Ionizing Radiation and Translation Control: A Link to Radiation Hormesis?



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
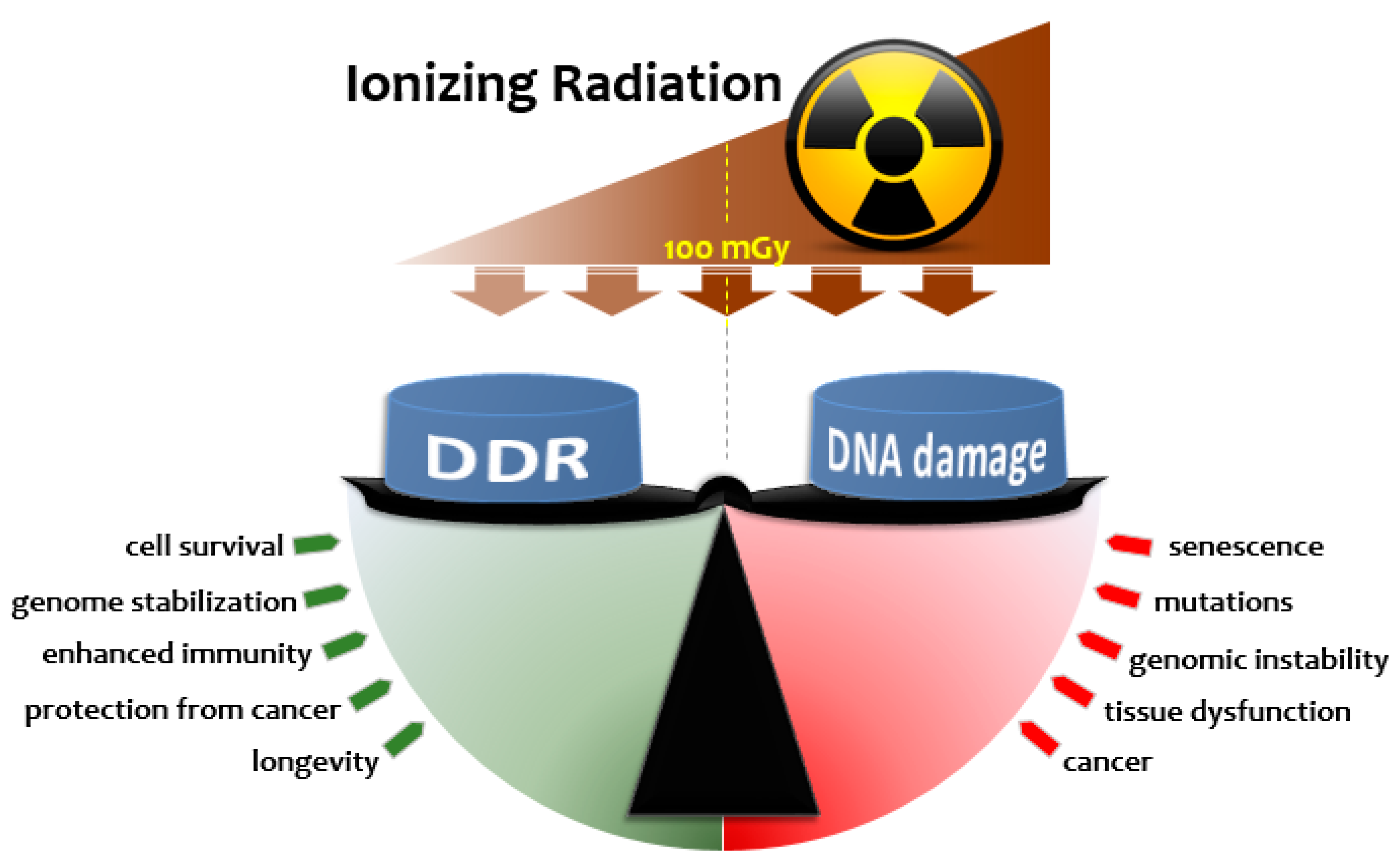
2. Radiation Hormesis
2.1. Radiation Hormesis and Dose-Response Considerations
2.2. Radiation Adaptive Responses
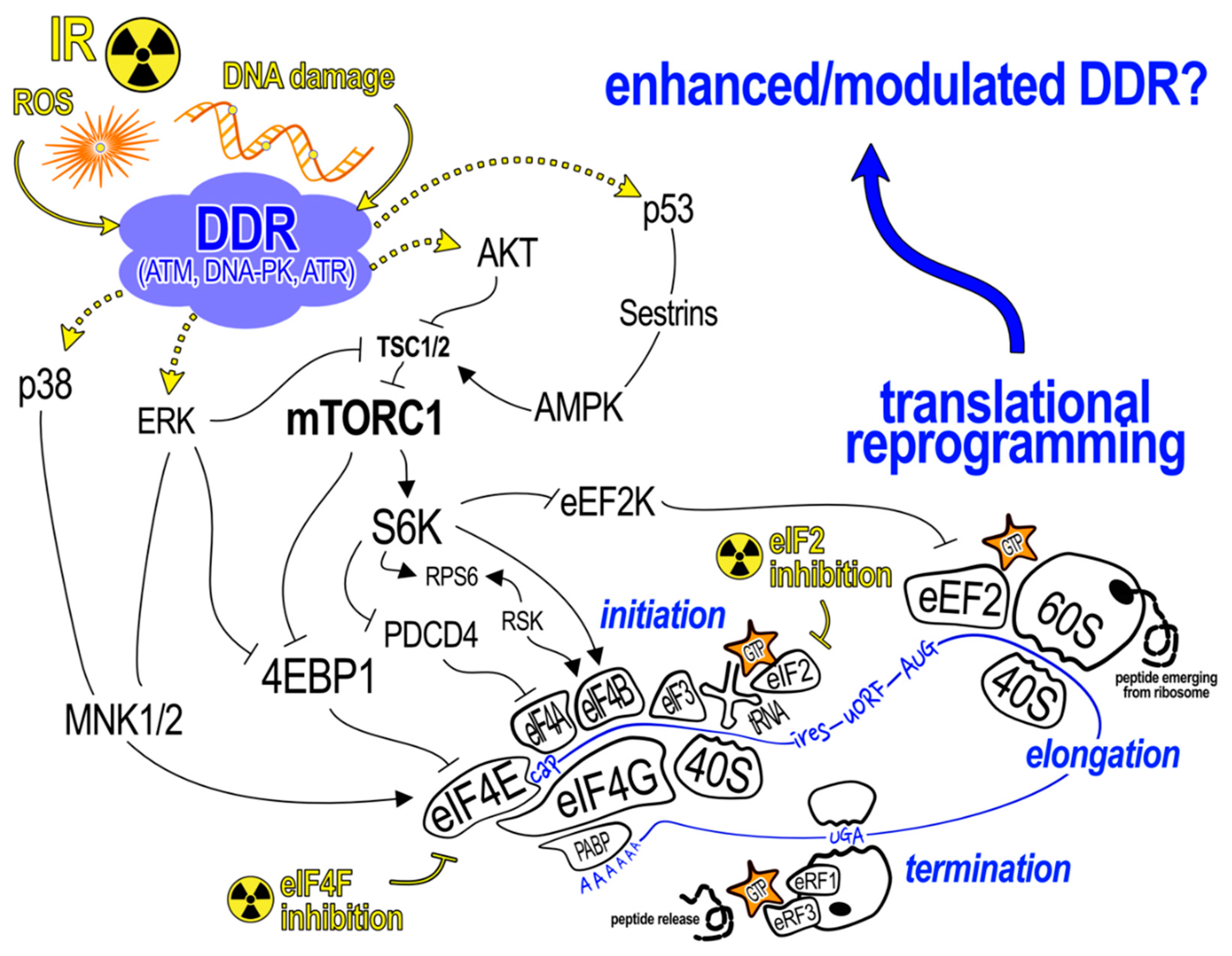
3. Translation and IR Stress Responses
4. Translation Control in Response to LDR
5. Summary and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 53BP1 | P53 binding protein |
| AKT | Protein kinase B |
| AMPK | Adenosine monophosphate-activated protein kinase |
| ATF4 | Activating transcription factor 4 |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related |
| ATRIP | ATR interacting protein |
| Bcl2 | B-cell lymphoma 2 |
| BRCA | Breast cancer |
| CHK | Checkpoint kinase |
| Clud1 | Glutamate receptor delta-1 |
| CT | Computed tomography |
| DDR | DNA damage response |
| DNA-PK | DNA-dependent protein kinase |
| DSB | Double-strand break |
| eIF | Eukaryotic initiation factor |
| ERK | Extracellular signal-regulated kinase |
| FOXO3A | Forkhead box O-3 A |
| GADD45α | Growth arrest and DNA damage inducible alpha |
| GCN2 | General control nonderepressible 2 |
| GTP | Guanosine triphosphate |
| HDR | High-dose ionizing radiation |
| HIF-1α | Hypoxia inducible factor 1 alpha |
| HR | Homologous recombination |
| Hsp70 | Heat shock protein 70 |
| HuR | Human antigen R |
| IR | Ionizing radiation |
| IRES | Internal ribosome entry sites |
| ITAF | Initiation trans-acting factors |
| LDR | Low-dose ionizing radiation |
| LNT | Linear-non-threshold |
| MAPK | Mitogen-activated protein kinase |
| MRE11 | Meiotic recombination 11 |
| mTOR | Mechanistic target of Rapamycin |
| mTORC | Mechanistic target of Rapamycin complex |
| NF-κB | Nuclear factor kappa B |
| NHEJ | Non-homologous end joining |
| NRF2 | Nuclear factor E2-related factor 2 |
| ORF | Open reading frame |
| PABP | Poly (A) binding protein |
| PARP | Poly (ADP-ribose) polymerase |
| PI3K | Phosphatidylinositol 3-kinase |
| PIC | 43S pre-initiation complex |
| PKC | Protein kinase C |
| RBP | RNA binding protein |
| RFC2-5 | Replication factor C subunit 2-5 |
| TGF-β | Transforming growth factor beta |
| TNFR1 | Tumour necrosis factor alpha receptor 1 |
| TNFα | Tumour necrosis factor alpha |
