iPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine



Abstract
1. Introduction
2. iPSCs Applications
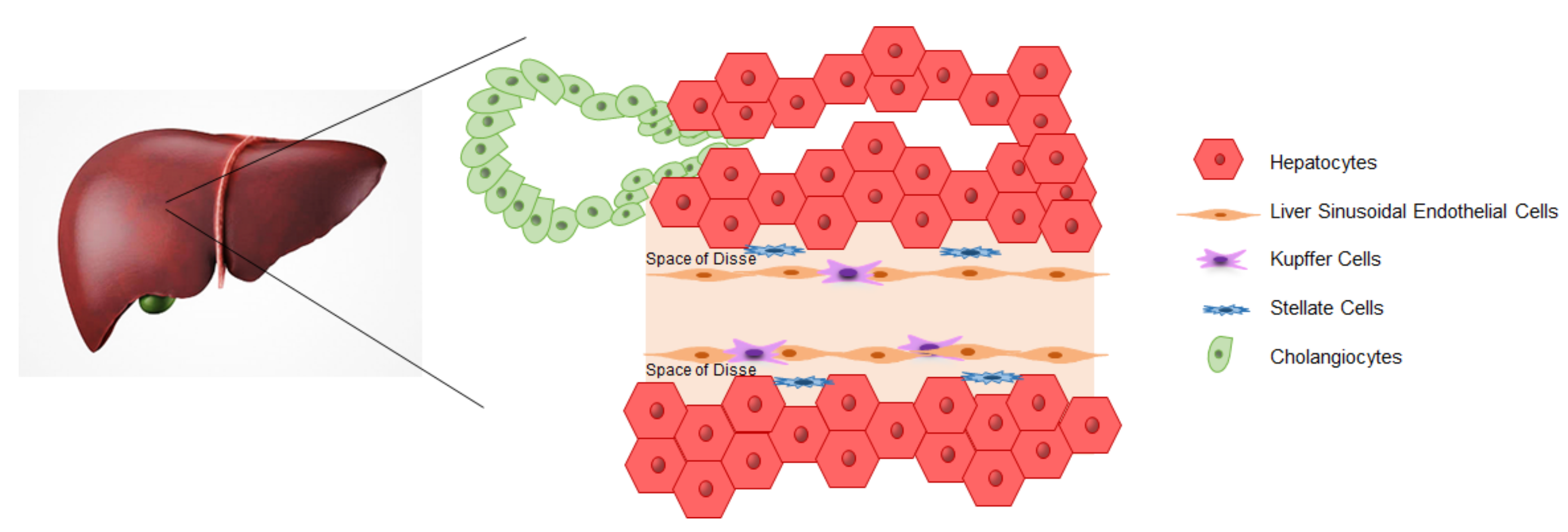
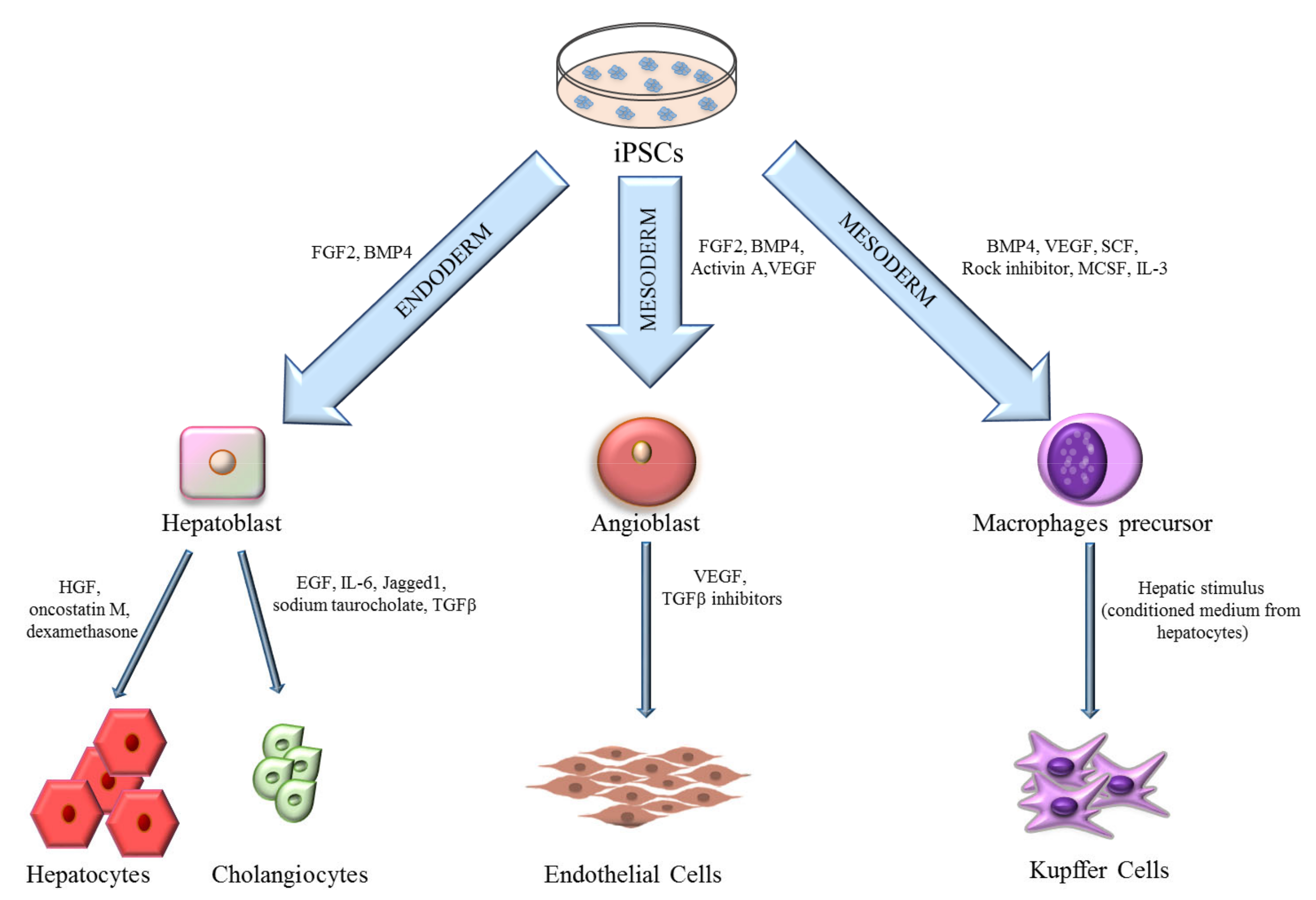
3. iPSCs-Derived Liver Cells
3.1. Hepatocytes
3.2. Cholangiocytes
3.3. Endothelial Cells
3.4. Kupffer Cells
4. Generation of Organoids
5. iPSCs-Derived Liver Organoids
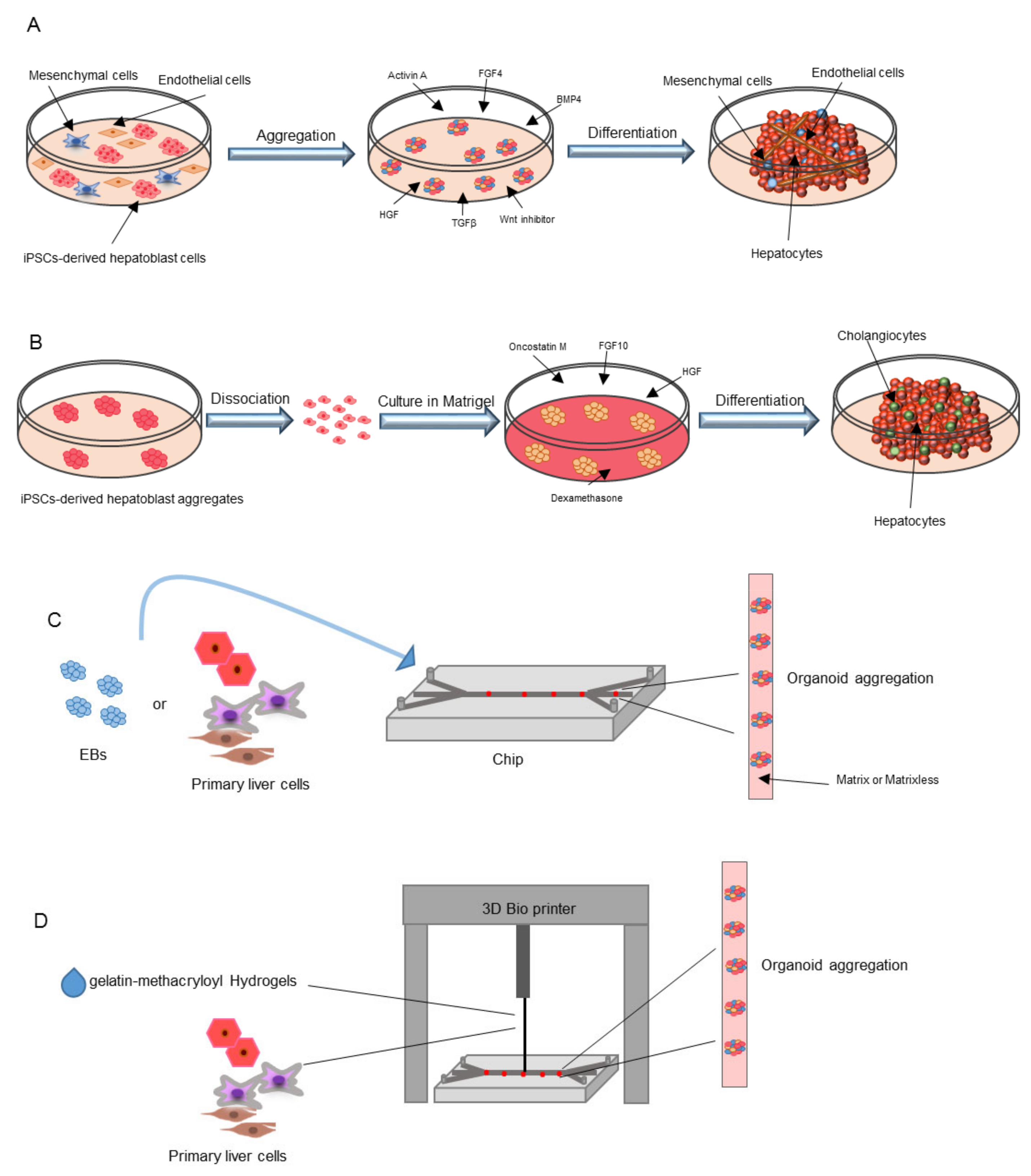
5.1. Co-Culture Methods
5.2. iPSC-Derived Organoids
5.3. Liver-on-a-Chip
5.4. 3D Printing Technology
6. Liver Organoids Applications
6.1. Regenerative Medicine
6.2. Drug Screening and Toxicity Test
6.3. Disease Modeling
6.4. Liver Cancer Organoids
7. Challenges and Limitations of Liver Organoids
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Furuta, T.; Furuya, K.; Zheng, Y.-W.; Oda, T. Novel alternative transplantation therapy for orthotopic liver transplantation in liver failure: A systematic review. World J. Transplant. 2020, 10, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Soltys, K.A.; Setoyama, K.; Tafaleng, E.N.; Soto Gutiérrez, A.; Fong, J.; Fukumitsu, K.; Nishikawa, T.; Nagaya, M.; Sada, R.; Haberman, K.; et al. Host conditioning and rejection monitoring in hepatocyte transplantation in humans. J. Hepatol. 2017, 66, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 107, 861–872. [Google Scholar] [CrossRef]
- Teshigawara, R.; Cho, J.; Kameda, M.; Tada, T. Mechanism of human somatic reprogramming to iPS cell. Lab. Investig. 2017, 97, 1152–1157. [Google Scholar] [CrossRef]
- Revilla, A.; González, C.; Iriondo, A.; Fernández, B.; Prieto, C.; Marín, C.; Liste, I. Current advances in the generation of human iPS cells: Implications in cell-based regenerative medicine. J. Tissue Eng. Regen. Med. 2016, 10, 893–907. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef]
- Koledova, Z. 3D cell culture: An introduction. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1612, pp. 1–11. [Google Scholar]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling physiological events in 2D vs. 3D cell culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, O.; Hesley, J.; Rusyn, I.; Cromwell, E.F. High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells. Assay Drug Dev. Technol. 2014, 12, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. DMM Dis. Models Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Paik, D.T.; Chandy, M.; Wu, J.C. Patient and disease—Specific induced pluripotent stem cells for discovery of personalized cardiovascular drugs and therapeutics. Pharmacol. Rev. 2020, 72, 320–342. [Google Scholar] [CrossRef]
- Csöbönyeiová, M.; Polák, Š.; Danišovič, L. Toxicity testing and drug screening using iPSC-derived hepatocytes, cardiomyocytes, and neural cells. Can. J. Physiol. Pharmacol. 2016, 94, 687–694. [Google Scholar] [CrossRef]
- Williams, T.M. Human leukocyte antigen gene polymorphism and the histocompatibility laboratory. J. Mol. Diagn. 2001, 3, 98–104. [Google Scholar] [CrossRef]
- Huang, C.Y.; Liu, C.L.; Ting, C.Y.; Chiu, Y.T.; Cheng, Y.C.; Nicholson, M.W.; Hsieh, P.C.H. Human iPSC banking: Barriers and opportunities. J. Biomed. Sci. 2019, 26, 1–14. [Google Scholar] [CrossRef]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient hla types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef]
- Solomon, S.; Pitossi, F.; Rao, M.S. Banking on iPSC- Is it Doable and is it Worthwhile. Stem Cell Rev. Rep. 2015, 11, 1–10. [Google Scholar] [CrossRef]
- Nakatsuji, N.; Nakajima, F.; Tokunaga, K. HLA-haplotype banking and iPS cells. Nat. Biotechnol. 2008, 26, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Rim, Y.A.; Park, N.; Nam, Y.; Ham, D.S.; Kim, J.W.; Ha, H.Y.; Jung, J.W.; Jung, S.M.; Baek, I.C.; Kim, S.Y.; et al. Recent progress of national banking project on homozygous HLA-typed induced pluripotent stem cells in South Korea. J. Tissue Eng. Regen. Med. 2018, 12, e1531–e1536. [Google Scholar] [CrossRef] [PubMed]
- Umekage, M.; Sato, Y.; Takasu, N. Overview: An iPS cell stock at CiRA. Inflamm. Regen. 2019, 39, 17. [Google Scholar] [CrossRef] [PubMed]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Norbnop, P.; Ingrungruanglert, P.; Israsena, N.; Suphapeetiporn, K.; Shotelersuk, V. Generation and characterization of HLA-universal platelets derived from induced pluripotent stem cells. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Suzuki, D.; Flahou, C.; Yoshikawa, N.; Stirblyte, I.; Hayashi, Y.; Sawaguchi, A.; Akasaka, M.; Nakamura, S.; Higashi, N.; Xu, H.; et al. Stem Cell Reports Article iPSC-Derived Platelets Depleted of HLA Class I Are Inert to Anti-HLA Class I and Natural Killer Cell Immunity. Stem Cell Rep. 2020, 14, 49–59. [Google Scholar] [CrossRef]
- Tremblay, K.D.; Zaret, K.S. Distinct populations of endoderm cells converge to generate the embryonic liver bud and ventral foregut tissues. Dev. Biol. 2005, 280, 87–99. [Google Scholar] [CrossRef]
