Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore




Abstract

1. Introduction
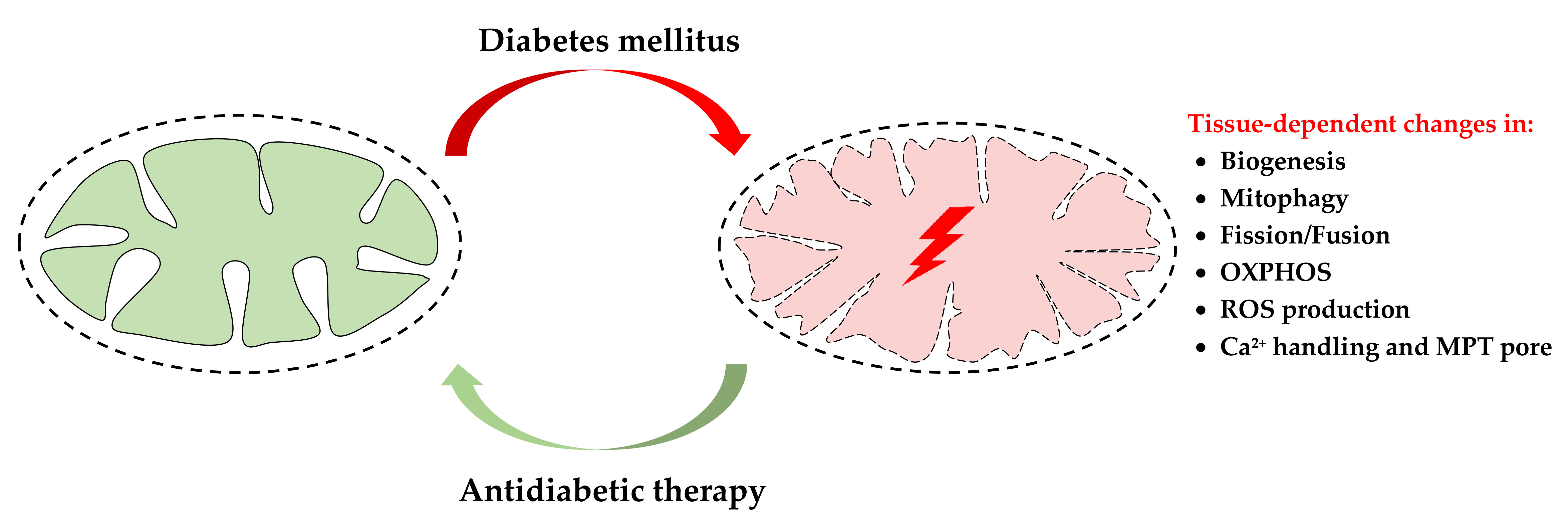
2. Mechanisms of the Diabetes-Induced Mitochondrial Dysfunction
2.1. Imbalance of Mitochondrial Biogenesis and Mitophagy upon Diabetes Mellitus
2.2. Impairment of Mitochondrial Dynamics in Diabetes Mellitus
2.3. Diabetes-Induced Changes in the Functional Activity of Mitochondria
2.4. Oxidative Stress and Diabetes Mellitus
3. Ca2+ Handling, MPT Pore and Diabetes Mellitus
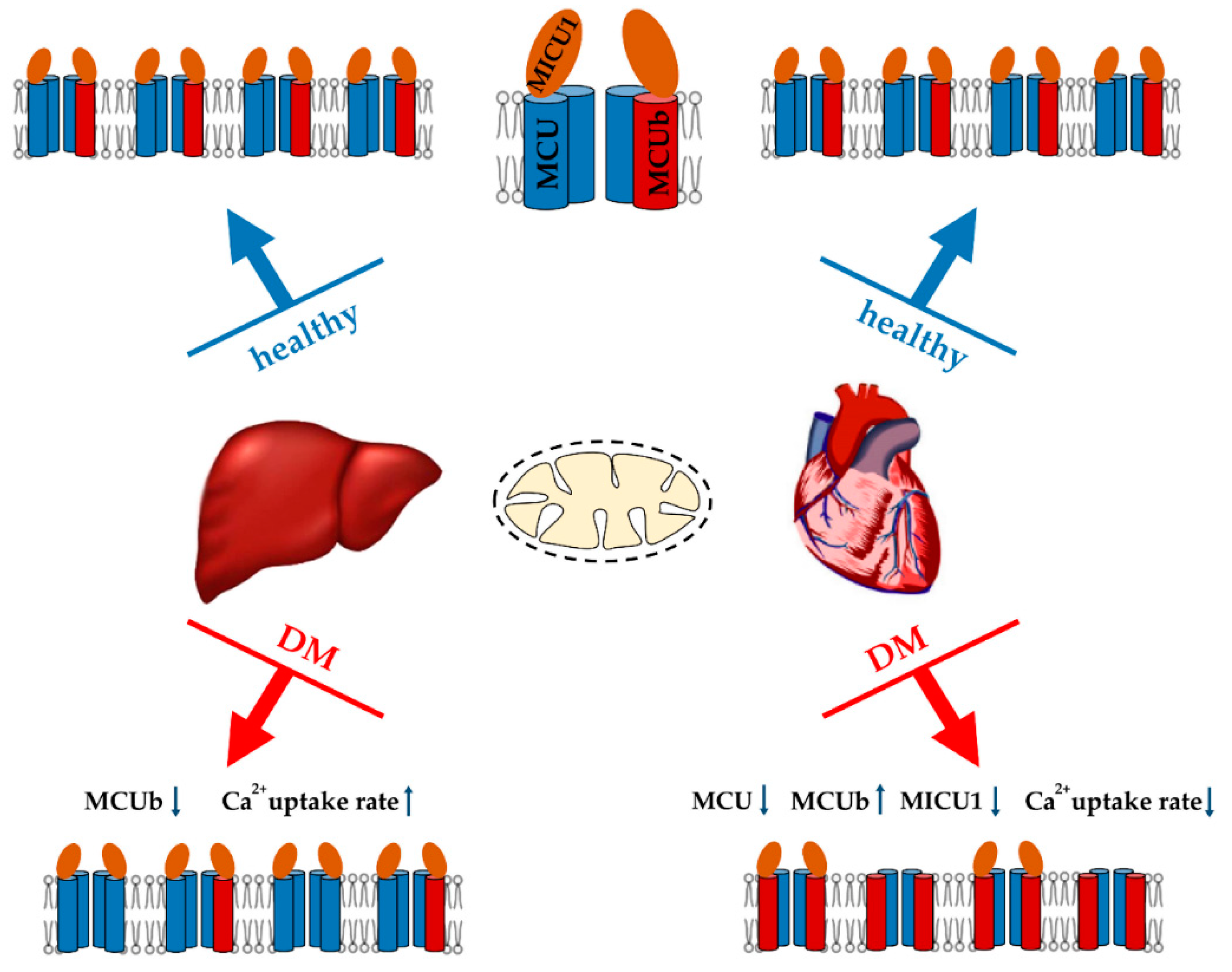
3.1. Mitochondrial Ca2+ Transport in Diabetes
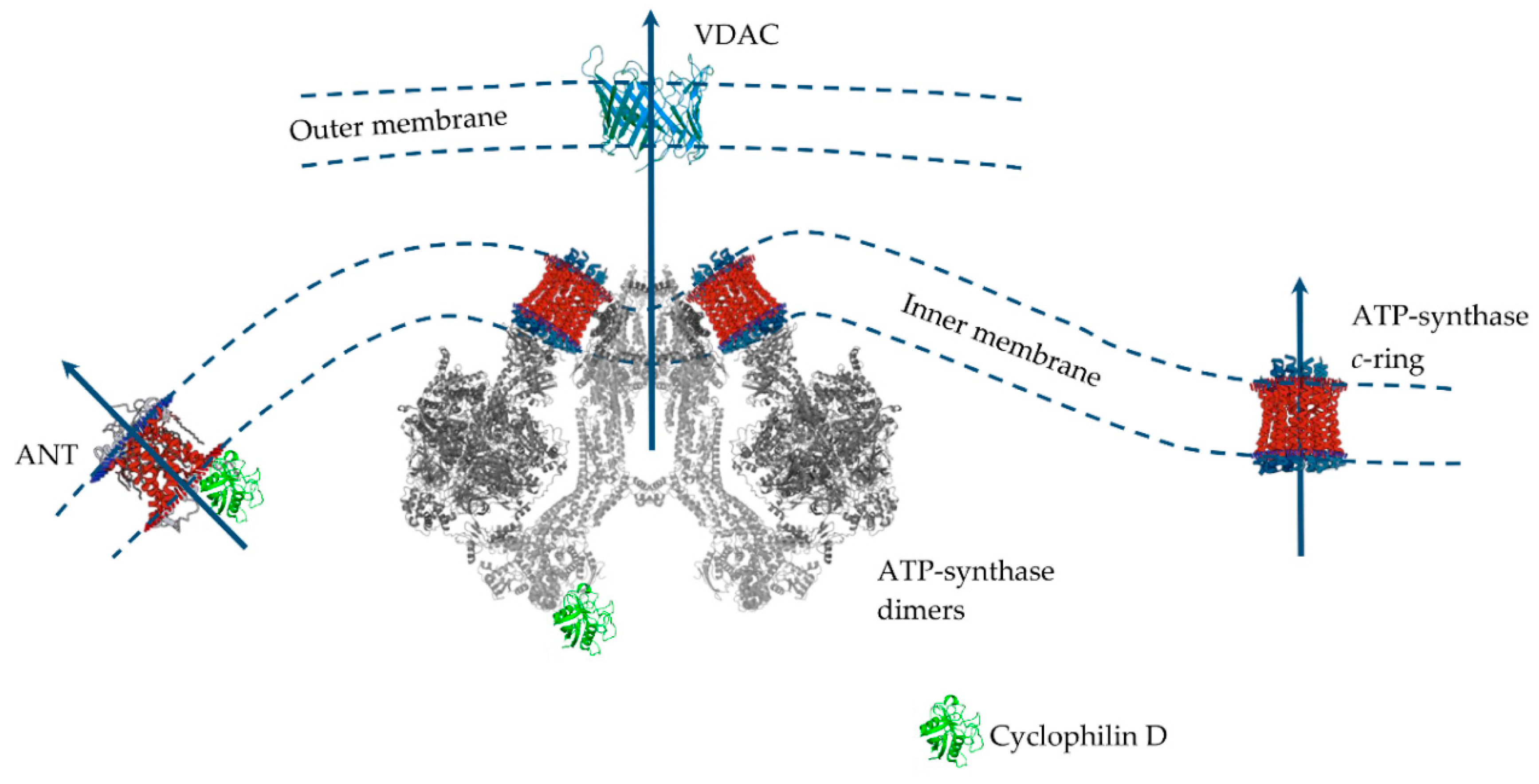
3.2. Mitochondrial Permeability Transition Pore
3.3. The Sensitivity of Mitochondria of Various Tissues to the MPT Pore in Diabetes
3.3.1. Pancreatic β-Cells
3.3.2. Heart
3.3.3. Skeletal Muscles
3.3.4. Neural Tissue
3.3.5. Kidney
3.3.6. Liver
3.4. MPT Pore as a Target for Diabetes Management
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANT | Adenine nucleotide translocator |
| DRP1 | Dynamin-related protein 1 |
| GK | Goto-Kakizaki (GK) rats |
| MAM | Mitochondria-associated ER membranes |
| MCU | Mitochondrial Ca2+ uniporter |
| MICU | Mitochondrial Ca2+ uptake protein |
| MPT | Mitochondrial permeability transition |
| NCLX | Na+/Li+/Ca2+ exchanger |
| Parkin | E3 ubiquitin ligase Parkin |
| PGC-1α | Peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1α |
| PINK1 | PTEN-induced putative kinase 1 |
| ROS | Reactive oxygen species |
| SGLT2 | Sodium-glucose linked transporter 2 |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| UCP | Uncoupling protein |
| VDAC | Voltage-dependent anion channel |
| ZDF | Zucker diabetic fatty (ZDF-Lepr fa/fa or fa/fa) rats |
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- World Health Organization. Global Report on Diabetes; WHO Press, World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Cristelo, C.; Azevedo, C.; Marques, J.M.; Nunes, R.; Sarmento, B. SARS-CoV-2 and diabetes: New challenges for the disease. Diabetes Res. Clin. Pract. 2020, 164, 108228. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43 (Suppl. S1), S14–S31. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Miller, R.G.; Costacou, T. Glucose management and the sex difference in excess cardiovascular disease risk in long-duration type 1 diabetes. Curr. Diab. Rep. 2019, 19, 139. [Google Scholar] [CrossRef]
- Groop, L.C.; Eriksson, J.G. The etiology and pathogenesis of non-insulin-dependent diabetes. Ann. Med. 1992, 24, 483–489. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Yamada, T.; Ida, T.; Yamaoka, Y.; Ozawa, K.; Takasan, H.; Honjo, I. Two distinct patterns of glucose intolerance in icteric rats and rabbits. Relationship to impaired liver mitochondria function. J. Lab. Clin. Med. 1975, 86, 38–45. [Google Scholar]
- Ling, C.; Del Guerra, S.; Lupi, R.; Ronn, T.; Granhall, C.; Luthman, H.; Masiello, P.; Marchetti, P.; Groop, L.; Del Prato, S. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008, 51, 615–622. [Google Scholar] [CrossRef]
- Yoon, J.C.; Xu, G.; Deeney, J.T.; Yang, S.N.; Rhee, J.; Puigserver, P.; Levens, A.R.; Yang, R.; Zhang, C.Y.; Lowell, B.B.; et al. Suppression of beta cell energy metabolism and insulin release by PGC-1alpha. Dev. Cell. 2003, 5, 73–83. [Google Scholar] [CrossRef]
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial Mechanisms in Diabetic Cardiomyopathy. Diabetes Metab. J. 2020, 44, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef]
- Choi, J.; Chandrasekaran, K.; Inoue, T.; Muragundla, A.; Russell, J.W. PGC-1α regulation of mitochondrial degeneration in experimental diabetic neuropathy. Neurobiol. Dis. 2014, 64, 118–130. [Google Scholar] [CrossRef]
- Guo, K.; Lu, J.; Huang, Y.; Wu, M.; Zhang, L.; Yu, H.; Zhang, M.; Bao, Y.; He, J.C.; Chen, H.; et al. Protective role of PGC-1α in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS ONE 2015, 10, e0125176. [Google Scholar] [CrossRef]
- Gao, C.L.; Zhu, C.; Zhao, Y.P.; Chen, X.H.; Ji, C.B.; Zhang, C.M.; Zhu, J.G.; Xia, Z.K.; Tong, M.L.; Guo, X.R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef]
- Holmstrom, M.H.; Iglesias-Gutierrez, E.; Zierath, J.R.; Garcia-Roves, P.M. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E731–E739. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in mitochondrial oxidative stress and mitophagy in subjects with prediabetes and type 2 diabetes mellitus. Front. Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef]
- Gundersen, A.E.; Kugler, B.A.; McDonald, P.M.; Veraksa, A.; Houmard, J.A.; Zou, K. Altered mitochondrial network morphology and regulatory proteins in mitochondrial quality control in myotubes from severely obese humans with or without type 2 diabetes. Appl. Physiol. Nutr. Metab. 2020, 45, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.X.; Correia, S.C.; Alves, M.G.; Oliveira, P.F.; Cardoso, S.; Carvalho, C.; Seiça, R.; Santos, M.S.; Moreira, P.I. Mitochondrial quality control systems sustain brain mitochondrial bioenergetics in early stages of type 2 diabetes. Mol. Cell Biochem. 2014, 394, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, M. Metformin rescues Parkin protein expression and mitophagy in high glucose-challenged human renal epithelial cells by inhibiting NF-κB via PP2A activation. Life Sci. 2020, 246, 117382. [Google Scholar] [CrossRef]
- Jiang, X.S.; Chen, X.M.; Hua, W.; He, J.L.; Liu, T.; Li, X.J.; Wan, J.M.; Gan, H.; Du, X.G. PINK1/Parkin mediated mitophagy ameliorates palmitic acid-induced apoptosis through reducing mitochondrial ROS production in podocytes. Biochem. Biophys. Res. Commun. 2020, 525, 954–961. [Google Scholar] [CrossRef]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef]
- Liu, P.; Lin, H.; Xu, Y.; Zhou, F.; Wang, J.; Liu, J.; Zhu, X.; Guo, X.; Tang, Y.; Yao, P. Frataxin-mediated PINK1-Parkin-dependent mitophagy in hepatic steatosis: The protective effects of quercetin. Mol. Nutr. Food Res. 2018, 62, e1800164. [Google Scholar] [CrossRef]