| TSC2 | Tuberous sclerosis complex 2 |
| UTR | Untranslated region |
| XIAP | X-linked inhibitor of apoptosis |
References
- List of Classifications—IARC Monographs on the Identification of Carcinogenic Hazards to Humans. Available online: https://monographs.iarc.fr/list-of-classifications/ (accessed on 19 July 2020).
- Grant, E.J.; Brenner, A.; Sugiyama, H.; Sakata, R.; Sadakane, A.; Utada, M.; Cahoon, E.K.; Milder, C.M.; Soda, M.; Cullings, H.M.; et al. Solid Cancer Incidence among the Life Span Study of Atomic Bomb Survivors: 1958–2009. Radiat. Res. 2017, 187, 513–537. [Google Scholar] [CrossRef] [PubMed]
- ICRP. The 2007 Recommendations of the International Commission on Radiological Protection. ICRP 2007, 103. [Google Scholar] [CrossRef]
- Charles, M. UNSCEAR report 2000: Sources and effects of ionizing radiation. United Nations Scientific Comittee on the Effects of Atomic Radiation. J. Radiol. Prot. 2001, 21, 83–86. [Google Scholar] [CrossRef] [PubMed]
- UNSCEAR. Biological Mechanisms of Radiation Actions at Low Doses; United Nations Scientific Committee on the Effects of Atomic Radiation: New York, NY, USA, 2012. [Google Scholar]
- Tao, Z.; Akiba, S.; Zha, Y.; Sun, Q.; Zou, J.; Li, J.; Liu, Y.; Yuan, Y.; Tokonami, S.; Morishoma, H.; et al. Cancer and non-cancer mortality among inhabitants in the high background radiation area of Yangjiang, China (1979-1998). Health Phys. 2012, 102, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyński, L.; Fornalski, K.W.; Feinendegen, L.E. Cancer Mortality Among People Living in Areas With Various Levels of Natural Background Radiation. Dose-Response 2015, 13, 155932581559239. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Hall, E.J. Computed tomography - An increasing source of radiation exposure. N. Engl. J. Med. 2007, 357, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Balter, S.; Dixon, R.G.; Nikolic, B.; Bartal, G.; Cardella, J.F.; Dauer, L.T.; Stecker, M.S. Quality improvement guidelines for recording patient radiation dose in the medical record for fluoroscopically guided procedures. J. Vasc. Interv. Radiol. 2012, 23, 11–18. [Google Scholar] [CrossRef]
- WHO Report on Cancer. Available online: https://www.who.int/publications/i/item/who-report-on-cancer-setting-priorities-investing-wisely-and-providing-care-for-all (accessed on 20 July 2020).