- Rossi, J.M.; Dunn, N.R.; Hogan, B.L.M.; Zaret, K.S. Distinct mesodermal signals, including BMPs from the septum, transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001, 15, 1998–2009. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yoshitomi, H.; Rossant, J.; Zaret, K.S. Liver organogenesis promoted by endothelial cells prior to vascular function. Science 2001, 294, 559–563. [Google Scholar] [CrossRef]
- Parviz, F.; Matullo, C.; Garrison, W.D.; Savatski, L.; Adamson, J.W.; Ning, G.; Kaestner, K.H.; Rossi, J.M.; Zaret, K.S.; Duncan, S.A. Hepatocyte nuclear factor 4α controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 2003, 34, 292–296. [Google Scholar] [CrossRef]
- Gordillo, M.; Evans, T.; Gouon-Evans, V. Orchestrating liver development. Development 2015, 142, 2094–2108. [Google Scholar] [CrossRef]
- Zaret, K.S. Regulatory phases of early liver development: Paradigms of organogenesis. Nat. Rev. Genet. 2002, 3, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Ober, E.A.; Lemaigre, F.P. Development of the liver: Insights into organ and tissue morphogenesis. J. Hepatol. 2018, 68, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Shinozaki, K.; Shannon, J.M.; Kouskoff, V.; Kennedy, M.; Woo, S.; Fehling, H.J.; Keller, G. Development of definitive endoderm from embryonic stem cells in culture. Development 2004, 131, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Kinoshita, T.; Ito, Y.; Matsui, T.; Morikawa, Y.; Senba, E.; Nakashima, K.; Taga, T.; Yoshida, K.; Kishimoto, T.; et al. Fetal liver development requires a paracrine action of oncostatin M through the gp130 signal transducer. EMBO J. 1999, 18, 2127–2136. [Google Scholar] [CrossRef]
- Kamiya, A.; Kinoshita, T.; Miyajima, A. Oncostatin M and hepatocyte growth factor induce hepatic maturation via distinct signaling pathways. FEBS Lett. 2001, 492, 90–94. [Google Scholar] [CrossRef]
- Lu, J.; Einhorn, S.; Venkatarangan, L.; Miller, M.; Mann, D.A.; Watkins, P.B.; Lecluyse, E. Morphological and Functional Characterization and Assessment of iPSC-Derived Hepatocytes for In Vitro Toxicity Testing. Toxicol. Sci. 2015, 147, 39–54. [Google Scholar] [CrossRef]
- Varghese, D.S.; Alawathugoda, T.T.; Ansari, S.A. Fine Tuning of Hepatocyte Differentiation from Human Embryonic Stem Cells: Growth Factor vs. Small Molecule-Based Approaches. Stem Cells Int. 2019. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.P.; Zhang, F.; Atkinson-Dell, R.; Rowe, C.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef]
- Raju, R.; Chau, D.; Notelaers, T.; Myers, C.L.; Verfaillie, C.M.; Hu, W.S. In vitro pluripotent stem cell differentiation to hepatocyte ceases further maturation at an equivalent stage of e15 in mouse embryonic liver development. Stem Cells Dev. 2018, 27, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Waring, J.F.; Ciurlionis, R.; Jolly, R.A.; Heindel, M.; Gagne, G.; Fagerland, J.A.; Ulrich, R.G. Isolated human hepatocytes in culture display markedly different gene expression patterns depending on attachment status. Toxicol. Vitr. 2003, 17, 693–701. [Google Scholar] [CrossRef]
- Park, K.M.; Hussein, K.H.; Hong, S.H.; Ahn, C.; Yang, S.R.; Park, S.M.; Kweon, O.K.; Kim, B.M.; Woo, H.M. Decellularized Liver Extracellular Matrix as Promising Tools for Transplantable Bioengineered Liver Promotes Hepatic Lineage Commitments of Induced Pluripotent Stem Cells. Tissue Eng. Part A 2016, 22, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.; Yeh, H.; Yarmush, M.L.; Uygun, B.E. Decellularized human liver extracellular matrix (hDLM)-mediated hepatic differentiation of human induced pluripotent stem cells (hIPSCs). J. Tissue Eng. Regen. Med. 2018, 12, e1962–e1973. [Google Scholar] [CrossRef]
- Grant, R.; Hallett, J.; Forbes, S.; Hay, D.; Callanan, A. Blended electrospinning with human liver extracellular matrix for engineering new hepatic microenvironments. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Luo, Y.; Lou, C.; Zhang, S.; Zhu, Z.; Xing, Q.; Wang, P.; Liu, T.; Liu, H.; Li, C.; Shi, W.; et al. Three-dimensional hydrogel culture conditions promote the differentiation of human induced pluripotent stem cells into hepatocytes. Cytotherapy 2018, 20, 95–107. [Google Scholar] [CrossRef]
- Mobarra, N.; Soleimani, M.; Ghayour-Mobarhan, M.; Safarpour, S.; Ferns, G.A.; Pakzad, R.; Pasalar, P. Hybrid poly-l-lactic acid/poly(ε-caprolactone) nanofibrous scaffold can improve biochemical and molecular markers of human induced pluripotent stem cell-derived hepatocyte-like cells. J. Cell. Physiol. 2019, 234, 11247–11255. [Google Scholar] [CrossRef]
- Zakikhan, K.; Pournasr, B.; Vosough, M.; Nassiri-Asl, M. In vitro generated hepatocyte-like cells: A novel tool in regenerative medicine and drug discovery. Cell J. 2017, 19, 204–217. [Google Scholar]
