4, 157–170 e114. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular alpha-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schondorf, D.C.; Wagner, L.; Glatza, M.; Hoing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Goke, J.; Cukuroglu, E.; Dranias, M.R.; VanDongen, A.M.; Stanton, L.W. Molecular Features Underlying Neurodegeneration Identified through In Vitro Modeling of Genetically Diverse Parkinson’s Disease Patients. Cell Rep. 2016, 15, 2411–2426. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Walter, J.; Jarazo, J.; Arias-Fuenzalida, J.; Hillje, A.L.; Schwamborn, J.C. CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and alpha-Synuclein modulation in dopaminergic neurons. Stem Cell Res. 2017, 24, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Korecka, J.A.; Talbot, S.; Osborn, T.M.; de Leeuw, S.M.; Levy, S.A.; Ferrari, E.J.; Moskites, A.; Atkinson, E.; Jodelka, F.M.; Hinrich, A.J.; et al. Neurite Collapse and Altered ER Ca(2+) Control in Human Parkinson Disease Patient iPSC-Derived Neurons with LRRK2 G2019S Mutation. Stem Cell Rep. 2019, 12, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Jiang, H.; Hu, Z.; Fan, K.; Wang, J.; Janoschka, S.; Wang, X.; Ge, S.; Feng, J. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 2015, 33, 68–78. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces alpha-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Kim, M.J.; Jeon, S.; Burbulla, L.F.; Krainc, D. Acid ceramidase inhibition ameliorates alpha-synuclein accumulation upon loss of GBA1 function. Hum. Mol. Genet. 2018, 27, 1972–1988. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef]
- Plowey, E.D.; Cherra, S.J., 3rd; Liu, Y.J.; Chu, C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008, 105, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Gardai, S.J.; Zago, W.; Bertoncini, C.W.; Cremades, N.; Roy, S.L.; Tambe, M.A.; Rochet, J.C.; Galvagnion, C.; Skibinski, G.; et al. Targeting the intrinsically disordered structural ensemble of alpha-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS ONE 2014, 9, e87133. [Google Scholar] [CrossRef] [PubMed]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson’s disease. Brain 2016, 139, 3217–3236. [Google Scholar] [CrossRef] [PubMed]
- Simmnacher, K.; Lanfer, J.; Rizo, T.; Kaindl, J.; Winner, B. Modeling Cell-Cell Interactions in Parkinson’s Disease Using Human Stem Cell-Based Models. Front. Cell. Neurosci. 2019, 13, 571. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 2015, 78, 21–38. [Google Scholar] [CrossRef]
- Zheng, W.; Thorne, N.; McKew, J.C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 2013, 18, 1067–1073. [Google Scholar] [CrossRef]
- Goodfellow, P.N. Genomics and drugs. An interview conducted by Holger Breithaupt and Caroline Hadley. EMBO Rep. 2004, 5, 843–846. [Google Scholar] [CrossRef]
- Burbaum, J.J.; Sigal, N.H. New technologies for high-throughput screening. Curr. Opin. Chem. Biol. 1997, 1, 72–78. [Google Scholar] [CrossRef]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, G.; Zhou, Y.; Wang, S.; Zhong, Z. Phenotypic screens targeting neurodegenerative diseases. J. Biomol. Screen. 2014, 19, 1–16. [Google Scholar] [CrossRef][Green Version]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Ishikawa, K.I.; Inoshita, T.; Shiba-Fukushima, K.; Saiki, S.; Hatano, T.; Mori, A.; Oji, Y.; Okuzumi, A.; Li, Y.; et al. Identifying Therapeutic Agents for Amelioration of Mitochondrial Clearance Disorder in Neurons of Familial Parkinson Disease. Stem Cell Rep. 2020, 14, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Tabata, Y.; Imaizumi, Y.; Sugawara, M.; Andoh-Noda, T.; Banno, S.; Chai, M.; Sone, T.; Yamazaki, K.; Ito, M.; Tsukahara, K.; et al. T-type Calcium Channels Determine the Vulnerability of Dopaminergic Neurons to Mitochondrial Stress in Familial Parkinson Disease. Stem Cell Rep. 2018, 11, 1171–1184. [Google Scholar] [CrossRef]