- Jezek, P.; Dlaskova, A. Dynamic of mitochondrial network, cristae, and mitochondrial nucleoids in pancreatic β-cells. Mitochondrion 2019, 49, 245–258. [Google Scholar] [CrossRef]
- Hu, L.; Ding, M.; Tang, D.; Gao, E.; Li, C.; Wang, K.; Qi, B.; Qiu, J.; Zhao, H.; Chang, P.; et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics 2019, 9, 3687–3706. [Google Scholar] [CrossRef] [PubMed]
- Fealy, C.E.; Mulya, A.; Lai, N.; Kirwan, J.P. Exercise training decreases activation of the mitochondrial fission protein dynamin-related protein-1 in insulin-resistant human skeletal muscle. J. Appl. Physiol. 2014, 117, 239–245. [Google Scholar] [CrossRef]
- Peyravi, A.; Yazdanpanahi, N.; Nayeri, H.; Hosseini, S.A. The effect of endurance training with crocin consumption on the levels of MFN2 and DRP1 gene expression and glucose and insulin indices in the muscle tissue of diabetic rats. J. Food Biochem. 2020, 44, e13125. [Google Scholar] [CrossRef]
- Chen, L.; Huang, J.; Li, X.C.; Liu, S.Y.; Li, Y.H.; Wang, Q.; Yang, J.J.; Cao, H.M.; Hu, Q.K.; He, L.J. High-glucose induced mitochondrial dynamics disorder of spinal cord neurons in diabetic rats and its effect on mitochondrial spatial distribution. Spine 2019, 44, E715–E722. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Chau, Y.Y.; Ng, H.Y.; Chen, C.H.; Wang, P.W.; Liou, C.W.; Lin, T.K.; Chen, J.B. Empagliflozin protects HK-2 cells from high glucose-mediated injuries via a mitochondrial mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; de Las Heras, N.; Lopez-Farre, A.; Manucha, W.; Ferder, L. Role of mitochondrial dysfunction in hypertension and obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, L.; Li, X.; Jiang, Z.; Sun, L.; Zhao, G.; Zhou, G.; Zhang, H.; Shang, J.; Wang, T. Mitochondrial fusion/fission process involved in the improvement of catalpol on high glucose-induced hepatic mitochondrial dysfunction. Acta Biochim. Biophys. Sin. 2015, 47, 730–740. [Google Scholar] [CrossRef]
- Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Quantification of mitochondrial oxidative phosphorylation in metabolic disease: Application to type 2 diabetes. Int. J. Mol. Sci. 2019, 20, 5271. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef]
- Szkudelska, K.; Okulicz, M.; Hertig, I.; Szkudelski, T. Resveratrol ameliorates inflammatory and oxidative stress in type 2 diabetic Goto-Kakizaki rats. Biomed. Pharmacother. 2020, 125, 110026. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Howarth, F.C. Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Talanov, E.Y.; Starinets, V.S.; Agafonov, A.V.; Dubinin, M.V.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+—Dependent permeability transition in rat liver mitochondria under the streptozotocin-induced type I diabetes. Cells 2019, 8, 1014. [Google Scholar] [CrossRef]
- Joost, H.-G.; Al-Hasani, H.; Schürmann, A. Animal Models in Diabetes Research; Humana Press: Totowa, NJ, USA, 2012; 325p. [Google Scholar] [CrossRef]
- Gvazava, I.G.; Rogovaya, O.S.; Borisov, M.A.; Vorotelyak, E.A.; Vasiliev, A.V. Pathogenesis of type 1 diabetes mellitus and rodent experimental models. Acta Nat. 2018, 10, 24–33. [Google Scholar] [CrossRef]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, A.L.; Pérez, S. PGC-1α, Inflammation, and oxidative stress: An integrative view in metabolism. Oxid. Med. Cell Longev. 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, Q.; Zhang, L.; Fang, Z.; Zhao, F.; Lv, Z.; Gu, Z.; Zhang, J.; Wang, J.; Zen, K.; et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010, 20, 676–687. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef]
- Schaeffer, P.J.; Wende, A.R.; Magee, C.J.; Neilson, J.R.; Leone, T.C.; Chen, F.; Kelly, D.P. Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J. Biol. Chem. 2004, 279, 39593–39603. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Di, W.; Lv, J.; Jiang, S.; Lu, C.; Yang, Z.; Ma, Z.; Hu, W.; Yang, Y.; Xu, B. PGC-1: The energetic regulator in cardiac metabolism. Curr. Issues Mol. Biol. 2018, 28, 29–46. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.J.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Kaambre, T.; Han, W.H.; de Goede, P.; et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2020, 228, e13430. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, X.; Shi, Y.; Su, Y.; Wei, J.; Duan, H. PGC-1α, glucose metabolism and type 2 diabetes mellitus. J. Endocrinol. 2016, 229, R99–R115. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Karakelides, H.; Asmann, Y.W.; Bigelow, M.L.; Short, K.R.; Dhatariya, K.; Coenen-Schimke, J.; Kahl, J.; Mukhopadhyay, D.; Nair, K.S. Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 2007, 56, 2683–2689. [Google Scholar] [CrossRef]
- Toledo, F.G.; Menshikova, E.V.; Ritov, V.B.; Azuma, K.; Radikova, Z.; DeLany, J.; Kelley, D.E. Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes 2007, 56, 2142–2147. [Google Scholar] [CrossRef]
- Fujisawa, K.; Nishikawa, T.; Kukidome, D.; Imoto, K.; Yamashiro, T.; Motoshima, H.; Matsumura, T.; Araki, E. TZDs reduce mitochondrial ROS production and enhance mitochondrial biogenesis. Biochem. Biophys. Res. Commun. 2009, 379, 43–48. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef]
- Docrat, T.F.; Nagiah, S.; Naicker, N.; Baijnath, S.; Singh, S.; Chuturgoon, A.A. The protective effect of metformin on mitochondrial dysfunction and endoplasmic reticulum stress in diabetic mice brain. Eur. J. Pharmacol. 2020, 875, 173059. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Masini, M.; Martino, L.; Marselli, L.; Bugliani, M.; Boggi, U.; Filipponi, F.; Marchetti, P.; De Tata, V. Ultrastructural alterations of pancreatic beta cells in human diabetes mellitus. Diabetes Metab. Res. Rev. 2017, 33, e2894. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Mitochondrial uncoupling: A key contributor to reduced cardiac efficiency in diabetes. Physiology 2006, 21, 250–258. [Google Scholar] [CrossRef]