- Tabibian, J.H.; Masyuk, A.I.; Masyuk, T.V.; O’Hara, S.P.; LaRusso, N.F. Physiology of cholangiocytes. Compr. Physiol. 2013, 3, 541–565. [Google Scholar] [CrossRef]
- Sampaziotis, F.; De Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef]
- Ogawa, M.; Ogawa, S.; Bear, C.E.; Ahmadi, S.; Chin, S.; Li, B.; Grompe, M.; Keller, G.; Kamath, B.M.; Ghanekar, A. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 853–861. [Google Scholar] [CrossRef] [PubMed]
- De Assuncao, T.M.; Sun, Y.; Jalan-Sakrikar, N.; Drinane, M.C.; Huang, B.Q.; Li, Y.; Davila, J.I.; Wang, R.; O’Hara, S.P.; Lomberk, G.A.; et al. Development and characterization of human-induced pluripotent stem cell-derived cholangiocytes. Lab. Investig. 2015, 95, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Mitani, S.; Nagamoto, Y.; Sakurai, F.; Tachibana, M.; Taniguchi, Y.; Sekiguchi, K.; Mizuguchi, H. Laminin 411 and 511 promote the cholangiocyte differentiation of human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2016, 474, 91–96. [Google Scholar] [CrossRef]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, S.; Hong, Y.K.; Harvey, N.; Schacht, V.; Matsuda, K.; Libermann, T.; Detmar, M. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am. J. Pathol. 2003, 162, 575–586. [Google Scholar] [CrossRef]
- Lindskog, H.; Kim, Y.H.; Jelin, E.B.; Kong, Y.; Guevara-Gallardo, S.; Kim, T.N.; Wang, R.A. Molecular identification of venous progenitors in the dorsal aorta reveals an aortic origin for the cardinal vein in mammals. Development 2014, 141, 1120–1128. [Google Scholar] [CrossRef]
- Dela Paz, N.G.; D’Amore, P.A. Arterial versus venous endothelial cells. Cell Tissue Res. 2009, 335, 5–16. [Google Scholar] [CrossRef]
- Hewett, P.W. Isolation and culture of human endothelial cells from micro- and macro-vessels. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2016; Volume 1430, pp. 61–76. [Google Scholar]
- Di Bernardini, E.; Campagnolo, P.; Margariti, A.; Zampetaki, A.; Karamariti, E.; Hu, Y.; Xu, Q. Endothelial Lineage differentiation from induced Pluripotent Stem Cells is regulated by MicroRNA-21 and transforming growth Factor β2 (TGF-β2) Pathways. J. Biol. Chem. 2014, 289, 3383–3393. [Google Scholar] [CrossRef]
- Choi, K.-D.; Yu, J.; Smuga-Otto, K.; Salvagiotto, G.; Rehrauer, W.; Vodyanik, M.; Thomson, J.; Slukvin, I. Hematopoietic and Endothelial Differentiation of Human Induced Pluripotent Stem Cells. Stem Cells 2009, 27, 559–567. [Google Scholar] [CrossRef]
- Minami, H.; Tashiro, K.; Okada, A.; Hirata, N.; Yamaguchi, T.; Takayama, K.; Mizuguchi, H.; Kawabata, K. Generation of Brain Microvascular Endothelial-Like Cells from Human Induced Pluripotent Stem Cells by Co-Culture with C6 Glioma Cells. PLoS ONE 2015, 10, e0128890. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.M.; Costa, E.B.O.; Orellana, M.D.; Picanço-Castro, V.; Covas, D.T. OP9 stromal cells proteins involved in hematoendothelial differentiation from human embryonic stem cells. Cell. Reprogramming 2015, 17, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Moon, S.H.; Lee, S.H.; Lee, D.R.; Koh, G.Y.; Chung, H.M. Effective isolation and culture of endothelial cells in embryoid body differentiated from human embryonic stem cells. Stem Cells Dev. 2007, 16, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Goldman, O.; Feraud, O.; Di Ponio, J.B.; Driancourt, C.; Clay, D.; Le Bousse-Kerdiles, M.C.; Bennaceur-Griscelli, A.; Uzan, G. A boost of BMP4 accelerates the commitment of human embryonic stem cells to the endothelial lineage. Stem Cells 2009, 27, 1750–1759. [Google Scholar] [CrossRef]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef]
- Olgasi, C.; Talmon, M.; Merlin, S.; Cucci, A.; Richaud-Patin, Y.; Ranaldo, G.; Colangelo, D.; Di Scipio, F.; Berta, G.N.; Borsotti, C.; et al. Patient-Specific iPSC-Derived Endothelial Cells Provide Long-Term Phenotypic Correction of Hemophilia A. Stem Cell Rep. 2018, 11. [Google Scholar] [CrossRef]
- Nourse, M.B.; Halpin, D.E.; Scatena, M.; Mortisen, D.J.; Tulloch, N.L.; Hauch, K.D.; Torok-Storb, B.; Ratner, B.D.; Pabon, L.; Murry, C.E. VEGF induces differentiation of functional endothelium from human embryonic stem cells: Implications for tissue engineering. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 80–89. [Google Scholar] [CrossRef]
- Wang, L.; Xiang, M.; Liu, Y.; Sun, N.; Lu, M.; Shi, Y.; Wang, X.; Meng, D.; Chen, S.; Qin, J. Human induced pluripotent stem cells derived endothelial cells mimicking vascular inflammatory response under flow. Biomicrofluidics 2016, 10. [Google Scholar] [CrossRef]
- Bai, H.; Gao, Y.; Hoyle, D.L.; Cheng, T.; Wang, Z.Z. Suppression of Transforming Growth Factor-β Signaling Delays Cellular Senescence and Preserves the Function of Endothelial Cells Derived from Human Pluripotent Stem Cells. Stem Cells Transl. Med. 2017, 6, 589–600. [Google Scholar] [CrossRef]