- Antoniou, N.; Prodromidou, K.; Kouroupi, G.; Samiotaki, M.; Panayotou, G.; Xilouri, M.; Stefanis, L.; Grailhe, R.; Taoufik, E.; Matsas, R. High Content Screening and Proteomic Analysis Identify the Kinase Inhibitor BX795 as a Potent Neuroprotective Compound in a Patient-Derived Model of Parkinson’s Disease. BioRxiv 2020. [Google Scholar] [CrossRef]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hamalainen, R.H.; Koistinaho, J. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar] [CrossRef]
- Heneka, M.T. Microglia take centre stage in neurodegenerative disease. Nat. Rev. Immunol. 2019, 19, 79–80. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Wagner, U.; Dumont, B.; Pikkarainen, M.; Osman, A.A.; Streichenberger, N.; Leisser, I.; Verchere, J.; Baron, T.; Alafuzoff, I.; et al. An antibody with high reactivity for disease-associated alpha-synuclein reveals extensive brain pathology. Acta Neuropathol. 2012, 124, 37–50. [Google Scholar] [CrossRef]
- Takeda, A.; Hashimoto, M.; Mallory, M.; Sundsumo, M.; Hansen, L.; Masliah, E. C-terminal alpha-synuclein immunoreactivity in structures other than Lewy bodies in neurodegenerative disorders. Acta Neuropathol. 2000, 99, 296–304. [Google Scholar] [CrossRef]
- Terada, S.; Ishizu, H.; Yokota, O.; Tsuchiya, K.; Nakashima, H.; Ishihara, T.; Fujita, D.; Ueda, K.; Ikeda, K.; Kuroda, S. Glial involvement in diffuse Lewy body disease. Acta Neuropathol. 2003, 105, 163–169. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Ros, C.M.; Annese, V.; Gomez, A.; Ros-Bernal, F.; Aguado-Yera, D.; Martinez-Pagan, M.E.; de Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. IFN-gamma signaling, with the synergistic contribution of TNF-alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2011, 2, e142. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Willemsen, A.T.; Doorduin, J.; de Vries, E.F.; Dierckx, R.A.; Leenders, K.L. [11C]-PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat. Disord. 2010, 16, 57–59. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5, e8784. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G.; Kawamata, T.; Yamada, T.; Akiyama, H. Reactions of the immune system in chronic degenerative neurological diseases. Can. J. Neurol. Sci. 1991, 18, 376–379. [Google Scholar] [CrossRef]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Kohutnicka, M.; Kurkowska-Jastrzebska, I.; Czlonkowski, A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 1996, 5, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Manocha, G.D.; Floden, A.M.; Puig, K.L.; Nagamoto-Combs, K.; Scherzer, C.R.; Combs, C.K. Defining the contribution of neuroinflammation to Parkinson’s disease in humanized immune system mice. Mol. Neurodegener. 2017, 12, 17. [Google Scholar] [CrossRef]
- Marinova-Mutafchieva, L.; Sadeghian, M.; Broom, L.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: A time course study in a 6-hydroxydopamine model of Parkinson’s disease. J. Neurochem. 2009, 110, 966–975. [Google Scholar] [CrossRef]
- Walsh, S.; Finn, D.P.; Dowd, E. Time-course of nigrostriatal neurodegeneration and neuroinflammation in the 6-hydroxydopamine-induced axonal and terminal lesion models of Parkinson’s disease in the rat. Neuroscience 2011, 175, 251–261. [Google Scholar] [CrossRef]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Mavroeidi, P.; Arvanitaki, F.; Karakitsou, A.K.; Vetsi, M.; Kloukina, I.; Zweckstetter, M.; Giller, K.; Becker, S.; Sorrentino, Z.A.; Giasson, B.I.; et al. Endogenous oligodendroglial alpha-synuclein and TPPP/p25alpha orchestrate alpha-synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol. 2019, 138, 415–441. [Google Scholar] [CrossRef]
