The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis


Abstract
1. Introduction
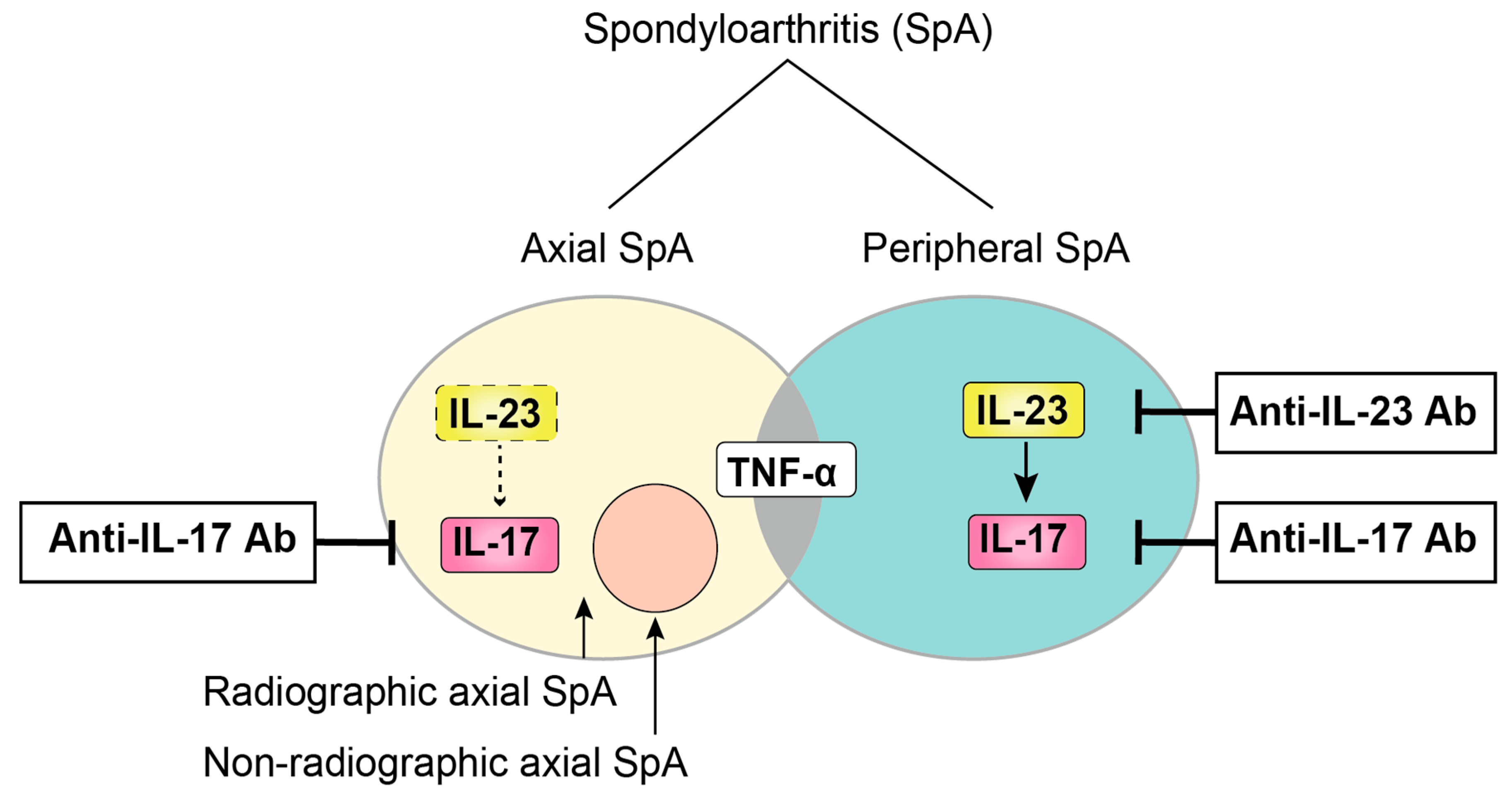
2. The Concept of SpA
3. Pathogenesis
3.1. Genetic Background
3.2. Mechanical Stress
3.3. Dysbiosis
4. IL-23
IL-23 in SpA Patients