- Shen, X.; Zheng, S.; Thongboonkerd, V.; Xu, M.; Pierce, W.M.; Klein, J.B.; Epstein, P.N. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E896–E905. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pan, W.; Xu, G.; Chen, L. Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clin. Chim. Acta 2020, 502, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in aging and disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef]
- Watada, H.; Fujitani, Y. Minireview: Autophagy in pancreatic β-cells and its implication in diabetes. Mol. Endocrinol. 2015, 29, 338–348. [Google Scholar] [CrossRef]
- Rocha, M.; Apostolova, N.; Diaz-Rua, R.; Muntane, J.; Victor, V.M. Mitochondria and T2D: Role of autophagy, ER stress, and inflammasome. Trends Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, B.; Lau, W.B.; Du, Y.; Guo, R.; Yan, Z.; Gan, L.; Yan, W.; Zhao, J.; Gao, E.; et al. Restoring diabetes-induced autophagic flux arrest in ischemic/reperfused heart by ADIPOR (adiponectin receptor) activation involves both AMPK-dependent and AMPK-independent signaling. Autophagy 2017, 13, 1855–1869. [Google Scholar] [CrossRef]
- Tao, A.; Xu, X.; Kvietys, P.; Kao, R.; Martin, C.; Rui, T. Experimental diabetes mellitus exacerbates ischemia/reperfusion-induced myocardial injury by promoting mitochondrial fission: Role of down-regulation of myocardial Sirt1 and subsequent Akt/Drp1 interaction. Int. J. Biochem. Cell Biol. 2018, 105, 94–103. [Google Scholar] [CrossRef]
- Packer, M. Autophagy-dependent and -independent modulation of oxidative and organellar stress in the diabetic heart by glucose-lowering drugs. Cardiovasc. Diabetol. 2020, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell Mol. Med. 2020, 24, 2832–2846. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Renal. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Buhlman, L.; Damiano, M.; Bertolin, G.; Ferrando-Miguel, R.; Lombès, A.; Brice, A.; Corti, O. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim. Biophys. Acta 2014, 1843, 2012–2026. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Sabouny, R.; Shutt, T.E. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem. Sci. 2020, 45, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jin, P.; Yu, L.; Wang, Y.; Han, L.; Shi, T.; Li, X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS ONE 2014, 9, e92810. [Google Scholar] [CrossRef]
- Makino, A.; Scottm, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar] [CrossRef]
- Yu, J.; Maimaitili, Y.; Xie, P.; Wu, J.J.; Wang, J.; Yang, Y.N.; Ma, H.P.; Zheng, H. High glucose concentration abrogates sevoflurane post-conditioning cardioprotection by advancing mitochondrial fission but dynamin-related protein 1 inhibitor restores these effects. Acta Physiol. 2017, 220, 83–98. [Google Scholar] [CrossRef]
- Ding, M.; Liu, C.; Shi, R.; Yu, M.; Zeng, K.; Kang, J.; Fu, F.; Mi, M. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol. 2020, 229, e13428. [Google Scholar] [CrossRef]
- Peng, L.; Men, X.; Zhang, W.; Wang, H.; Xu, S.; Fang, Q.; Liu, H.; Yang, W.; Lou, J. Involvement of dynamin-related protein 1 in free fatty acid-induced INS-1-derived cell apoptosis. PLoS ONE 2012, 7, e49258. [Google Scholar] [CrossRef]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xiang, H.; Zhao, S.; Sang, H.; Lv, F.; Chen, R.; Shu, Z.; Chen, A.F.; Chen, S.; Lu, H. Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J. Cell. Mol. Med. 2019, 23, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, I.; Rothman, D.L.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J. Clin. Investig. 1992, 90, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Price, T.B.; Bergeron, R. Regulation of net hepatic glycogenolysis and gluconeogenesis during exercise: Impact of type 1 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 4656–4664. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Dean, D. Malonyl CoA, long chain fatty acyl CoA and insulin resistance in skeletal muscle. J. Basic Clin. Physiol. Pharmacol. 1998, 9, 295–308. [Google Scholar] [CrossRef]
- Adams, S.H. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv. Nutr. 2011, 2, 445–456. [Google Scholar] [CrossRef]
- Yin, J.; Ren, W.; Chen, S.; Li, Y.; Han, H.; Gao, J.; Liu, G.; Wu, X.; Li, T.; Woo Kim, S.; et al. Metabolic regulation of methionine restriction in diabetes. Mol. Nutr. Food Res. 2018, 62, e1700951. [Google Scholar] [CrossRef]
- Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Koentges, C.; Konig, A.; Pfeil, K.; Holscher, M.E.; Schnick, T.; Wende, A.R.; Schrepper, A.; Cimolai, M.C.; Kersting, S.; Hoffmann, M.M.; et al. Myocardial mitochondrial dysfunction in mice lacking adiponectin receptor 1. Basic Res. Cardiol. 2015, 110, 37. [Google Scholar] [CrossRef] [PubMed]
- Bombicino, S.S.; Iglesias, D.E.; Mikusic, I.; D’Annunzio, V.; Gelpi, R.J.; Boveris, A.; Valdez, L.B. Diabetes impairs heart mitochondrial function without changes in resting cardiac performance. Int. J. Biochem. Cell Biol. 2016, 81, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Rojo, C.; Vargas-Vargas, M.A.; Olmos-Orizaba, B.E.; Rodríguez-Orozco, A.R.; Calderón-Cortés, E. Interplay between NADH oxidation by complex I, glutathione redox state and sirtuin-3, and its role in the development of insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165801. [Google Scholar] [CrossRef]
- Ni, R.; Zheng, D.; Xiong, S.; Hill, D.J.; Sun, T.; Gardiner, R.B.; Fan, G.C.; Lu, Y.; Abel, E.D.; Greer, P.A.; et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: A novel mechanism contributing to diabetic cardiomyopathy. Diabetes 2016, 65, 255–268. [Google Scholar] [CrossRef]
- Ong, S.B.; Lee, W.H.; Shao, N.Y.; Ismail, N.I.; Katwadi, K.; Lim, M.M.; Kwek, X.Y.; Michel, N.A.; Li, J.; Newson, J.; et al. Calpain inhibition restores autophagy and prevents mitochondrial fragmentation in a human iPSC model of diabetic endotheliopathy. Stem. Cell Reports. 2019, 12, 597–610. [Google Scholar] [CrossRef]
- Antoun, G.; McMurray, F.; Thrush, A.B.; Patten, D.A.; Peixoto, A.C.; Slack, R.S.; McPherson, R.; Dent, R.; Harper, M.E. Impaired mitochondrial oxidative phosphorylation and supercomplex assembly in rectus abdominis muscle of diabetic obese individuals. Diabetologia 2015, 58, 2861–2866. [Google Scholar] [CrossRef]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nuñez, S.; Nuño-Lámbarri, N.; Enrich, C.; García-Ruiz, C.; Fernández-Checa, J.C. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef]