- Alieva, A.K.; Zyrin, V.S.; Rudenok, M.M.; Kolacheva, A.A.; Shulskaya, M.V.; Ugryumov, M.V.; Slominsky, P.A.; Shadrina, M.I. Whole-Transcriptome Analysis of Mouse Models with MPTP-Induced Early Stages of Parkinson’s Disease Reveals Stage-Specific Response of Transcriptome and a Possible Role of Myelin-Linked Genes in Neurodegeneration. Mol. Neurobiol. 2018, 55, 7229–7241. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Wessely, F.; Connor-Robson, N.; Rinaldi, F.; Vowles, J.; Browne, C.; Evetts, S.G.; Hu, M.T.; Cowley, S.A.; Webber, C.; et al. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson’s iPSC-derived astrocytes. Neurobiol.Dis. 2019, 129, 56–66. [Google Scholar] [CrossRef]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Munoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Aflaki, E.; Stubblefield, B.K.; McGlinchey, R.P.; McMahon, B.; Ory, D.S.; Sidransky, E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef]
- Sonninen, T.M.; Hamalainen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef]
- Haenseler, W.; Zambon, F.; Lee, H.; Vowles, J.; Rinaldi, F.; Duggal, G.; Houlden, H.; Gwinn, K.; Wray, S.; Luk, K.C.; et al. Excess alpha-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci. Rep. 2017, 7, 9003. [Google Scholar] [CrossRef]
- Lee, H.; Flynn, R.; Sharma, I.; Haberman, E.; Carling, P.J.; Nicholls, F.J.; Stegmann, M.; Vowles, J.; Haenseler, W.; Wade-Martins, R.; et al. LRRK2 Is Recruited to Phagosomes and Co-recruits RAB8 and RAB10 in Human Pluripotent Stem Cell-Derived Macrophages. Stem Cell Rep. 2020, 14, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Speidel, A.; Felk, S.; Reinhardt, P.; Sterneckert, J.; Gillardon, F. Leucine-Rich Repeat Kinase 2 Influences Fate Decision of Human Monocytes Differentiated from Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0165949. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Song, H.; Ming, G.L. Brain organoids: Advances, applications and challenges. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Bolognin, S.; Fossepre, M.; Qing, X.; Jarazo, J.; Scancar, J.; Moreno, E.L.; Nickels, S.L.; Wasner, K.; Ouzren, N.; Walter, J.; et al. 3D Cultures of Parkinson’s Disease-Specific Dopaminergic Neurons for High Content Phenotyping and Drug Testing. Adv. Sci. 2019, 6, 1800927. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 2019, 5, 5. [Google Scholar] [CrossRef]
- Brull, M.; Spreng, A.S.; Gutbier, S.; Loser, D.; Krebs, A.; Reich, M.; Kraushaar, U.; Britschgi, M.; Patsch, C.; Leist, M. Incorporation of stem cell-derived astrocytes into neuronal organoids to allow neuro-glial interactions in toxicological studies. ALTEX 2020, 37, 409–428. [Google Scholar] [CrossRef]
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 2020, 26, 766–781 e769. [Google Scholar] [CrossRef]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pasca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef]
- Madhavan, M.; Nevin, Z.S.; Shick, H.E.; Garrison, E.; Clarkson-Paredes, C.; Karl, M.; Clayton, B.L.L.; Factor, D.C.; Allan, K.C.; Barbar, L.; et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Kwak, T.H.; Kang, J.H.; Hali, S.; Kim, J.; Kim, K.P.; Park, C.; Lee, J.H.; Ryu, H.K.; Na, J.E.; Jo, J.; et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson’s disease modeling. Stem Cells 2020, 38, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Seth, B.; Chaturvedi, R.K. Brain Organoids: Tiny Mirrors of Human Neurodevelopment and Neurological Disorders. Neuroscientist 2020, 1073858420943192. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Cui, K.; Guo, Y.; Zhang, X.; Qin, J. Advances in Hydrogels in Organoids and Organs-on-a-Chip. Adv. Mater. 2019, 31, e1902042. [Google Scholar] [CrossRef] [PubMed]