- Einstein, A.J. Beyond the bombs: Cancer risks of low-dose medical radiation. Lancet 2012, 380, 455–457. [Google Scholar] [CrossRef]
- Storrs, C. Do CT scans cause cancer? Sci. Am. 2013, 309, 30–32. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Bachmann, K.A.; Bailer, A.J.; Bolger, P.M.; Borak, J.; Cai, L.; Cedergreen, N.; Cherian, M.G.; Chiueh, C.C.; Clarkson, T.W.; et al. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 2007, 222, 122–128. [Google Scholar] [CrossRef]
- Tubiana, M. Computed tomography and radiation exposure. N. Engl. J. Med. 2008, 358, 850–853. [Google Scholar] [PubMed]
- Sykes, P.J. Until There Is a Resolution of the Pro-LNT/Anti-LNT Debate, We Should Head Toward a More Sensible Graded Approach for Protection From Low-Dose Ionizing Radiation. Dose-Response 2020, 18, 155932582092165. [Google Scholar] [CrossRef] [PubMed]
- Feinendegen, L.E. Evidence for beneficial low level radiation effects and radiation hormesis. BJR 2005, 78, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Shibamoto, Y.; Nakamura, H. Overview of biological, epidemiological, and clinical evidence of radiation hormesis. Int. J. Mol. Sci. 2018, 19, 2387. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Mathur, R.; Hu, X.; Zhang, X.; Lu, X. MiRNA response to DNA damage. Trends Biochem. Sci. 2011, 36, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Mikolaskova, B.; Jurcik, M.; Cipakova, I.; Kretova, M.; Chovanec, M.; Cipak, L. Maintenance of genome stability: The unifying role of interconnections between the DNA damage response and RNA-processing pathways. Curr. Genet. 2018, 64, 971–983. [Google Scholar] [CrossRef]
- Trivigno, D.; Bornes, L.; Huber, S.M.; Rudner, J. Regulation of protein translation initiation in response to ionizing radiation. Radiat. Oncol. 2013, 8, 35. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational Regulation of Gene Expression during Conditions of Cell Stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef]
- Kasteri, J.; Das, D.; Zhong, X.; Persaud, L.; Francis, A.; Muharam, H.; Sauane, M. Translation control by p53. Cancers 2018, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Lü, X.; De La Peña, L.; Barker, C.; Camphausen, K.; Tofilon, P.J. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res. 2006, 66, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Nantajit, D.; Fan, M.; Murley, J.S.; Grdina, D.J.; Li, J.J. Coactivation of ATM/ERK/NF-κB in the low-dose radiation-induced radioadaptive response in human skin keratinocytes. Free Radic. Biol. Med. 2009, 46, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Broome, E.J.; Brown, D.L.; Mitchel, R.E.J. Dose Responses for Adaption to Low Doses of 60Co γ Rays and 3H β Particles in Normal Human Fibroblasts. Radiat. Res. 2002, 158, 181–186. [Google Scholar] [CrossRef]
- Hou, J.; Wang, F.; Kong, P.; Yu, P.K.N.; Wang, H.; Han, W. Gene Profiling Characteristics of Radioadaptive Response in AG01522 Normal Human Fibroblasts. PLoS ONE 2015, 10, e0123316. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Yan-Sanders, Y.; Lyn-Cook, B.D.; Wang, T.; Tamae, D.; Ogi, J.; Khaletskiy, A.; Li, Z.; Weydert, C.; Longmate, J.A.; et al. Manganese Superoxide Dismutase-Mediated Gene Expression in Radiation-Induced Adaptive Responses. Mol. Cell. Biol. 2003, 23, 2362–2378. [Google Scholar] [CrossRef] [PubMed]
- Murley, J.S.; Baker, K.L.; Miller, R.C.; Darga, T.E.; Weichselbaum, R.R.; Grdina, D.J. SOD2-mediated adaptive responses induced by low-dose ionizing radiation via TNF signaling and amifostine. Free Radic. Biol. Med. 2011, 51, 1918–1925. [Google Scholar] [CrossRef]
- Shelke, S.; Das, B. Dose response and adaptive response of non-homologous end joining repair genes and proteins in resting human peripheral blood mononuclear cells exposed to γ radiation. Mutagenesis 2015, 30, 365–379. [Google Scholar] [CrossRef]
- Lall, R.; Ganapathy, S.; Yang, M.; Xiao, S.; Xu, T.; Su, H.; Shadfan, M.; Asara, J.M.; Ha, C.S.; Ben-Sahra, I.; et al. Low-dose radiation exposure induces a HIF-1-mediated adaptive and protective metabolic response. Cell Death Differ. 2014, 21, 836–844. [Google Scholar] [CrossRef]
- Alexandrou, A.T.; Li, J.J. Cell cycle regulators guide mitochondrial activity in radiation-induced adaptive response. Antioxidants Redox Signal. 2014, 20, 1463–1480. [Google Scholar] [CrossRef]
- Eldridge, A.; Fan, M.; Woloschak, G.; Grdina, D.J.; Chromy, B.A.; Jian Li, J. Manganese superoxide dismutase interacts with a large scale of cellular and mitochondrial proteins in low-dose radiation-induced adaptive radioprotection. Free Radic. Biol. Med. 2012, 53, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Youngblom, J.H.; Wiencke, J.K.; Wolff, S. Inhibition of the adaptive response of human lymphocytes to very low doses of ionizing radiation by the protein synthesis inhibitor cycloheximide. Mutat. Res. Lett. 1989, 227, 257–261. [Google Scholar] [CrossRef]
- Cai, L.; Liu, S. Effect of Cycloheximide on the Adaptive Response Induced by Low Dose Radiation—PubMed. Biomed Env. Sci. 1992, 5, 46–52. [Google Scholar]