- Starinets, V.S.; Lebedeva, E.V.; Mikheeva, I.B.; Belosludtseva, N.V.; Dubinin, M.V.; Belosludtsev, K.N. Ultrastructural and functional changes in liver mitochondria in a rat model of type I diabetes mellitus. Biophysics 2019, 64, 755–760. [Google Scholar] [CrossRef]
- Ferreira, F.M.; Palmeira, C.M.; Seiça, R.; Santos, M.S. Alterations of liver mitochondrial bioenergetics in diabetic Goto-Kakizaki rats. Metabolism 1999, 48, 1115–1119. [Google Scholar] [CrossRef][Green Version]
- Ferreira, F.M.; Seiça, R.; Oliveira, P.J.; Coxito, P.M.; Moreno, A.J.; Palmeira, C.M.; Santos, M.S. Diabetes induces metabolic adaptations in rat liver mitochondria: Role of coenzyme Q and cardiolipin contents. Biochim. Biophys. Acta 2003, 1639, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Martín-Rodríguez, S.; de Pablos-Velasco, P.; Calbet, J.A.L. Mitochondrial complex I inhibition by metformin: Drug-exercise interactions. Trends Endocrinol. Metab. 2020, 31, 269–271. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Pioglitazone leads to an inactivation and disassembly of complex I of the mitochondrial respiratory chain. BMC Biol. 2013, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Seydi, E.; Servati, T.; Samiei, F.; Naserzadeh, P.; Pourahmad, J. Toxicity of pioglitazone on mitochondria isolated from brain and heart: An analysis for probable drug-induced neurotoxicity and cardiotoxicity. Drug Res. 2020, 70, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef]
- Samartsev, V.N. Fatty acids as uncouplers of oxidative phosphorylation. Biochemistry 2000, 65, 991–1005. [Google Scholar]
- Hidaka, S.; Kakuma, T.; Yoshimatsu, H.; Sakino, H.; Fukuchi, S.; Sakata, T. Streptozotocin treatment upregulates uncoupling protein 3 expression in the rat heart. Diabetes 1999, 48, 430–435. [Google Scholar] [CrossRef]
- He, Y.; Luan, Z.; Fu, X.; Xu, X. Overexpression of uncoupling protein 2 inhibits the high glucose-induced apoptosis of human umbilical vein endothelial cells. Int. J. Mol. Med. 2016, 37, 631–638. [Google Scholar] [CrossRef]
- Kim, J.D.; Yoon, N.A.; Jin, S.; Diano, S. Microglial UCP2 mediates inflammation and obesity induced by high-fat feeding. Cell Metab. 2019, 30, 952–962. [Google Scholar] [CrossRef]
- Boudina, S.; Han, Y.H.; Pei, S.; Tidwell, T.J.; Henrie, B.; Tuinei, J.; Olsen, C.; Sena, S.; Abel, E.D. UCP3 regulates cardiac efficiency and mitochondrial coupling in high fat-fed mice but not in leptin-deficient mice. Diabetes 2012, 61, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid. Med. Cell Longev. 2020, 8609213. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Mattox, T.A.; Thayne, K.; Katunga, L.A.; La Favor, J.D.; Neufer, P.D.; Hickner, R.C.; Wingard, C.J.; Anderson, E.J. Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol. 2013, 591, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Malaguti, C.; La Guardia, P.G.; Leite, A.C.; Oliveira, D.N.; de Lima Zollner, R.L.; Catharino, R.R.; Vercesi, A.E.; Oliveira, H.C. Oxidative stress and susceptibility to mitochondrial permeability transition precedes the onset of diabetes in autoimmune non-obese diabetic mice. Free Radic. Res. 2014, 48, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS metabolism: 10 years later. Biochemistry 2015, 80, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Sirtuins and type 2 diabetes: Role in inflammation, oxidative stress, and mitochondrial function. Front. Endocrinol. 2019, 10, 187. [Google Scholar] [CrossRef]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Huang, Y.; Chi, J.; Wei, F.; Zhou, Y.; Cao, Y.; Wang, Y. Mitochondrial DNA: A new predictor of diabetic kidney disease. Int. J. Endocrinol. 2020, 3650937. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A. Molecular interactions between reactive oxygen species and autophagy in kidney disease. Int. J. Mol. Sci. 2019, 20, 3791. [Google Scholar] [CrossRef] [PubMed]
- El-Daly, M.; Pulakazhi Venu, V.K.; Saifeddine, M.; Mihara, K.; Kang, S.; Fedak, P.; Alston, L.A.; Hirota, S.A.; Ding, H.; Triggle, C.R.; et al. Hyperglycaemic impairment of PAR2-mediated vasodilation: Prevention by inhibition of aortic endothelial sodium-glucose-co-Transporter-2 and minimizing oxidative stress. Vasc. Pharmacol. 2018, 109, 56–71. [Google Scholar] [CrossRef]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, L.; Shen, M.; Liu, Y.; Liu, G.; Wu, Y.; Ding, F.; Ma, K.; Wang, W.; Zhang, Y.; et al. Pioglitazone protects compression-mediated apoptosis in nucleus pulposus mesenchymal stem cells by suppressing oxidative stress. Oxid. Med. Cell Longev. 2019, 4764071. [Google Scholar] [CrossRef] [PubMed]
- De Blasio, M.J.; Huynh, K.; Qin, C.; Rosli, S.; Kiriazis, H.; Ayer, A.; Cemerlang, N.; Stocker, R.; Du, X.J.; McMullen, J.R.; et al. Therapeutic targeting of oxidative stress with coenzyme Q10 counteracts exaggerated diabetic cardiomyopathy in a mouse model of diabetes with diminished PI3K(p110α) signaling. Free Radic. Biol. Med. 2015, 87, 137–147. [Google Scholar] [CrossRef]
- Fink, B.D.; Guo, D.F.; Kulkarni, C.A.; Rahmouni, K.; Kerns, R.J.; Sivitz, W.I. Metabolic effects of a mitochondrial-targeted coenzyme Q analog in high fat fed obese mice. Pharmacol. Res. Perspect. 2017, 5, e00301. [Google Scholar] [CrossRef]
- Kalinovich, A.V.; Mattsson, C.L.; Youssef, M.R.; Petrovic, N.; Ost, M.; Skulachev, V.P.; Shabalina, I.G. Mitochondria-targeted dodecyltriphenylphosphonium (C12TPP) combats high-fat-diet-induced obesity in mice. Int. J. Obes. 2016, 40, 1864–1874. [Google Scholar] [CrossRef]
- Axelrod, C.L.; King, W.T.; Davuluri, G.; Noland, R.C.; Hall, J.; Hull, M.; Dantas, W.S.; Zunica, E.R.; Alexopoulos, S.J.; Hoehn, K.L.; et al. BAM15-mediated mitochondrial uncoupling protects against obesity and improves glycemic control. EMBO Mol. Med. 2020, 12, e12088. [Google Scholar] [CrossRef]