- Orlova, V.V.; Van Den Hil, F.E.; Petrus-Reurer, S.; Drabsch, Y.; Ten Dijke, P.; Mummery, C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014, 9, 1514–1531. [Google Scholar] [CrossRef]
- Palpant, N.J.; Pabon, L.; Friedman, C.E.; Roberts, M.; Hadland, B.; Zaunbrecher, R.J.; Bernstein, I.; Zheng, Y.; Murry, C.E. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat. Protoc. 2017, 12, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Dutton, J.R.; Su, L.; Zhang, J.; Ye, L. The influence of a spatiotemporal 3D environment on endothelial cell differentiation of human induced pluripotent stem cells. Biomaterials 2014, 35, 3786–3793. [Google Scholar] [CrossRef] [PubMed]
- Ditadi, A.; Sturgeon, C.M.; Tober, J.; Awong, G.; Kennedy, M.; Yzaguirre, A.D.; Azzola, L.; Ng, E.S.; Stanley, E.G.; French, D.L.; et al. Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat. Cell Biol. 2015, 17, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Su, J.; Liang, P. Modeling cadmium-induced endothelial toxicity using human pluripotent stem cell-derived endothelial cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Sances, S.; Ho, R.; Vatine, G.; West, D.; Laperle, A.; Meyer, A.; Godoy, M.; Kay, P.S.; Mandefro, B.; Hatata, S.; et al. Human iPSC-Derived Endothelial Cells and Microengineered Organ-Chip Enhance Neuronal Development. Stem Cell Rep. 2018, 10, 1222–1236. [Google Scholar] [CrossRef]
- Masumoto, H.; Yamashita, J.K. Human iPS cell-engineered three-dimensional cardiac tissues perfused by capillary networks between host and graft. Inflamm. Regen. 2018, 38. [Google Scholar] [CrossRef]
- Koui, Y.; Kido, T.; Ito, T.; Oyama, H.; Chen, S.W.; Katou, Y.; Shirahige, K.; Miyajima, A. An In Vitro Human Liver Model by iPSC-Derived Parenchymal and Non-parenchymal Cells. Stem Cell Rep. 2017, 9, 490–498. [Google Scholar] [CrossRef]
- Danoy, M.; Poulain, S.; Koui, Y.; Tauran, Y.; Scheidecker, B.; Kido, T.; Miyajima, A.; Sakai, Y.; Plessy, C.; Leclerc, E. Transcriptome profiling of hiPSC-derived LSECs with nanoCAGE. Mol. Omics 2020, 16, 138–146. [Google Scholar] [CrossRef]
- Selmi, C.; Mackay, I.R.; Gershwin, M.E. The immunological milieu of the liver. Semin. Liver Dis. 2007, 27, 129–139. [Google Scholar] [CrossRef]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, Inflammation, and Insulin Resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.A.; Holman, N.S.; Green, A.M.; Andersen, M.E.; Lecluyse, E.L. Co-culture of Hepatocytes and Kupffer Cells as an in Vitro Model of Inflammation and Drug-Induced Hepatotoxicity. J. Pharm. Sci. 2016, 105, 950–964. [Google Scholar] [CrossRef]
- Pfeiffer, E.; Kegel, V.; Zeilinger, K.; Hengstler, J.G.; Nüssler, A.K.; Seehofer, D.; Damm, G. Featured Article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells. Exp. Biol. Med. 2015, 240, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Merlin, S.; Bhargava, K.K.; Ranaldo, G.; Zanolini, D.; Palestro, C.J.; Santambrogio, L.; Prat, M.; Follenzi, A.; Gupta, S. Kupffer cell transplantation in mice for elucidating monocyte/macrophage biology and for potential in cell or gene therapy. Am. J. Pathol. 2016, 186, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Landmann-Suter, R. Generation and use of a mouse Kupffer cell line. ALTEX 2007, 24, 42–45. [Google Scholar] [PubMed]
- Heuff, G.; Van De Loosdrecht, A.A.; Betjes, M.G.H.; Beelen, R.H.J.; Meijer, S. Isolation and purification of large quantities of fresh human Kupffer cells, which are cytotoxic against colon carcinoma. Hepatology 1995, 21, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xue, C.; Shah, R.; Bermingham, K.; Hinkle, C.C.; Li, W.; Rodrigues, A.; Tabita-Martinez, J.; Millar, J.S.; Cuchel, M.; et al. Functional Analysis and Transcriptomic Profiling of iPSC-Derived Macrophages and Their Application in Modeling Mendelian Disease. Circ. Res. 2015, 117, 17–28. [Google Scholar] [CrossRef]
- van Wilgenburg, B.; Browne, C.; Vowles, J.; Cowley, S.A. Efficient, Long Term Production of Monocyte-Derived Macrophages from Human Pluripotent Stem Cells under Partly-Defined and Fully-Defined Conditions. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef] [PubMed]
- Panicker, L.M.; Miller, D.; Awad, O.; Bose, V.; Lun, Y.; Park, T.S.; Zambidis, E.T.; Sgambato, J.A.; Feldman, R.A. Gaucher iPSC-Derived Macrophages Produce Elevated Levels of Inflammatory Mediators and Serve as a New Platform for Therapeutic Development. Stem Cells 2014, 32, 2338–2349. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef]
- Buchrieser, J.; James, W.; Moore, M.D. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny with MYB-Independent Tissue-Resident Macrophages. Stem Cell Rep. 2017, 8, 334–345. [Google Scholar] [CrossRef]