- Franco, N.; Lamartine, J.; Frouin, V.; Le Minter, P.; Petat, C.; Leplat, J.-J.; Libert, F.; Gidrol, X.; Martin, M.T. Low-Dose Exposure to γ Rays Induces Specific Gene Regulations in Normal Human Keratinocytes. Radiat. Res. 2005, 163, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Velegzhaninov, I.O.; Ermakova, A.V.; Klokov, D.Y. Low dose ionizing irradiation suppresses cellular senescence in normal human fibroblasts. Int. J. Radiat. Biol. 2018, 94, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Klokov, D.; Leskov, K.; Araki, S.; Zou, Y.; Goetz, E.M.; Luo, X.; Willson, D.; Boothman, D.A. Low dose IR-induced IGF-1-sCLU expression: A p53-repressed expression cascade that interferes with TGFβ1 signaling to confer a pro-survival bystander effect. Oncogene 2013, 32, 479–490. [Google Scholar] [CrossRef]
- Osipov, A.N.; Pustovalova, M.; Grekhova, A.; Eremin, P.; Vorobyova, N.; Pulin, A.; Zhavoronkov, A.; Roumiantsev, S.; Klokov, D.Y.; Eremin, I. Low doses of X-rays induce prolonged and ATM-independent persistence of γH2AX foci in human gingival mesenchymal stem cells. Oncotarget 2015, 6, 27275–27287. [Google Scholar] [CrossRef][Green Version]
- Calabrese, E.J.; Baldwin, L.A. Radiation hormesis: Its historical foundations as a biological hypothesis. Hum. Exp. Toxicol. 2000, 19, 41–75. [Google Scholar] [CrossRef]
- Luckey, T.D. Radiation Hormesis: The Good, the Bad, and the Ugly. Dose-Response 2006, 4, dose-response.06-102. [Google Scholar] [CrossRef]
- Davey, W.P. Prolongation of life of Tribolium confusum apparently due to small doses of x-rays. J. Exp. Zool. 1919, 28, 447–458. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Mattson, M.P. How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech. Dis. 2017, 3, 13. [Google Scholar] [CrossRef]
- Marini, A.M.; Jiang, X.; Wu, X.; Pan, H.; Guo, Z.; Mattson, M.P.; Blondeau, N.; Novelli, A.; Lipsky, R.H. Preconditioning and neurotrophins: A model for brain adaptation to seizures, ischemia and other stressful stimuli. Amino Acids 2007, 32, 299–304. [Google Scholar] [CrossRef]
- Korde, A.S.; Pettigrew, L.C.; Craddock, S.D.; Maragos, W.F. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J. Neurochem. 2005, 94, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, A. Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Parsons, P.A. Evolutionary rates: Effects of stress upon recombination. Biol. J. Linn. Soc. 1988, 35, 49–68. [Google Scholar] [CrossRef]
- Sarup, P.; Sørensen, P.; Loeschcke, V. The long-term effects of a life-prolonging heat treatment on the Drosophila melanogaster transcriptome suggest that heat shock proteins extend lifespan. Exp. Gerontol. 2014, 50, 34–39. [Google Scholar] [CrossRef]
- Sani, E.; Herzyk, P.; Perrella, G.; Colot, V.; Amtmann, A. Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 2013, 14. [Google Scholar] [CrossRef]
- Badawi, Y.; Pal, R.; Hui, D.; Michaelis, E.K.; Shi, H. Ischemic tolerance in an in vivo model of glutamate preconditioning. J. Neurosci. Res. 2015, 93, 623–632. [Google Scholar] [CrossRef]
- Steinberg, C.E.W. Stress Ecology: Environmental Stress as Ecological Driving Force and Key Player in Evolution; Springer: Dordrecht, The Netherlands, 2012; ISBN 9789400720725. [Google Scholar]
- Costantini, D. Hormesis Promotes Evolutionary Change. Dose-Response An Int. J. 2019, 1–4. [Google Scholar] [CrossRef]
- Karam, P.A.; Leslie, S.A. Calculations of background beta-gamma radiation dose through geologic time. Health Phys. 1999, 77, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Karam, P.A.; Leslie, S.A. Changes in terrestrial natural radiation levels over the history of life. Radioact. Environ. 2005, 7, 107–117. [Google Scholar] [CrossRef]
- Hickey, R.J.; Bowers, E.J.; Clelland, R.C. Radiation hormesis, public health, and public policy: A commentary. Health Phys. 1983, 44, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Boice, J.D. The linear nonthreshold (LNT) model as used in radiation protection: An NCRP update. Int. J. Radiat. Biol. 2017, 93, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.J. Artificial transmutation of the gene. Science 1927, 66, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, S.J.; Krestinina, L.Y.; Epifanova, S.; Degteva, M.O.; Akleyev, A.V.; Preston, D.L. Solid Cancer Mortality in the Techa River Cohort (1950–2007). Radiat. Res. 2013, 179, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Leuraud, K.; Richardson, D.B.; Cardis, E.; Daniels, R.D.; Gillies, M.; O’Hagan, J.A.; Hamra, G.B.; Haylock, R.; Laurier, D.; Moissonnier, M.; et al. Ionising radiation and risk of death from leukaemia and lymphoma in radiation-monitored workers (INWORKS): An international cohort study. Lancet Haematol. 2015, e276–e281. [Google Scholar] [CrossRef]
- Sutou, S. Low-dose radiation from A-bombs elongated lifespan and reduced cancer mortality relative to un-irradiated individuals. Genes Environ. 2018, 40, 26. [Google Scholar] [CrossRef]
- Doss, M. Are we approaching the end of the linear no-threshold era? J. Nucl. Med. 2018, 59, 1786–1793. [Google Scholar] [CrossRef]