- Bozi, L.H.M.; Campos, J.C.; Zambelli, V.O.; Ferreira, N.D.; Ferreira, J.C.B. Mitochondrially-targeted treatment strategies. Mol. Aspects Med. 2020, 71, 100836. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Arkat, S.; Umbarkar, P.; Singh, S.; Sitasawad, S.L. Mitochondrial peroxiredoxin-3 protects against hyperglycemia induced myocardial damage in diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 97, 489–500. [Google Scholar] [CrossRef]
- Shen, X.; Zheng, S.; Metreveli, N.S.; Epstein, P.N. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 2006, 55, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Idevall-Hagren, O.; Tengholm, A. Metabolic regulation of calcium signaling in beta cells. Semin. Cell Dev. Biol. 2020, 103, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Draznin, B.; Sussman, K.E.; Eckel, R.H.; Kao, M.; Yost, T.; Sherman, N.A. Possible role of cytosolic free calcium concentrations in mediating insulin resistance of obesity and hyperinsulinemia. J. Clin. Investig. 1988, 82, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Silva, D.; Wüst, R.; Conceição, G.; Gonçalves-Rodrigues, P.; Gonçalves, N.; Gonçalves, A.; Kuster, D.; Leite-Moreira, A.F.; van der Velden, J.; de Sousa Beleza, J.M.; et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol. 2020, 228, e13378. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Junger, K.D. The effect of streptozocin-induced diabetes on the plasma membrane calcium uptake activity of rat liver. Diabetes 1984, 33, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Checchetto, V.; Biasutto, L.; Rossa, A.; Costa, R.; Bachmann, M.; Zoratti, M.; Szabo, I. Pharmacological modulation of mitochondrial ion channels. Br. J. Pharmacol. 2019, 176, 4258–4283. [Google Scholar] [CrossRef]
- Tarasova, N.V.; Vishnyakova, P.A.; Logashina, Y.A.; Elchaninov, A.V. Mitochondrial calcium uniporter structure and function in different types of muscle tissues in health and disease. Int. J. Mol. Sci. 2019, 20, 4823. [Google Scholar] [CrossRef]
- Georgiadou, E.; Haythorne, E.; Dickerson, M.T.; Lopez-Noriega, L.; Pullen, T.J.; da Silva Xavier, G.; Davis, S.; Martinez-Sanchez, A.; Semplici, F.; Rizzuto, R.; et al. The pore-forming subunit MCU of the mitochondrial Ca2+ uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 2020, 63, 1368–1381. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Semplici, F.; Ravier, M.A.; Bellomo, E.A.; Pullen, T.J.; Gilon, P.; Sekler, I.; Rizzuto, R.; Rutter, G.A. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS ONE 2012, 7, e39722. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Ly, D.D.; Nguyen, N.T.; Kim, J.H.; Yoo, H.; Chung, J.; Lee, M.S.; Cha, S.K.; Park, K.S. Mitochondrial Ca2+ uptake relieves palmitate-induced cytosolic Ca2+ overload in MIN6 cells. Mol. Cells 2020, 43, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, P.V.; Speckmann, T.; Lynn, F.C. Friend and foe: β-cell Ca2+ signaling and the development of diabetes. Mol. Metab. 2019, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef]
- Fauconnier, J.; Lanner, J.T.; Zhang, S.J.; Tavi, P.; Bruton, J.D.; Katz, A.; Westerblad, H. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes 2005, 54, 2375–2381. [Google Scholar] [CrossRef]
- Oliveira, P.J.; Seiça, R.; Coxito, P.M.; Rolo, A.P.; Palmeira, C.M.; Santos, M.S.; Moreno, A.J. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003, 554, 511–514. [Google Scholar] [CrossRef]
- Diaz-Juarez, J.; Suarez, J.; Cividini, F.; Scott, B.T.; Diemer, T.; Dai, A.; Dillmann, W.H. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am. J. Physiol. Cell Physiol. 2016, 311, C1005–C1013. [Google Scholar] [CrossRef]
- Suarez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Suarez, J.A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef]
- Ji, L.; Liu, F.; Jing, Z.; Huang, Q.; Zhao, Y.; Cao, H.; Li, J.; Yin, C.; Xing, J.; Li, F. MICU1 alleviates diabetic cardiomyopathy through mitochondrial Ca2+—Dependent antioxidant response. Diabetes 2017, 66, 1586–1600. [Google Scholar] [CrossRef]
- Garrick, R.A.; Hall, J.C. Adenosine diphosphate and calcium stimulation of respiration in mitochondria from alloxan diabetic rats. J. Cell Physiol. 1974, 84, 261–268. [Google Scholar] [CrossRef]
- Wright, L.E.; Vecellio Reane, D.; Milan, G.; Terrin, A.; Di Bello, G.; Belligoli, A.; Sanna, M.; Foletto, M.; Favaretto, F.; Raffaello, A.; et al. Increased mitochondrial calcium uniporter in adipocytes underlies mitochondrial alterations associated with insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E641–E650. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Wan, L.J.; Cui, S.Y.; Gong, Y.P.; Li, C.L. The mitochondrial Na(+)/Ca(2+) exchanger may reduce high glucose-induced oxidative stress and nucleotide-binding oligomerization domain receptor 3 inflammasome activation in endothelial cells. J. Geriatr. Cardiol. 2015, 12, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Leem, J.; Koh, E.H. Interaction between mitochondria and the endoplasmic reticulum: Implications for the pathogenesis of type 2 diabetes mellitus. Exp. Diabetes Res. 2012, 242984. [Google Scholar] [CrossRef] [PubMed]
- Thivolet, C.; Vial, G.; Cassel, R.; Rieusset, J.; Madec, A.M. Reduction of endoplasmic reticulum-mitochondria interactions in beta cells from patients with type 2 diabetes. PLoS ONE 2017, 12, e0182027. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Mohammed Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving insulin secretion in diabetes by inhibiting VDAC1 overexpression and surface translocation in β cells. Cell Metab. 2019, 29, 64–77. [Google Scholar] [CrossRef]