- Miccoli, B.; Braeken, D.; Li, Y.E. Brain-on-a-chip Devices for Drug Screening and Disease Modeling Applications. Curr. Pharm. Des. 2018, 24, 5419–5436. [Google Scholar] [CrossRef]
- Fernandes, J.T.; Chutna, O.; Chu, V.; Conde, J.P.; Outeiro, T.F. A Novel Microfluidic Cell Co-culture Platform for the Study of the Molecular Mechanisms of Parkinson’s Disease and Other Synucleinopathies. Front. Neurosci. 2016, 10, 511. [Google Scholar] [CrossRef]
- Seidi, A.; Kaji, H.; Annabi, N.; Ostrovidov, S.; Ramalingam, M.; Khademhosseini, A. A microfluidic-based neurotoxin concentration gradient for the generation of an in vitro model of Parkinson’s disease. Biomicrofluidics 2011, 5, 22214. [Google Scholar] [CrossRef]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef]
- Mank, M.; Griesbeck, O. Genetically encoded calcium indicators. Chem. Rev. 2008, 108, 1550–1564. [Google Scholar] [CrossRef]
- Schwab, A.J.; Ebert, A.D. Neurite Aggregation and Calcium Dysfunction in iPSC-Derived Sensory Neurons with Parkinson’s Disease-Related LRRK2 G2019S Mutation. Stem Cell Rep. 2015, 5, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Zygogianni, O.; Antoniou, N.; Kalomoiri, M.; Kouroupi, G.; Taoufik, E.; Matsas, R. In Vivo Phenotyping of Familial Parkinson’s Disease with Human Induced Pluripotent Stem Cells: A Proof-of-Concept Study. Neurochem. Res. 2019, 44, 1475–1493. [Google Scholar] [CrossRef] [PubMed]
- Negri, J.; Menon, V.; Young-Pearse, T.L. Assessment of Spontaneous Neuronal Activity In Vitro Using Multi-Well Multi-Electrode Arrays: Implications for Assay Development. ENeuro 2020, 7. [Google Scholar] [CrossRef]
- Spira, M.E.; Hai, A. Multi-electrode array technologies for neuroscience and cardiology. Nat. Nanotechnol. 2013, 8, 83–94. [Google Scholar] [CrossRef]
- Obien, M.E.; Deligkaris, K.; Bullmann, T.; Bakkum, D.J.; Frey, U. Revealing neuronal function through microelectrode array recordings. Front. Neurosci. 2014, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Woodard, C.M.; Campos, B.A.; Kuo, S.H.; Nirenberg, M.J.; Nestor, M.W.; Zimmer, M.; Mosharov, E.V.; Sulzer, D.; Zhou, H.; Paull, D.; et al. iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep. 2014, 9, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Laperle, A.H.; Sances, S.; Yucer, N.; Dardov, V.J.; Garcia, V.J.; Ho, R.; Fulton, A.N.; Jones, M.R.; Roxas, K.M.; Avalos, P.; et al. iPSC modeling of young-onset Parkinson’s disease reveals a molecular signature of disease and novel therapeutic candidates. Nat. Med. 2020, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Amin, H.; Nieus, T.; Lonardoni, D.; Maccione, A.; Berdondini, L. High-resolution bioelectrical imaging of Abeta-induced network dysfunction on CMOS-MEAs for neurotoxicity and rescue studies. Sci. Rep. 2017, 7, 2460. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef]
- Durens, M.; Nestor, J.; Williams, M.; Herold, K.; Niescier, R.F.; Lunden, J.W.; Phillips, A.W.; Lin, Y.C.; Dykxhoorn, D.M.; Nestor, M.W. High-throughput screening of human induced pluripotent stem cell-derived brain organoids. J. Neurosci. Methods 2020, 335, 108627. [Google Scholar] [CrossRef]
- Schwartz, M.P.; Hou, Z.; Propson, N.E.; Zhang, J.; Engstrom, C.J.; Santos Costa, V.; Jiang, P.; Nguyen, B.K.; Bolin, J.M.; Daly, W.; et al. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 12516–12521. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Hemmer, K.; Kaoma, T.; Smits, L.M.; Bolognin, S.; Lucarelli, P.; Rosety, I.; Zagare, A.; Antony, P.; Nickels, S.L.; et al. Machine learning-assisted neurotoxicity prediction in human midbrain organoids. Parkinsonism Relat. Disord. 2020, 75, 105–109. [Google Scholar] [CrossRef] [PubMed]