- Boice, J.D.; Cohen, S.S.; Mumma, M.T.; Chen, H.; Golden, A.P.; Beck, H.L.; Till, J.E. Mortality among US military participants at eight aboveground nuclear weapons test series. Int. J. Radiat. Biol. 2020, 1–64. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Converging concepts: Adaptive response, preconditioning, and the Yerkes-Dodson Law are manifestations of hormesis. Ageing Res. Rev. 2008, 7, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Day, T.K.; Zeng, G.; Hooker, A.M.; Bhat, M.; Scott, B.R.; Turner, D.R.; Sykes, P.J. Adaptive Response for Chromosomal Inversions in pKZ1 Mouse Prostate Induced by Low Doses of X Radiation Delivered after a High Dose. Radiat. Res. 2007, 167, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Dieriks, B.; De Vos, W.; Baatout, S.; Van Oostveldt, P. Repeated exposure of human fibroblasts to ionizing radiation reveals an adaptive response that is not mediated by interleukin-6 or TGF-β. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 2011, 715, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Mitchel, R.E.J.; Jackson, J.S.; McCann, R.A.; Boreham, D.R. The Adaptive Response Modifies Latency for Radiation-Induced Myeloid Leukemia in CBA/H Mice. Radiat. Res. 1999, 152, 273. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Plyusnina, E.N.; Shaposhnikov, M.V. Radiation hormesis and radioadaptive response in Drosophila melanogaster flies with different genetic backgrounds: The role of cellular stress-resistance mechanisms. Biogerontology 2011, 12, 253–263. [Google Scholar] [CrossRef]
- Olivieri, G.; Bodycote, J.; Wolff, S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science 1984, 223, 594–597. [Google Scholar] [CrossRef]
- Shadley, J.D. Chromosomal Adaptive Response in Human Lymphocytes. Radiat. Res. 1994, 138, S9. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhong, R.; Sun, L.; Jia, J.; Ma, S.; Liu, X. Ionizing Radiation-Induced Adaptive Response in Fibroblasts under Both Monolayer and 3-Dimensional Conditions. PLoS ONE 2015, 10, e0121289. [Google Scholar] [CrossRef] [PubMed]
- Høilund-Carlsen, P.F. The good rays: Let them shine! Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.N.; Grekhova, A.; Pustovalova, M.; Ozerov, I.V.; Eremin, P.; Vorobyeva, N.; Lazareva, N.; Pulin, A.; Zhavoronkov, A.; Roumiantsev, S.; et al. Activation of homologous recombination DNA repair in human skin fibroblasts continuously exposed to X-ray radiation. Oncotarget 2015, 6, 26876–26885. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tapio, S.; Jacob, V. Radioadaptive response revisited. Radiat. Environ. Biophys. 2007, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nenoi, M.; Wang, B.; Vares, G. In vivo radioadaptive response: A review of studies relevant to radiation-induced cancer risk. Hum. Exp. Toxicol. 2015, 34, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Afzal, V.; Wiencke, J.K.; Olivieri, G.; Michaeli, A. Human lymphocytes exposed to low doses of ionizing radiations become refractory to high doses of radiation as well as to chemical mutagens that induce double-Strand breaks in DNA. Int. J. Radiat. Biol. 1988, 53, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; You, G.E.; Yang, K.H.; Kim, J.Y.; An, S.; Song, J.Y.; Lee, S.J.; Lim, Y.K.; Nam, S.Y. Role of AKT and ERK pathways in controlling sensitivity to ionizing radiation and adaptive response induced by low-dose radiation in human immune cells. Eur. J. Cell Biol. 2015, 94, 653–660. [Google Scholar] [CrossRef]
- Cai, L.; Liu, S.Z. Induction of cytogenetic adaptive response of somatic and germ cells in vivo and in vitro by low-dose x-irradiation. Int. J. Radiat. Biol. 1990, 58, 187–194. [Google Scholar] [CrossRef]
- Azzam, E.I.; Raaphorst, G.P.; Mitchel, R.E.J. Radiation-Induced Adaptive Response for Protection against Micronucleus Formation and Neoplastic Transformation in C3H 10T1/2 Mouse Embryo Cells. Radiat. Res. 1994, 138, S28. [Google Scholar] [CrossRef]
- Toprani, S.M.; Das, B. Radio-adaptive response of base excision repair genes and proteins in human peripheral blood mononuclear cells exposed to gamma radiation. Mutagenesis 2015, 30, 663–676. [Google Scholar] [CrossRef]
- Wiencke, J.K.; Afzal, V.; Olivieri, G.; Wolff, S. Evidence that the [3H]thymidine-induced adaptive response of human lymphocytes to subsequent doses of X-rays involves the induction of a chromosomal repair mechanism. Mutagenesis 1986, 1, 375–380. [Google Scholar] [CrossRef]
- Hafer, K.; Iwamoto, K.K.; Scuric, Z.; Schiestl, R.H. Adaptive Response to Gamma Radiation in Mammalian Cells Proficient and Deficient in Components of Nucleotide Excision Repair. Radiat. Res. 2007, 168, 168–174. [Google Scholar] [CrossRef]
- Ikushima, T.; Aritomi, H.; Morisita, J. Radioadaptive response: Efficient repair of radiation-induced DNA damage in adapted cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1996, 358, 193–198. [Google Scholar] [CrossRef]
- Sasaki, M.S.; Ejima, Y.; Tachibana, A.; Yamada, T.; Ishizaki, K.; Shimizu, T.; Nomura, T. DNA damage response pathway in radioadaptive response. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2002, 504, 101–118. [Google Scholar] [CrossRef]