- Sasaki, K.; Donthamsetty, R.; Heldak, M.; Cho, Y.E.; Scott, B.T.; Makino, A. VDAC: Old protein with new roles in diabetes. Am. J. Physiol. Cell Physiol. 2012, 303, C1055–C1060. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Y.; Ge, W.; Zhou, X.; Pan, M. High glucose induces the apoptosis of HUVECs in mitochondria dependent manner by enhancing VDAC1 expression. Pharmazie 2018, 73, 725–728. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Šileikytė, J.; Forte, M. The mitochondrial permeability transition in mitochondrial disorders. Oxid. Med. Cell Longev. 2019, 3403075. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, M.; Szabò, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Efimov, S.V.; Dubinin, M.V.; Kobchikova, P.P.; Zgadzay, Y.O.; Khodov, I.A.; Belosludtsev, K.N.; Klochkov, V.V. Comparison of cyclosporin variants B-E based on their structural properties and activity in mitochondrial membranes. Biochem. Biophys. Res. Commun. 2020, 526, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.; Baggett, A.; Csordas, G.; Sheu, S.S. SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca2+ influx, and regulation of mitochondrial permeability transition pore opening. J. Biol. Chem. 2019, 294, 10807–10818. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Rück, A.; Dolder, M.; Wallimann, T.; Brdiczka, D. Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS Lett. 1998, 426, 97–101. [Google Scholar] [CrossRef]
- Beutner, G.; Rück, A.; Riede, B.; Brdiczka, D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta 1998, 1368, 7–18. [Google Scholar] [CrossRef]
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998, 258, 729–735. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef]
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323. [Google Scholar] [CrossRef] [PubMed]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef]
- Carraro, M.; Giorgio, V.; Šileikytė, J.; Sartori, G.; Forte, M.; Lippe, G.; Zoratti, M.; Szabò, I.; Bernardi, P. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J. Biol. Chem. 2014, 289, 15980–15985. [Google Scholar] [CrossRef]
- von Stockum, S.; Giorgio, V.; Trevisan, E.; Lippe, G.; Glick, G.D.; Forte, M.A.; Da-Rè, C.; Checchetto, V.; Mazzotta, G.; Costa, R.; et al. F-ATPase of drosophila melanogaster forms 53-picosiemen (53-pS) channels responsible for mitochondrial Ca2+-induced Ca2+ release. J. Biol. Chem. 2015, 290, 4537–4544. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Checchetto, V.; Sartori, G.; Kucharczyk, R.; di Rago, J.P.; Minervini, G.; Franchin, C.; Arrigoni, G.; Giorgio, V.; Petronilli, V.; et al. High-conductance channel formation in yeast mitochondria is mediated by F-ATP synthase e and g subunits. Cell Physiol. Biochem. 2018, 50, 1840–1855. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. A An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The mitochondrial permeability transition pore: Channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Zhou, W.; Marinelli, F.; Nief, C.; Faraldo-Gómez, J.D. Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. eLife 2017, 6, e23781. [Google Scholar] [CrossRef] [PubMed]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP synthase C-subunit-deficient mitochondria have a small cyclosporine A-sensitive channel, but lack the permeability transition pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Gutiérrez-Aguilar, M. The mitochondrial permeability transition pore: Is it formed by the ATP synthase, adenine nucleotide translocators or both? Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148249. [Google Scholar] [CrossRef] [PubMed]
- Agafonov, A.; Gritsenko, E.; Belosludtsev, K.; Kovalev, A.; Gateau-Roesch, O.; Saris, N.E.; Mironova, G.D. A permeability transition in liposomes induced by the formation of Ca2+/palmitic acid complexes. Biochim. Biophys. Acta 2003, 1609, 153–160. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Agafonov, A.V.; Astashev, M.E.; Kazakov, A.S.; Saris, N.E.; Mironova, G.D. Ca2+-dependent permeabilization of mitochondria and liposomes by palmitic and oleic acids: A comparative study. Biochim. Biophys. Acta 2014, 1838, 2600–2606. [Google Scholar] [CrossRef]
- Mironova, G.D.; Saris, N.E.; Belosludtseva, N.V.; Agafonov, A.V.; Elantsev, A.B.; Belosludtsev, K.N. Involvement of palmitate/Ca2+(Sr2+)-induced pore in the cycling of ions across the mitochondrial membrane. Biochim. Biophys. Acta 2015, 1848, 488–495. [Google Scholar] [CrossRef][Green Version]
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 β-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Death Dis. 2011, 2, e134. [Google Scholar] [CrossRef]
- Lablanche, S.; Cottet-Rousselle, C.; Argaud, L.; Laporte, C.; Lamarche, F.; Richard, M.J.; Berney, T.; Benhamou, P.Y.; Fontaine, E. Respective effects of oxygen and energy substrate deprivation on beta cell viability. Biochim. Biophys. Acta 2015, 1847, 629–639. [Google Scholar] [CrossRef]
- Fujimoto, K.; Chen, Y.; Polonsky, K.S.; Dorn, G.W., 2nd. Targeting cyclophilin D and the mitochondrial permeability transition enhances beta-cell survival and prevents diabetes in Pdx1 deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 10214–10219. [Google Scholar] [CrossRef]
- Monaco, C.; Hughes, M.C.; Ramos, S.V.; Varah, N.E.; Lamberz, C.; Rahman, F.A.; McGlory, C.; Tarnopolsky, M.A.; Krause, M.P.; Laham, R.; et al. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia 2018, 61, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Lumini-Oliveira, J.; Ascensão, A.; Pereira, C.V.; Magalhães, S.; Marques, F.; Oliveira, P.J.; Magalhães, J. Long-term hyperglycaemia decreases gastrocnemius susceptibility to permeability transition. Eur. J. Clin. Investig. 2010, 40, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Couturier, K.; Hininger, I.; Poulet, L.; Anderson, R.A.; Roussel, A.M.; Canini, F.; Batandier, C. Cinnamon intake alleviates the combined effects of dietary-induced insulin resistance and acute stress on brain mitochondria. J. Nutr. Biochem. 2016, 28, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Proença, T.; Seiça, R.; Oliveira, C.R. Effect of streptozotocin-induced diabetes on rat brain mitochondria. J. Neuroendocrinol. 2004, 16, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.J.; Esteves, T.C.; Seiça, R.; Moreno, A.J.; Santos, M.S. Calcium-dependent mitochondrial permeability transition is augmented in the kidney of Goto-Kakizaki diabetic rat. Diabetes Metab. Res. Rev. 2004, 20, 131–136. [Google Scholar] [CrossRef]