- Tasnim, F.; Xing, J.; Huang, X.; Mo, S.; Wei, X.; Tan, M.H.; Yu, H. Generation of mature kupffer cells from human induced pluripotent stem cells. Biomaterials 2019, 192, 377–391. [Google Scholar] [CrossRef]
- Simian, M.; Bissell, M.J. Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef]
- Lou, Y.R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef]
- Weaver, V.M.; Lelièvre, S.; Lakins, J.N.; Chrenek, M.A.; Jones, J.C.R.; Giancotti, F.; Werb, Z.; Bissell, M.J. β4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2002, 2, 205–216. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J. The Differentiated State of Normal and Malignant Cells or How to Define a “Normal” Cell in Culture. Int. Rev. Cytol. 1981, 70, 27–100. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Aggeler, J.; Farson, D.A.; Hatier, C.; Hassell, J.; Bissell, M.J. Influence of a reconstituted basement membrane and its components on casein gene expression and secretion in mouse mammary epithelial cells. Proc. Natl. Acad. Sci. USA 1987, 84, 136–140. [Google Scholar] [CrossRef]
- Roskelley, C.D.; Desprez, P.Y.; Bissell, M.J. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc. Natl. Acad. Sci. USA 1994, 91, 12378–12382. [Google Scholar] [CrossRef] [PubMed]
- Ootani, A.; Toda, S.; Fujimoto, K.; Sugihara, H. Foveolar differentiation of mouse gastric mucosa in vitro. Am. J. Pathol. 2003, 162, 1905–1912. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve Stem Cells Drive Self-Renewal in the Stomach and Build Long-Lived Gastric Units In Vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Bowen, W.C.; Mulè, K.; Stolz, D.B. Histological organization in hepatocyte organoid cultures. Am. J. Pathol. 2001, 159, 1877–1887. [Google Scholar] [CrossRef]
- Huch, M.; Dorrell, C.; Boj, S.F.; Van Es, J.H.; Li, V.S.W.; Van De Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5 + liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Zhang, R.R.; Koike, H.; Kimura, M.; Yoshizawa, E.; Enomura, M.; Koike, N.; Sekine, K.; Taniguchi, H. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat. Protoc. 2014, 9, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Ehashi, T.; Koyama, T.; Ookawa, K.; Ohshima, N.; Miyoshi, H. Effects of oncostatin M on secretion of vascular endothelial growth factor and reconstruction of liver-like structure by fetal liver cells in monolayer and three-dimensional cultures. J. Biomed. Mater. Res. Part A 2007, 82, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Kojima, N.; Kinoshita, T.; Sakai, Y.; Miyaijma, A. Maturation of fetal hepatocytes in vitro by extracellular matrices and oncostatin M: Induction of tryptophan oxygenase. Hepatology 2002, 35, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Clotman, F.; Jacquemin, P.; Plumb-Rudewiez, N.; Pierreux, C.E.; Van Der Smissen, P.; Dietz, H.C.; Courtoy, P.J.; Rousseau, G.G.; Lemaigre, F.P. Control of liver cell fate decision by a gradient of TGFβ signaling modulated by Onecut transcription factors. Genes Dev. 2005, 19, 1849–1854. [Google Scholar] [CrossRef]
- Gelly, J.L.; Richoux, J.P.; Grignon, G.; Bouhnik, J.; Baussant, T.; Alhenc-Gelas, F.; Corvol, P. Immunocytochemical localization of albumin, transferrin, angiotensinogen and kininogens during the initial stages of the rat liver differentiation. Histochemistry 1991, 96, 7–12. [Google Scholar] [CrossRef]
- Shanmukhappa, K.; Matte, U.; Degen, J.L.; Bezerra, J.A. Plasmin-mediated proteolysis is required for hepatocyte growth factor activation during liver repair. J. Biol. Chem. 2009, 284, 12917–12923. [Google Scholar] [CrossRef]
- Tiggelman, A.M.B.C.; Linthorst, C.; Boers, W.; Brand, H.S.; Chamuleau, R.A.F.M. Transforming growth factor-β-induced collagen synthesis by human liver myofibroblasts is inhibited by α2-macroglobulin. J. Hepatol. 1997, 26, 1220–1228. [Google Scholar] [CrossRef]
- Asai, A.; Aihara, E.; Watson, C.; Mourya, R.; Mizuochi, T.; Shivakumar, P.; Phelan, K.; Mayhew, C.; Helmrath, M.; Takebe, T.; et al. Paracrine signals regulate human liver organoid maturation from induced pluripotent stem cells. Development 2017, 144, 1056–1064. [Google Scholar] [CrossRef]
- Hu, J.; Srivastava, K.; Wieland, M.; Runge, A.; Mogler, C.; Besemfelder, E.; Terhardt, D.; Vogel, M.J.; Cao, L.; Korn, C.; et al. Endothelial cell-derived Angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science 2014, 343, 416–419. [Google Scholar] [CrossRef]