- Rothkamm, K.; Löbrich, M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. USA 2003, 100, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Blimkie, M.S.J.; Fung, L.C.W.; Petoukhov, E.S.; Girard, C.; Klokov, D. Repair of DNA Double-Strand Breaks is Not Modulated by Low-Dose Gamma Radiation in C57BL/6J Mice. Radiat. Res. 2014, 181, 548. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.L. Variability: The common factor linking low dose-induced genomic instability, adaptation and bystander effects. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2007, 616, 196–200. [Google Scholar] [CrossRef]
- Wojcik, A.; Bonk, K.; Müller, W.U.; Streffer, C.; Weissenborn, U.; Obe, G. Absence of adaptive response to low doses of x-rays in preimplantation embryos and spleen lymphocytes of an inbred mouse strain as compared to human peripheral lymphocytes: A cytogenetic study. Int. J. Radiat. Biol. 1992, 62, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Guéguen, Y.; Bontemps, A.; Ebrahimian, T.G. Adaptive responses to low doses of radiation or chemicals: Their cellular and molecular mechanisms. Cell. Mol. Life Sci. 2019, 76, 1255–1273. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.R. Radiation-hormesis phenotypes, the related mechanisms and implications for disease prevention and therapy. J. Cell Commun. Signal. 2014, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Mathers, J.; Fraser, J.-A.; McMahon, M.; Saunders, R.-D.-C.; Hayes, J.-D.; McLellan, L.-I. Antioxidant and Cytoprotective Responses to Redox Stress; The Biochemical Society: London, UK, 2004; Volume 71, pp. 157–176. [Google Scholar]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxidants Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Löbrich, M. Contribution of DNA repair and cell cycle checkpoint arrest to the maintenance of genomic stability. DNA Repair 2006, 5, 1192–1198. [Google Scholar] [CrossRef]
- de Toledo, S.M.; Asaad, N.; Venkatachalam, P.; Li, L.; Howell, R.W.; Spitz, D.R.; Azzam, E.I. Adaptive Responses to Low-Dose/Low-Dose-Rate γ Rays in Normal Human Fibroblasts: The Role of Growth Architecture and Oxidative Metabolism. Radiat. Res. 2006, 166, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Paraswani, N.; Thoh, M.; Bhilwade, H.N.; Ghosh, A. Early antioxidant responses via the concerted activation of NF-κB and Nrf2 characterize the gamma-radiation-induced adaptive response in quiescent human peripheral blood mononuclear cells. Mutat. Res. - Genet. Toxicol. Environ. Mutagen. 2018, 831, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Bravard, A.; Luccioni, C.; Moustacchi, E.; Rigaud, O. Contribution of antioxidant enzymes to the adaptive response to ionizing radiation of human lymphoblasts. Int. J. Radiat. Biol. 1999, 75, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/autophagy/Nrf2 pathway mediated low-dose radiation induced radio-resistance in human lung adenocarcinoma A549 cell. Int. J. Biol. Sci. 2015, 11, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K. Activation of Antioxidant System by Low Dose Radiation and Its Applicable Possibility for Treatment of Reactive Oxygen Species-Related Diseases. J. Clin. Biochem. Nutr. 2006, 39, 114–133. [Google Scholar] [CrossRef]
- Mustonen, V.; Kesäniemi, J.; Lavrinienko, A.; Tukalenko, E.; Mappes, T.; Watts, P.C.; Jurvansuu, J. Fibroblasts from bank voles inhabiting Chernobyl have increased resistance against oxidative and DNA stresses. BMC Cell Biol. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.R.; Loke, W.K. Molecular mechanisms of low dose ionizing radiation-induced hormesis, adaptive responses, radioresistance, bystander effects, and genomic instability. Int. J. Radiat. Biol. 2015, 91, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Mitchel, R.E.J. Adaption by low dose radiation exposure: A look at scope and limitations for radioprotection. Dose-Response 2015, 13. [Google Scholar] [CrossRef]
- Grdina, D.J.; Murley, J.S.; Miller, R.C.; Mauceri, H.J.; Sutton, H.G.; Thirman, M.J.; Li, J.J.; Woloschak, G.E.; Weichselbaum, R.R. A Manganese Superoxide Dismutase (SOD2)-Mediated Adaptive Response. Radiat. Res. 2013, 179, 115–124. [Google Scholar] [CrossRef]
- Tarrade, S.; Bhardwaj, T.; Flegal, M.; Bertrand, L.; Velegzhaninov, I.; Moskalev, A.; Klokov, D. Histone H2AX Is Involved in FoxO3a-Mediated transcriptional responses to ionizing radiation to maintain genome stability. Int. J. Mol. Sci. 2015, 16, 29996–30014. [Google Scholar] [CrossRef]
- Portess, D.I.; Bauer, G.; Hill, M.A.; O’Neill, P. Low-dose irradiation of nontransformed cells stimulates the selective removal of precancerous cells via intercellular induction of apoptosis. Cancer Res. 2007, 67, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Rashi-Elkeles, S.; Elkon, R.; Shavit, S.; Lerenthal, Y.; Linhart, C.; Kupershtein, A.; Amariglio, N.; Rechavi, G.; Shamir, R.; Shiloh, Y. Transcriptional modulation induced by ionizing radiation: P53 remains a central player. Mol. Oncol. 2011, 5, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Balakin, V.E.; Zaichkina, S.I.; Klokov, D.I.; Aptikaeva, G.F.; Akhmadieva, A.K.; Rozanova, O.M.; Smirnova, E.N. Determination of the effect of long-term maintenance of the radiation adaptive response in murine bone marrow. Dokl Akad Nauk 1998, 363, 843–845. [Google Scholar] [PubMed]