- Williamson, C.L.; Dabkowski, E.R.; Baseler, W.A.; Croston, T.L.; Always, S.E.; Hollander, J.M. Enhanced apoptotic propensity in diabetic cardiac mitochondria: Influence of subcellular spatial location. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H633–H642. [Google Scholar] [CrossRef]
- Yan, S.; Du, F.; Wu, L.; Zhang, Z.; Zhong, C.; Yu, Q.; Wang, Y.; Lue, L.F.; Walker, D.G.; Douglas, J.T.; et al. F1F0 ATP synthase-cyclophilin D interaction contributes to diabetes-induced synaptic dysfunction and cognitive decline. Diabetes 2016, 65, 3482–3494. [Google Scholar] [CrossRef]
- Lindblom, R.; Higgins, G.C.; Nguyen, T.V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. 2020, 134, 239–259. [Google Scholar] [CrossRef]
- Dabkowski, E.R.; Baseler, W.A.; Williamson, C.L.; Powell, M.; Razunguzwa, T.T.; Frisbee, J.C.; Hollander, J.M. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H529–H540. [Google Scholar] [CrossRef]
- Sparks, L.M.; Gemmink, A.; Phielix, E.; Bosma, M.; Schaart, G.; Moonen-Kornips, E.; Jörgensen, J.A.; Nascimento, E.B.; Hesselink, M.K.; Schrauwen, P.; et al. ANT1-mediated fatty acid-induced uncoupling as a target for improving myocellular insulin sensitivity. Diabetologia 2016, 59, 1030–1039. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, C.; Vasquez, F.E.; Galeva, N.; Onyango, I.; Swerdlow, R.H.; Dobrowsky, R.T. Hyperglycemia alters the schwann cell mitochondrial proteome and decreases coupled respiration in the absence of superoxide production. J. Proteome Res. 2010, 9, 458–471. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andelova, N.; Waczulikova, I.; Talian, I.; Sykora, M.; Ferko, M. mPTP proteins regulated by streptozotocin-induced diabetes mellitus are effectively involved in the processes of maintaining myocardial metabolic adaptation. Int. J. Mol. Sci. 2020, 21, 2622. [Google Scholar] [CrossRef]
- Munusamy, S.; Saba, H.; Mitchell, T.; Megyesi, J.K.; Brock, R.W.; Macmillan-Crow, L.A. Alteration of renal respiratory Complex-III during experimental type-1 diabetes. BMC Endocr. Dis. 2009, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Sloan, R.C.; Moukdar, F.; Frasier, C.R.; Patel, H.D.; Bostian, P.A.; Lust, R.M.; Brown, D.A. Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell Cardiol. 2012, 52, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, E.P.; Laker, R.C.; Breen, D.S.; Akhtar, Y.N.; Kenwood, B.M.; Liao, J.A.; Zhang, M.; Fazakerley, D.J.; Tomsig, J.L.; Harris, T.E.; et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 2013, 3, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; Uchino, H.; Elmér, E.; Siesjö, B.K. Amelioration by cyclosporin A of brain damage following 5 or 10 min of ischemia in rats subjected to preischemic hyperglycemia. Brain Res. 1997, 753, 133–140. [Google Scholar] [CrossRef]
- Qi, R.; Wang, D.; Xing, L.; Wu, Z. Cyclosporin A inhibits mitochondrial biogenesis in Hep G2 cells. Biochem. Biophys. Res. Commun. 2018, 496, 941–946. [Google Scholar] [CrossRef]
- Koshkin, V.; Bikopoulos, G.; Chan, C.B.; Wheeler, M.B. The characterization of mitochondrial permeability transition in clonal pancreatic beta-cells. Multiple modes and regulation. J. Biol. Chem. 2004, 279, 41368–41376. [Google Scholar] [CrossRef]
- Lu, A.; Chu, C.; Mulvihill, E.; Wang, R.; Liang, W. ATP-sensitive K+ channels and mitochondrial permeability transition pore mediate effects of hydrogen sulfide on cytosolic Ca2+ homeostasis and insulin secretion in β-cells. Pflugers Arch. 2019, 471, 1551–1564. [Google Scholar] [CrossRef]
- Kim, J.W.; Yang, J.H.; Park, H.S.; Sun, C.; Lee, S.H.; Cho, J.H.; Yang, C.W.; Yoon, K.H. Rosiglitazone protects the pancreatic beta-cell death induced by cyclosporine A. Biochem. Biophys. Res. Commun. 2009, 390, 763–768. [Google Scholar] [CrossRef]
- Taddeo, E.P.; Alsabeeh, N.; Baghdasarian, S.; Wikstrom, J.D.; Ritou, E.; Sereda, S.; Erion, K.; Li, J.; Stiles, L.; Abdulla, M.; et al. Mitochondrial proton leak regulated by cyclophilin D elevates insulin secretion in islets at nonstimulatory glucose levels. Diabetes 2020, 69, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Bugliani, M.; Masini, M.; Liechti, R.; Marselli, L.; Xenarios, I.; Boggi, U.; Filipponi, F.; Masiello, P.; Marchetti, P. The direct effects of tacrolimus and cyclosporin A on isolated human islets: A functional, survival and gene expression study. Islets 2009, 1, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, K.; Fukunaga, K.; Matsumoto, K.; Shichiri, M.; Miyamoto, E. Cyclosporin A stimulation of glucose-induced insulin secretion in MIN6 cells. Endocrinology 1996, 137, 5255–5263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riojas-Hernández, A.; Bernal-Ramírez, J.; Rodríguez-Mier, D.; Morales-Marroquín, F.E.; Domínguez-Barragán, E.M.; Borja-Villa, C.; Rivera-Álvarez, I.; García-Rivas, G.; Altamirano, J.; García, N. Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from Zucker Fa/fa rats with type 2 diabetes. Life Sci. 2015, 141, 32–43. [Google Scholar] [CrossRef]
- Itoh, T.; Kouzu, H.; Miki, T.; Tanno, M.; Kuno, A.; Sato, T.; Sunaga, D.; Murase, H.; Miura, T. Cytoprotective regulation of the mitochondrial permeability transition pore is impaired in type 2 diabetic Goto-Kakizaki rat hearts. J. Mol. Cell Cardiol. 2012, 53, 870–879. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef]
- Baseler, W.A.; Dabkowski, E.R.; Williamson, C.L.; Croston, T.L.; Thapa, D.; Powell, M.J.; Razunguzwa, T.T.; Hollander, J.M. Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: Contribution of protein import dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R186–R200. [Google Scholar] [CrossRef]
- Lumini-Oliveira, J.; Magalhães, J.; Pereira, C.V.; Moreira, A.C.; Oliveira, P.J.; Ascensão, A. Endurance training reverts heart mitochondrial dysfunction, permeability transition and apoptotic signaling in long-term severe hyperglycemia. Mitochondrion 2011, 11, 54–63. [Google Scholar] [CrossRef]