- Nonaka, H.; Watabe, T.; Saito, S.; Miyazono, K.; Miyajima, A. Development of stabilin2+ endothelial cells from mouse embryonic stem cells by inhibition of TGFβ/activin signaling. Biochem. Biophys. Res. Commun. 2008, 375, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wu, D.; Ren, Y.; Huang, Y.; Feng, B.; Zhao, N.; Zhang, T.; Chen, X.; Chen, S.; Xu, A. Generation of hepatobiliary organoids from human induced pluripotent stem cells. J. Hepatol. 2019, 70, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Pettinato, G.; Lehoux, S.; Ramanathan, R.; Salem, M.M.; He, L.X.; Muse, O.; Flaumenhaft, R.; Thompson, M.T.; Rouse, E.A.; Cummings, R.D.; et al. Generation of fully functional hepatocyte-like organoids from human induced pluripotent stem cells mixed with Endothelial Cells. Sci. Rep. 2019, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Xu, D.; Garfin, P.M.; Ehmer, U.; Hurwitz, M.; Enns, G.; Michie, S.; Wu, M.; Zheng, M.; Nishimura, T.; et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef]
- Toh, Y.C.; Lim, T.C.; Tai, D.; Xiao, G.; Van Noort, D.; Yu, H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef]
- Jang, M.; Neuzil, P.; Volk, T.; Manz, A.; Kleber, A. On-chip three-dimensional cell culture in phaseguides improves hepatocyte functions in vitro. Biomicrofluidics 2015, 9. [Google Scholar] [CrossRef]
- Zhu, L.; Fan, X.; Wang, B.; Liu, L.; Yan, X.; Zhou, L.; Zeng, Y.; Poznansky, M.C.; Wang, L.; Chen, H.; et al. Biomechanically primed liver microtumor array as a high-throughput mechanopharmacological screening platform for stroma-reprogrammed combinatorial therapy. Biomaterials 2017, 124, 12–24. [Google Scholar] [CrossRef]
- Norona, L.M.; Nguyen, D.G.; Gerber, D.A.; Presnell, S.C.; Lecluyse, E.L. Modeling Compound-Induced Fibrogenesis In Vitro Using Three-Dimensional Bioprinted Human Liver Tissues. Toxicol. Sci. 2016, 154, 354–367. [Google Scholar] [CrossRef]
- Nguyen, D.G.; Funk, J.; Robbins, J.B.; Crogan-Grundy, C.; Presnell, S.C.; Singer, T.; Roth, A.B. Bioprinted 3D primary liver tissues allow assessment of organ-level response to clinical drug induced toxicity in vitro. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Zhang, Y.S.; Shin, S.R.; Calzone, G.; et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Ortega-Ribera, M.; Guimerà, X.; Sowade, E.; Zea, M.; Illa, X.; Ramon, E.; Villa, R.; Gracia-Sancho, J.; Gabriel, G. Online oxygen monitoring using integrated inkjet-printed sensors in a liver-on-a-chip system. Lab Chip 2018, 18, 2023–2035. [Google Scholar] [CrossRef] [PubMed]
- Grix, T.; Ruppelt, A.; Thomas, A.; Amler, A.K.; Noichl, B.P.; Lauster, R.; Kloke, L. Bioprinting perfusion-enabled liver equivalents for advanced organ-on-a-chip applications. Genes 2018, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Goulart, E.; De Caires-Junior, L.C.; Telles-Silva, K.A.; Araujo, B.H.S.; Rocco, S.A.; Sforca, M.; De Sousa, I.L.; Kobayashi, G.S.; Musso, C.M.; Assoni, A.F.; et al. 3D bioprinting of liver spheroids derived from human induced pluripotent stem cells sustain liver function and viability in vitro. Biofabrication 2020. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wei, W.; Chen, Z.; Lin, B.; Zhao, W.; Luo, Y.; Zhang, X. Engineered liver-on-a-chip platform to mimic liver functions and its biomedical applications: A review. Micromachines 2019, 10, 676. [Google Scholar] [CrossRef]
- Frey, O.; Misun, P.M.; Fluri, D.A.; Hengstler, J.G.; Hierlemann, A. Reconfigurable microfluidic hanging drop network for multi-tissue interaction and analysis. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Ikeuchi, M.; Noguchi, H.; Yagi, T.; Hayashi, S. Spheroid Formation and Evaluation of Hepatic Cells in a Three-Dimensional Culture Device. Cell Med. 2015, 8, 47–56. [Google Scholar] [CrossRef]
- Misun, P.M.; Rothe, J.; Schmid, Y.R.F.; Hierlemann, A.; Frey, O. Multi-analyte biosensor interface for real-time monitoring of 3D microtissue spheroids in hanging-drop networks. Microsyst. Nanoeng. 2016, 2. [Google Scholar] [CrossRef]
- Aeby, E.A.; Misun, P.M.; Hierlemann, A.; Frey, O. Microfluidic Hydrogel Hanging-Drop Network for Long-Term Culturing of 3D Microtissues and Simultaneous High-Resolution Imaging. Adv. Biosyst. 2018, 2, 1800054. [Google Scholar] [CrossRef]
- Ma, L.D.; Wang, Y.T.; Wang, J.R.; Wu, J.L.; Meng, X.S.; Hu, P.; Mu, X.; Liang, Q.L.; Luo, G.A. Design and fabrication of a liver-on-a-chip platform for convenient, highly efficient, and safe: In situ perfusion culture of 3D hepatic spheroids. Lab Chip 2018, 18, 2547–2562. [Google Scholar] [CrossRef]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; Van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; Van Wenum, M.; Fuchs, S.A.; De Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e19. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, H.; Luu, N.T.; Alwahsh, S.M.; Ginai, M.; Alhaque, S.; Dong, H.; Tomaz, R.A.; Vernay, B.; Vigneswara, V.; Hallett, J.M.; et al. 3D human liver tissue from pluripotent stem cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch. Toxicol. 2018, 92, 3117–3129. [Google Scholar] [CrossRef]