- Zaichkina, S.I.; Rozanova, O.M.; Klokov, D.I.; Aptikaeva, G.F.; Akhmadieva, A.K.; Smirnova, E.N. Low doses of radiation decrease the level of spontaneous and gamma-induced chromosomal mutagenesis in bone marrow cells of mice in vivo. Radiatsionnaya Biol. Radioekol. 2003, 43, 153–155. [Google Scholar]
- Cui, J.; Yang, G.; Pan, Z.; Zhao, Y.; Liang, X.; Li, W.; Cai, L. Hormetic response to low-dose radiation: Focus on the immune system and its clinical implications. Int. J. Mol. Sci. 2017, 18, 280. [Google Scholar] [CrossRef]
- Cheda, A.; Wrembel-Wargocka, J.; Lisiak, E.; Nowosielska, E.M.; Marciniak, M.; Janiak, M.K. Single Low Doses of X Rays Inhibit the Development of Experimental Tumor Metastases and Trigger the Activities of NK Cells in Mice. Radiat. Res. 2004, 161, 335–340. [Google Scholar] [CrossRef]
- Mitchel, R.E.J.; Jackson, J.S.; Morrison, D.P.; Carlisle, S.M. Low Doses of Radiation Increase the Latency of Spontaneous Lymphomas and Spinal Osteosarcomas in Cancer-Prone, Radiation-Sensitive Trp53 Heterozygous Mice. Radiat. Res. 2003, 159, 320–327. [Google Scholar] [CrossRef]
- Calabrese, E.-J.; Baldwin, L.-A. Radiation Hormesis and Cancer. Hum. Ecol. Risk Assess. Int. J. 2010, 8, 327–353. [Google Scholar] [CrossRef]
- Ina, Y.; Sakai, K. Prolongation of Life Span Associated with Immunological Modification by Chronic Low-Dose-Rate Irradiation in MRL- lpr/lpr Mice. Radiat. Res. 2004, 161, 168–173. [Google Scholar] [CrossRef]
- Zhikrevetskaya, S.; Peregudova, D.; Danilov, A.; Plyusnina, E.; Krasnov, G.; Dmitriev, A.; Kudryavtseva, A.; Shaposhnikov, M.; Moskalev, A. Effect of low doses (5–40 cGy) of gamma-irradiation on lifespan and stress-related genes expression profile in Drosophila melanogaster. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Caratero, A.; Courtade, M.; Bonnet, L.; Planel, H.; Caratero, C. Effect of a continuous gamma irradiation at a very low dose on the life span of mice. Gerontology 1998, 44, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Li, G.W.; Burkhardt, D.; Gross, C.; Weissman, J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 2014, 157, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Hoang, H.D.; Graber, T.E.; Alain, T. Battling for Ribosomes: Translational Control at the Forefront of the Antiviral Response. J. Mol. Biol. 2018, 430, 1965–1992. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Braunstein, S.; Badura, M.L.; Xi, Q.; Formenti, S.C.; Schneider, R.J. Regulation of Protein Synthesis by Ionizing Radiation. Mol. Cell. Biol. 2009, 29, 5645–5656. [Google Scholar] [CrossRef]
- Badura, M.; Braunstein, S.; Zavadil, J.; Schneider, R.J. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 18767–18772. [Google Scholar] [CrossRef]
- Hayman, T.J.; Wahba, A.; Rath, B.H.; Bae, H.; Kramp, T.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin. Cancer Res. 2014, 20, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Conn, C.S.; Shi, Z.; Pang, X.; Tokuyasu, T.; Coady, A.M.; Seo, Y.; Barna, M.; Ruggero, D. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 2015, 162, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Graber, T.E.; Holcik, M. Cap-independent regulation of gene expression in apoptosis. Mol. Biosyst. 2007, 3, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Comparative genetics: Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351. [Google Scholar] [CrossRef]
- Sherrill, K.W.; Byrd, M.P.; Van Eden, M.E.; Lloyd, R.E. BCL-2 translation is mediated via internal ribosome entry during cell stress. J. Biol. Chem. 2004, 279, 29066–29074. [Google Scholar] [CrossRef]
- Dobbyn, H.C.; Hill, K.; Hamilton, T.L.; Spriggs, K.A.; Pickering, B.M.; Coldwell, M.J.; De Moor, C.H.; Bushell, M.; Willis, A.E. Regulation of BAG-1 IRES-mediated translation following chemotoxic stress. Oncogene 2008, 27, 1167–1174. [Google Scholar] [CrossRef]
- Holcik, M.; Lefebvre, C.; Yeh, C.; Chow, T.; Korneluk, R.G. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell Biol. 1999, 1, 190–192. [Google Scholar] [CrossRef]
- Cui, J.-J.; Wang, L.-Y.; Yin, J.-Y. Translational regulation of RPA2 via IRES by UNR and eIF3a. In Proceedings of the AACR Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017; Volume 77, p. 1407. [Google Scholar]
- Shatsky, I.N.; Terenin, I.M.; Smirnova, V.V.; Andreev, D.E. Cap-Independent Translation: What’s in a Name? Trends Biochem. Sci. 2018, 43, 882–895. [Google Scholar] [CrossRef]
- Powley, I.R.; Kondrashov, A.; Young, L.A.; Dobbyn, H.C.; Hill, K.; Cannell, I.G.; Stoneley, M.; Kong, Y.W.; Cotes, J.A.; Smith, G.C.M.; et al. Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev. 2009, 23, 1207–1220. [Google Scholar] [CrossRef]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef]
- Ron, D.; Harding, H.P. eIF2 Phosphorylation in Cellular Stress Responses and Disease; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006; ISBN 9780879697679. [Google Scholar]