- Najafi, M.; Farajnia, S.; Mohammadi, M.; Badalzadeh, R.; Ahmadi Asl, N.; Baradaran, B.; Amani, M. Inhibition of mitochondrial permeability transition pore restores the cardioprotection by postconditioning in diabetic hearts. J. Diabetes Metab. Disord. 2014, 13, 106. [Google Scholar] [CrossRef]
- Huhn, R.; Heinen, A.; Hollmann, M.W.; Schlack, W.; Preckel, B.; Weber, N.C. Cyclosporine A administered during reperfusion fails to restore cardioprotection in prediabetic Zucker obese rats in vivo. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 706–712. [Google Scholar] [CrossRef]
- Morrow, R.M.; Picard, M.; Derbeneva, O.; Leipzig, J.; McManus, M.J.; Gouspillou, G.; Barbat-Artigas, S.; Dos Santos, C.; Hepple, R.T.; Murdock, D.G.; et al. Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Seiça, R.; Oliveira, C.R. Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes 2003, 52, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Krügel, K.; Wurm, A.; Pannicke, T.; Hollborn, M.; Karl, A.; Wiedemann, P.; Reichenbach, A.; Kohen, L.; Bringmann, A. Involvement of oxidative stress and mitochondrial dysfunction in the osmotic swelling of retinal glial cells from diabetic rats. Exp. Eye Res. 2011, 92, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, J.; Pennington, A.; Klawitter, J.; Thurman, J.M.; Christians, U. Mitochondrial cyclophilin D ablation is associated with the activation of Akt/p70S6K pathway in the mouse kidney. Sci. Rep. 2017, 7, 10540. [Google Scholar] [CrossRef] [PubMed]
- Kristal, B.S.; Matsuda, M.; Yu, B.P. Abnormalities in the mitochondrial permeability transition in diabetic rats. Biochem. Biophys. Res. Commun. 1996, 222, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.A.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B.; et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2016, 59, 614–623. [Google Scholar] [CrossRef]
- Sobel, D.O.; Henzke, A.; Abbassi, V. Cyclosporin and methotrexate therapy induces remission in type 1 diabetes mellitus. Acta Diabetol. 2010, 47, 243–250. [Google Scholar] [CrossRef]
- Devalaraja-Narashimha, K.; Diener, A.M.; Padanilam, B.J. Cyclophilin D deficiency prevents diet-induced obesity in mice. FEBS Lett. 2011, 585, 677–682. [Google Scholar] [CrossRef]
- Guigas, B.; Detaille, D.; Chauvin, C.; Batandier, C.; De Oliveira, F.; Fontaine, E.; Leverve, X. Metformin inhibits mitochondrial permeability transition and cell death: A pharmacological in vitro study. Biochem. J. 2004, 382, 877–884. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Detaille, D.; Villanueva, G.R.; Delgado-Esteban, M.; Guigas, B.; Attia, S.; Fontaine, E.; Almeida, A.; Leverve, X. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J. Mol. Neurosci. 2008, 34, 77–87. [Google Scholar] [CrossRef]
- Carvalho, C.; Correia, S.; Santos, M.S.; Seiça, R.; Oliveira, C.R.; Moreira, P.I. Metformin promotes isolated rat liver mitochondria impairment. Mol. Cell Biochem. 2008, 308, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Kano, S.; Horie, T. Mitochondrial permeability transition as a potential determinant of hepatotoxicity of antidiabetic thiazolidinediones. Toxicology 2006, 222, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Segawa, M.; Sekine, S.; Sato, T.; Ito, K. Increased susceptibility to troglitazone-induced mitochondrial permeability transition in type 2 diabetes mellitus model rat. J. Toxicol. Sci. 2018, 43, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.T.; Qian, L.B.; Zhang, F.J.; Wang, J.; Ai, H.; Tang, L.H.; Wang, H.P. Cardioprotective effects of luteolin on ischemia/reperfusion injury in diabetic rats are modulated by eNOS and the mitochondrial permeability transition pathway. J. Cardiovasc. Pharmacol. 2015, 65, 349–356. [Google Scholar] [CrossRef]
- Zhong, Y.; Jin, J.; Liu, P.; Song, Y.; Zhang, H.; Sheng, L.; Zhou, H.; Jiang, B. Berberine attenuates hyperglycemia by inhibiting the hepatic glucagon pathway in diabetic mice. Oxid. Med. Cell Longev. 2020, 6210526. [Google Scholar] [CrossRef]
- Pereira, C.V.; Machado, N.G.; Oliveira, P.J. Mechanisms of berberine (natural yellow 18)-induced mitochondrial dysfunction: Interaction with the adenine nucleotide translocator. Toxicol. Sci. 2008, 105, 408–417. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pancreatic Islets | Heart | Skeletal Muscles | Brain | Kidney | Adipose | Liver | |
|---|---|---|---|---|---|---|---|
| Biogenesis | ↓ [11] ↑ [12] | ↓↑ [13] | ↓ [14] | ↓ [15] | ↓ [16] | ↓ [17] | ↓ [18] ↑ [19] |
| Mitophagy | ↓ [20] ↑ [21] | ↓ [22] | ↓ [23] | ↓ [24] | ↓ [25] ↑ [26] | ↑ [27] | ↓ [28] |
| Fission/Fusion | ↑/↓ [29] | ↑/↓ [30] | ↑ [31] ↓ [32] | ↑/↓ [33] | ↑/↓ [34] | ↑ [35] ↓? | ↑/↓ [36] |
| OXPHOS | ↑ or ↓ or not changed, see review [37] | ||||||
| ROS production | ↑ [20] | ↑ [38] | ↑ [39] | ↑ [40] | ↑ [34] | ↑ [17] | ↑ [41] |
| Pancreatic β-Cells | Heart | Skeletal Muscles | Brain | Kidney | Liver | ||
|---|---|---|---|---|---|---|---|
| MPT resistance | ↓ [201] | ↓ [159] | ↓ [204] ↑ [205] | ↓ [206] ↑ [207] | ↓ [208] | ↑ [114] | |
| MPT proteins | |||||||
| cyclophilin D | n.e. | ↑ [209] | n.e. | ↑ [210] | − [211] | − [41] | |
| ANT | n.e. | ↓ [212] | − [213] | ↑ [214] | n.e. | ↓ [41] | |
| F0F1-ATPase | n.e. | ↓ [212] ↑ [215] | n.e | n.e. | ↓ [211] ↑ [216] | ↓ [41] | |
| MPT pore inhibitors | + [201] | + [217] | + [218] | + [219] | no effect [211] | − [220] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. https://doi.org/10.3390/ijms21186559
Belosludtsev KN, Belosludtseva NV, Dubinin MV. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. International Journal of Molecular Sciences. 2020; 21(18):6559. https://doi.org/10.3390/ijms21186559
Chicago/Turabian StyleBelosludtsev, Konstantin N., Natalia V. Belosludtseva, and Mikhail V. Dubinin. 2020. "Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore" International Journal of Molecular Sciences 21, no. 18: 6559. https://doi.org/10.3390/ijms21186559
APA StyleBelosludtsev, K. N., Belosludtseva, N. V., & Dubinin, M. V. (2020). Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. International Journal of Molecular Sciences, 21(18), 6559. https://doi.org/10.3390/ijms21186559