- Blackford, S.J.I.; Ng, S.S.; Segal, J.M.; King, A.J.F.; Austin, A.L.; Kent, D.; Moore, J.; Sheldon, M.; Ilic, D.; Dhawan, A.; et al. Validation of Current Good Manufacturing Practice Compliant Human Pluripotent Stem Cell-Derived Hepatocytes for Cell-Based Therapy. Stem Cells Transl. Med. 2019, 8, 124–137. [Google Scholar] [CrossRef]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells from Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Choi, S.M.; Kim, Y.; Shim, J.S.; Park, J.T.; Wang, R.H.; Leach, S.D.; Liu, J.O.; Deng, C.; Ye, Z.; Jang, Y.Y. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology 2013, 57, 2458–2468. [Google Scholar] [CrossRef]
- Cayo, M.A.; Mallanna, S.K.; Di Furio, F.; Jing, R.; Tolliver, L.B.; Bures, M.; Urick, A.; Noto, F.K.; Pashos, E.E.; Greseth, M.D.; et al. A Drug Screen using Human iPSC-Derived Hepatocyte-like Cells Reveals Cardiac Glycosides as a Potential Treatment for Hypercholesterolemia. Cell Stem Cell 2017, 20, 478–489.e5. [Google Scholar] [CrossRef]
- Jing, R.; Corbett, J.L.; Cai, J.; Beeson, G.C.; Beeson, C.C.; Chan, S.S.; Dimmock, D.P.; Lazcares, L.; Geurts, A.M.; Lemasters, J.J.; et al. A Screen Using iPSC-Derived Hepatocytes Reveals NAD+ as a Potential Treatment for mtDNA Depletion Syndrome. Cell Rep. 2018, 25, 1469–1484.e5. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Artegiani, B.; Clevers, H. Use and application of 3D-organoid technology. Hum. Mol. Genet. 2018, 27, R99–R107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Sampaziotis, F.; De Brito, M.C.; Geti, I.; Bertero, A.; Hannan, N.R.F.; Vallier, L. Directed differentiation of human induced pluripotent stem cells into functional cholangiocyte-like cells. Nat. Protoc. 2017, 12, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Akbari, S.; Sevinç, G.G.; Ersoy, N.; Basak, O.; Kaplan, K.; Sevinç, K.; Ozel, E.; Sengun, B.; Enustun, E.; Ozcimen, B.; et al. Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Rep. 2019, 13, 627–641. [Google Scholar] [CrossRef]
- Hu, J.; Lin, Y.Y.; Chen, P.J.; Watashi, K.; Wakita, T. Cell and Animal Models for Studying Hepatitis B Virus Infection and Drug Development. Gastroenterology 2019, 156, 338–354. [Google Scholar] [CrossRef]
- Nie, Y.Z.; Zheng, Y.W.; Miyakawa, K.; Murata, S.; Zhang, R.R.; Sekine, K.; Ueno, Y.; Takebe, T.; Wakita, T.; Ryo, A.; et al. Recapitulation of hepatitis B virus–host interactions in liver organoids from human induced pluripotent stem cells. EBioMedicine 2018, 35, 114–123. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Andersen, J.B. Liver cancer oncogenomics: Opportunities and dilemmas for clinical applications. Hepatic Oncol. 2015, 2, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Aberle, M.R.; Burkhart, R.A.; Tiriac, H.; Olde Damink, S.W.M.; Dejong, C.H.C.; Tuveson, D.A.; van Dam, R.M. Patient-derived organoid models help define personalized management of gastrointestinal cancer. Br. J. Surg. 2018, 105, e48–e60. [Google Scholar] [CrossRef] [PubMed]
- Takai, A.; Fako, V.; Dang, H.; Forgues, M.; Yu, Z.; Budhu, A.; Wang, X.W. Three-dimensional Organotypic Culture Models of Human Hepatocellular Carcinoma. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Zhang, B.; Sun, H.; Xu, Q.; Tan, Y.; Wang, G.; Luo, Q.; Xu, W.; Yang, S.; Li, J.; et al. Genomic characterization of a large panel of patient-derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget 2015, 6, 20160–20176. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- He, S.; Hu, B.; Li, C.; Lin, P.; Tang, W.G.; Sun, Y.F.; Feng, F.Y.M.; Guo, W.; Li, J.; Xu, Y.; et al. PDXliver: A database of liver cancer patient derived xenograft mouse models. BMC Cancer 2018, 18, 550. [Google Scholar] [CrossRef]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef]
- Saito, Y.; Nakaoka, T.; Muramatsu, T.; Ojima, H.; Sukeda, A.; Sugiyama, Y.; Uchida, R.; Furukawa, R.; Kitahara, A.; Sato, T.; et al. Induction of differentiation of intrahepatic cholangiocarcinoma cells to functional hepatocytes using an organoid culture system. Sci. Rep. 2018, 8, 2821. [Google Scholar] [CrossRef]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, S.; Kim, K.; Kim, S.H.; Chung, Y.J.; Lee, C. Studying cancer immunotherapy using patient-derived xenografts (PDXs) in humanized mice. Exp. Mol. Med. 2018, 50, 99. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front. Immunol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.; Melero, I.; Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 67, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Artegiani, B.; van Voorthuijsen, L.; Lindeboom, R.G.H.; Seinstra, D.; Heo, I.; Tapia, P.; López-Iglesias, C.; Postrach, D.; Dayton, T.; Oka, R.; et al. Probing the Tumor Suppressor Function of BAP1 in CRISPR-Engineered Human Liver Organoids. Cell Stem Cell 2019, 24, 927–943.e6. [Google Scholar] [CrossRef]
- Liver Biopsy Market Research Report. Available online: https://www.marketresearchfuture.com/reports/liver-biopsy-market-5534 (accessed on 3 July 2020).