- Zhang, H.; Wang, Y.; Lu, J. Function and Evolution of Upstream ORFs in Eukaryotes. Trends Biochem. Sci. 2019, 44, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Ketley, R.F.; Gullerova, M. Beyond the Trinity of ATM, ATR, and DNA-PK: Multiple Kinases Shape the DNA Damage Response in Concert With RNA Metabolism. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Zucal, C.; D’Agostino, V.; Loffredo, R.; Mantelli, B.; Thongon, N.; Lal, P.; Latorre, E.; Provenzani, A. Targeting the Multifaceted HuR Protein, Benefits and Caveats. Curr. Drug Targets 2015, 16, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Abdelmohsen, K.; Gorospe, M. Regulation of HuR by DNA Damage Response Kinases. J. Nucleic Acids 2010, 2010, 981487. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Hagner, P.R.; Zhang, Y.; Dai, B.; Lehrmann, E.; Becker, K.G.; Keene, J.D.; Gorospe, M.; Liu, Z.; Gartenhaus, R.B. ATM regulates a DNA damage response posttranscriptional RNA operon in lymphocytes. Blood 2011, 117, 2441–2450. [Google Scholar] [CrossRef]
- Masuda, K.; Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Lee, E.K.; Tominaga, K.; Selimyan, R.; Martindale, J.L.; Yang, X.; Lehrmann, E.; et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011, 30, 1040–1053. [Google Scholar] [CrossRef]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef]
- Gardiner, M.; Toth, R.; Vandermoere, F.; Morrice, N.A.; Rouse, J. Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J. 2008, 415, 297–307. [Google Scholar] [CrossRef]
- Chaudhury, A.; Chander, P.; Howe, P.H. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA 2010, 16, 1449–1462. [Google Scholar] [CrossRef]
- Zhang, S.; Schlott, B.; Go, M.; Rlach, È.; Grosse, F. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res. 2004, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Dacheux, E.; Vincent, A.; Nazaret, N.; Combet, C.; Wierinckx, A.; Mazoyer, S.; Diaz, J.-J.; Lachuer, J.; Venezia, N.D. BRCA1-Dependent Translational Regulation in Breast Cancer Cells. PLoS ONE 2013, 8, e67313. [Google Scholar] [CrossRef] [PubMed]
- Berthel, E.; Vincent, A.; Eberst, L.; Torres, A.G.; Dacheux, E.; Rey, C.; Marcel, V.; Paraqindes, H.; Lachuer, J.; Catez, F.; et al. Uncovering the Translational Regulatory Activity of the Tumor Suppressor BRCA1. Cells 2020, 9, 941. [Google Scholar] [CrossRef]
- Tsvetkova, A.; Ozerov, I.V.; Pustovalova, M.; Grekhova, A.; Eremin, P.; Vorobyeva, N.; Eremin, I.; Pulin, A.; Zorin, V.; Kopnin, P.; et al. γH2AX, 53BP1 and Rad51 protein foci changes in mesenchymal stem cells during prolonged X-ray irradiation. Oncotarget 2017, 8, 64317–64329. [Google Scholar] [CrossRef]
- Odebunmi, O. Effect of Low Dose Ionizing Radiation on DNA Damage and Repair Response in Proliferating Muscle Stem Cells. Master’s Thesis, University of Ottawa, Ottawa, ON, Canada, 2020. [Google Scholar]
- Crawford, D.R.; Davies, K.J.A. Adaptive Response and Oxidative Stress. Environ. Health Perspect. 1994, 102 (Suppl. S10), 25–28. [Google Scholar]
- Kang, C.-M.; Park, K.-P.; Cho, C.-K.; Seo, J.-S.; Park, W.-Y.; Lee, S.-J.; Lee, Y.-S. Hspa4 (HSP70) is Involved in the Radioadaptive Response: Results from Mouse Splenocytes. Radiat. Res. 2002, 157, 650–655. [Google Scholar] [CrossRef]
- Park, S.-H.; Lee, S.-J.; Chung, H.-Y.; Kim, T.-H.; Cho, C.-K.; Yoo, S.-Y.; Lee, Y.-S. Inducible Heat-Shock Protein 70 Is Involved in the Radioadaptive Response. Radiat. Res. 2000, 153, 318–326. [Google Scholar] [CrossRef]
- Moskalev, A.; Shaposhnikov, M.; Turysheva, E. Life span alteration after irradiation in Drosophila melanogaster strains with mutations of Hsf and Hsps. Biogerontology 2009, 10, 3–11. [Google Scholar] [CrossRef]
- Saunders, L.R.; Verdin, E. Cell biology: Stress response and aging. Science 2009, 323, 1021–1022. [Google Scholar] [CrossRef] [PubMed]
- Tago, F.; Tsukimoto, M.; Nakatsukasa, H.; Kojima, S. Repeated 0.5-Gy Gamma Irradiation Attenuates Autoimmune Disease in MRL- lpr/lpr Mice with Suppression of CD3 + CD4 − CD8 − B220 + T-Cell Proliferation and with Up-regulation of CD4 + CD25 + Foxp3 + Regulatory T Cells. Radiat. Res. 2008, 169, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. MTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; De Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; Tullet, J.M.A.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabilan, U.; Graber, T.E.; Alain, T.; Klokov, D. Ionizing Radiation and Translation Control: A Link to Radiation Hormesis? Int. J. Mol. Sci. 2020, 21, 6650. https://doi.org/10.3390/ijms21186650
Kabilan U, Graber TE, Alain T, Klokov D. Ionizing Radiation and Translation Control: A Link to Radiation Hormesis? International Journal of Molecular Sciences. 2020; 21(18):6650. https://doi.org/10.3390/ijms21186650
Chicago/Turabian StyleKabilan, Usha, Tyson E. Graber, Tommy Alain, and Dmitry Klokov. 2020. "Ionizing Radiation and Translation Control: A Link to Radiation Hormesis?" International Journal of Molecular Sciences 21, no. 18: 6650. https://doi.org/10.3390/ijms21186650
APA StyleKabilan, U., Graber, T. E., Alain, T., & Klokov, D. (2020). Ionizing Radiation and Translation Control: A Link to Radiation Hormesis? International Journal of Molecular Sciences, 21(18), 6650. https://doi.org/10.3390/ijms21186650