| Gene | Phenotypes Described | Compound Testing | Phenotype Restored | Reference |
|---|---|---|---|---|
| GBA mutations | increased levels of αSyn; reduced lysosomal GCase levels, reduced lysosomal GCase activity | small-molecule noninhibitory chaperone of GCase NCGC607 | reduced αSyn levels and associated toxicity | [76] |
| GBA (N370S/c.84dupG), SNCA- triplication | presence of amyloidogenic αSyn within cell bodies and neurites; accumulation of insoluble αSyn; reduced neuronal viability; reduced lysosomal activity of GCase | small-molecule GCase modulator 758 | improved GCase activity; reduced αSyn levels | [77] |
| GBA (c.84dupG frameshift mutation) LRRK2 and Parkin mutations | reduced amounts of GCase; decreased GCase enzymatic activity; accumulation of oxidized dopamine | small-molecule GCase modulator S-181 | increased amounts of lysosomal GCase; enhanced GCase enzymatic activity; decreased dopamine oxidation | [78] |
| GBA (c.84dupG frameshift mutation) | increased acid ceramidase activity in the context of decreased GCase, leading to intracellular accumulation of αSyn | carmofur, acid ceramidase inhibitor | reduced αSyn levels | [79] |
| DJ-1 mutations | mitochondrial oxidant stress causing lysosomal dysfunction and αSyn accumulation | mitochondrial antioxidants | diminished accumulation of oxidized dopamine; improved lysosomal GCase activity and proteolysis | [80] |
| LRRK2 (G2019S) | reduced neurite outgrowth; increased sensitivity to oxidative stress | ERK phosphorylation inhibitor PD0325901 or LRRK2 kinase inhibitor LRRK2-IN1 | increased neurite growth; reduced cytotoxicity | [71] |
| LRRK2 (G2019S) | neurite collapse; altered ER calcium homeostasis | LRRK2 kinase inhibitor Mli-2 or LRRK2-ASO | rescued neurite collapse | [74] |
| SNCA (G209A) | αSyn aggregation; mitochondrial dysfunction; increased susceptibility to mitochondrial toxins | small molecule targeting MEF2C-PGC1α (isoxazole) | reduced apoptosis | [36] |
| SNCA (G209A) | αSyn aggregation; compromised neurite outgrowth and axonal neuropathology; defective synaptic connectivity | small molecules targeting αSyn (NPT100-18A, NPT100-14A or ELN484228) | improved neurite outgrowth; rescue of axonal pathology and morphological restoration of the neuronal network | [38] |
| OPA1 mutation | mitochondrial dysfunction; impaired oxidative phosphorylation and high oxidative stress levels leading to neuronal cell loss | necrostatin-1, specific necroptosis inhibitor | increased survival | [58] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouroupi, G.; Antoniou, N.; Prodromidou, K.; Taoufik, E.; Matsas, R. Patient-Derived Induced Pluripotent Stem Cell-Based Models in Parkinson’s Disease for Drug Identification. Int. J. Mol. Sci. 2020, 21, 7113. https://doi.org/10.3390/ijms21197113
Kouroupi G, Antoniou N, Prodromidou K, Taoufik E, Matsas R. Patient-Derived Induced Pluripotent Stem Cell-Based Models in Parkinson’s Disease for Drug Identification. International Journal of Molecular Sciences. 2020; 21(19):7113. https://doi.org/10.3390/ijms21197113
Chicago/Turabian StyleKouroupi, Georgia, Nasia Antoniou, Kanella Prodromidou, Era Taoufik, and Rebecca Matsas. 2020. "Patient-Derived Induced Pluripotent Stem Cell-Based Models in Parkinson’s Disease for Drug Identification" International Journal of Molecular Sciences 21, no. 19: 7113. https://doi.org/10.3390/ijms21197113
APA StyleKouroupi, G., Antoniou, N., Prodromidou, K., Taoufik, E., & Matsas, R. (2020). Patient-Derived Induced Pluripotent Stem Cell-Based Models in Parkinson’s Disease for Drug Identification. International Journal of Molecular Sciences, 21(19), 7113. https://doi.org/10.3390/ijms21197113