{kind=link}
{kind=link}
{kind=link}
| Methodology | Author | Approach | In Vivo Transplantation and Survival | Advancement | Reference |
|---|---|---|---|---|---|
| Co-Culture Method | Takebe et al. | Co-culture of iPSCs-derived hepatic endoderm like cells with mesenchymal and human umbilical vein endothelial cells | Transplantation in TK-NOG mice after induced liver failure: survival up to 30 days | Vascularization of liver bud organoids and maturation in hepatocytes after in vivo transplantation | [113,114] |
| Asai et al. | Co-culture of hepatic-specified endoderm iPSCs with mesenchymal and human umbilical vein endothelial cells | Implantation under the kidney capsule of immunodeficient mice: high serum levels of human albumin up to 8 weeks and hepatic maturation of the liver organoid in vivo | Paracrine factors secreted by mesenchymal cells and ECs (HGF, ANG, A2M, PLG) induce the formation of liver organoids | [121] | |
| Pettinato et al. | Co-culture of iPSCs-derived embryoid bodies with Human Adipose Microvascular Endothelial Cells (HAMEC) | An immune-deficient rat model for acute liver failure was transplanted with iPSCs-derived embryoid bodies + HAMEC: survival rate of 66.7% 14 days after induction of liver failure | The addition of HAMEC during hepatic differentiation of iPSCs induces liver-specific gene expression improving hepatic cell functionality | [125] | |
| iPSC Derived Organoids | Koui et al. | iPSCs were differentiated into LSECs and hepatic stem cells by modulating TGFβ and Rho signaling pathways | ND | Self-renewal properties of iPSCs derived liver cells in 2D culture systems | [79] |
| Wu et al. | iPSCs were differentiated into hepatobiliary organoids using Activin A, BMP4, BMP2, FGF4, HGF, OSM and dexamethasone | Hepatobiliary organoids were transplanted under the splenic capsule of immune-deficient mice: 4 weeks after transplantation biliary duct-like structures positive for human albumin were identified; 8 weeks after transplantation the hepatic structure was almost lost | By differentiating iPSCs into hepatobiliary organoids, there is no requirement of supportive cells, thus reducing costs and avoiding immune-rejection | [124] | |
| Guan et al. |
| ND |
| [126] | |
| Ouchi et al. | First iPSCs were differentiated into foregut spheroids and then, in the presence of a hepatocyte specific medium, into a liver organoid containing hepatocytes, Kupffer, stellate and biliary cells | ND | The obtained organoids showed a transcriptomic profile comparable to hepatic tissue | [127] | |
| Toh et al., Jang et al., Zhu et al. | Hepatocytes or embryoid bodies were cultured in microfluidic 3D hepatocyte chip on collagen, Matrigel or hydrogel | ND | Matrices support the formation of 3D aggregates and can be used for drug testing | [128,129,130] | |
| Liver-on-a-Chip | Norona et al., Nguyen et al., Bhise et al., Moya et al., Grix et al. | Gelatin-methacryloyl Hydrogels are used as ink. Primary hepatic cells were printed in transwell microwells to induce the generation of a 3D liver | ND | The coating of microfluidic chips supports the formation of 3D aggregates | [131,132,133,134,135] |
| 3D Printing Technology | Goulart et al. | Bio compatible ink were used to print iPSC-derived parenchymal and non-parenchymal cells to generate a 3D liver organoid | ND | 3D liver organoid showed hepatic functions | [136] |
| Author | Approach | Disease Mouse Model | Reconstitution | Follow Up | Reference |
|---|---|---|---|---|---|
| Huch et al. | Bile-duct derived organoids from Lgr5+ stem cells | Fumarylacetoacetate hydrolase (FAH)−/− mutant mice (model for Tyrosinemia type I liver disease) | 0.1% of total liver volume | 60–90 days | [112] |
| Huch et al. | EpCAM+ ductal cells from human liver biopsies induced to differentiate in hepatocytes and to form organoids | Balb/c nude mice treated with CCl4-retrorsine to induce acute liver damage | 50–100 ng/mL of blood human albumin levels | 120 days | [144] |
| Hu et al. | 3D organoids from mouse and human primary hepatocytes | Fah−/− NOD Rag1−/− Il2rg−/− (FNRG) mice (model of tyrosinemia type I) | 200 µg/mL on average of blood human albumin levels | 90 days | [145] |
| Rashidi et al. | Organoid from iPSC-derived hepatocytes | Fumarylacetoacetate hydrolase (FAH)−/− mutant mice (model for Tyrosinemia type I liver disease) and Fah−/− NOD Rag1−/− Il2rg−/− (FNRG) mice (model tyrosinemia type I) | Detectable levels of human albumin | 14 days | [146] |
| Blackford et al. | iPSC-derived hepatocytes generated with a cGMP compliant method was established to generate and seeded on a 3D poly-ethylene glycol-diacrylate scaffold to generate an organoid | Immune competent (C57BL/6 and Crl:CD1) and immune deficient (Rag2γ) mice | Detectable levels of human albumin | 12 days | [147] |
| Author | Approach | Disease model | Gene | Aim | Reference |
|---|---|---|---|---|---|
| Sampaziotis et al. | Human iPSCs from healthy donors and cystic fibrosis patients were differentiated into cholangiocyte-like cells | Cystic fibrosis associated biliary disease | Cystic fibrosis transmembrane conductance regulator gene (CFTR) | To test the effects of the drug VX809 on organoids | [51,157] |
| Guan et al. | iPSCs from healthy donors and Alagille syndrome patients were differentiated into 3D human hepatic organoids | Alagille syndrome | JAG1 |
| [126] |
| Akbani et al. | iPSC-derived-EpCAM-positive endodermal cells differentiated into hepatic organoids | Citrullinemia type 1 | Argininosuccinate synthetase (ASS1) gene |
| [158] |
| Nie et al. | Human iPSC-derived endodermal, mesenchymal, and endothelial cells were cultured in specific medium to obtain liver organoids | Hepatitis B virus (HBV) infection | ND |
| [160] |
| Starting Material | Author | Approach | Aim | Result | Limitation | Reference |
|---|---|---|---|---|---|---|
| Needle Biopsies | Takai et al. | Hepatocellular carcinoma cells cultured in porous alginate scaffolds generated spheroids | Mimic numerous features of glandular epithelium | Mimic numerous features of glandular epithelium | Tumor microenvironment interactions are not recapitulated | [165] |
| PDX in Immunocompromised Mice | Broutier et al. | PDX organoids from HCC | Drug sensitivity experiment | ERK inhibition could have an effect of HCC progression | Not suitable for immunotherapeutic approaches | [153] |
| Gu et al. | PDX organoids from HCC | To generate a cohort of liver cancers to test a multi-kinase inhibitor | The drug was effective and used as a treatment for patients with advanced HCC | Not suitable for immunotherapeutic approaches | [167] | |
| Nie et al. | PDX organoids from HCC | To generate a cohort of liver cancers containing information about the expression profiles and the genetic alterations of all considered tumors | Identification of biomarkers for personalized medicine | Not suitable for immunotherapeutic approaches | [160] | |
| Saito et al. | PDX organoids from cholangiocarcinoma cells | To demonstrate that cholangiocarcinoma derives from differentiated hepatocytes | Restored hepatic functions | Not suitable for immunotherapeutic approaches | [171] | |
| PDX in Immunocompetent Mice | Jiang et al. | HCC PDXs were generated in NSG gamma null mice repopulated with CAR-T cells | To study cancer immunotherapy | CAR-T cells directed against an HCC tumor-associated antigen suppressed tumor growth | Low engraftment of hematopoietic stem cells in the bone marrow of transplanted mice | [174] |
| Choi et al. | HCCs generated in NSG mice with human leukocyte antigen-matched human immune systems | To study cancer immunotherapy | Organoids models were responsive to immunotherapies | Low engraftment of hematopoietic stem cells in the bone marrow of transplanted mice | [173] | |
| Organoids from Normal Tissues | Artegiani et al. | healthy iPSCs or normal tissues | Introduce BAP1 and cholangiocarcinoma mutations by CRIPSR Cas9 | Acquisition of malignant features | - | [177] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olgasi, C.; Cucci, A.; Follenzi, A. iPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine. Int. J. Mol. Sci. 2020, 21, 6215. https://doi.org/10.3390/ijms21176215
Olgasi C, Cucci A, Follenzi A. iPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine. International Journal of Molecular Sciences. 2020; 21(17):6215. https://doi.org/10.3390/ijms21176215
Chicago/Turabian StyleOlgasi, Cristina, Alessia Cucci, and Antonia Follenzi. 2020. "iPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine" International Journal of Molecular Sciences 21, no. 17: 6215. https://doi.org/10.3390/ijms21176215
APA StyleOlgasi, C., Cucci, A., & Follenzi, A. (2020). iPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine. International Journal of Molecular Sciences, 21(17), 6215. https://doi.org/10.3390/ijms21176215