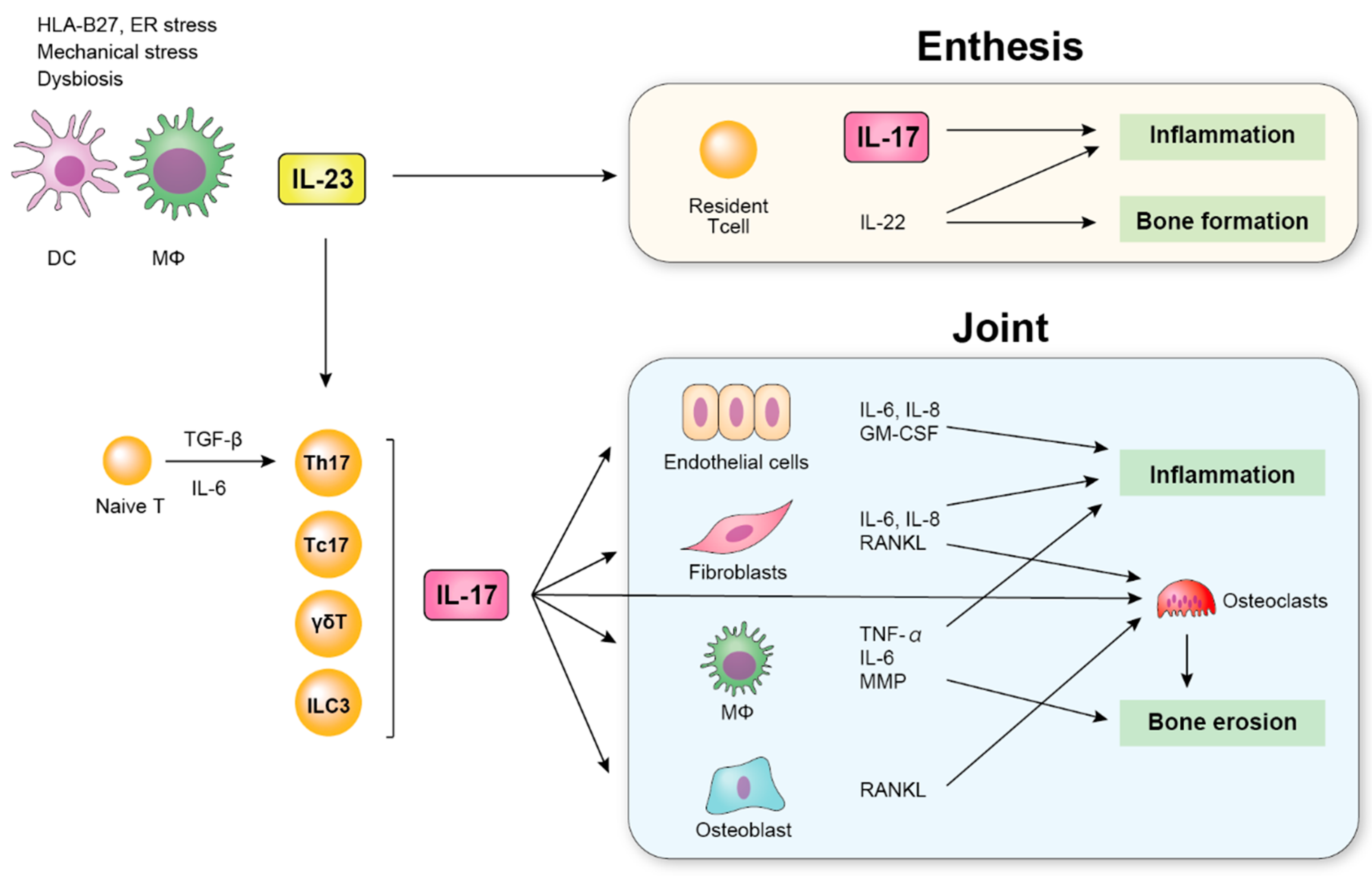
5. IL-17
5.1. IL-17 in SpA Patients
5.2. IL-17-Producing Cells
5.2.1. Th17 Cells (IL-17+ CD4+ T Cells)
5.2.2. γδT Cells
5.2.3. Mucosa-Associated Invariant T (MAIT) Cells
5.2.4. Type 3 Innate Lymphoid Cells (ILC3)
5.2.5. Other IL-17-Producing Cells
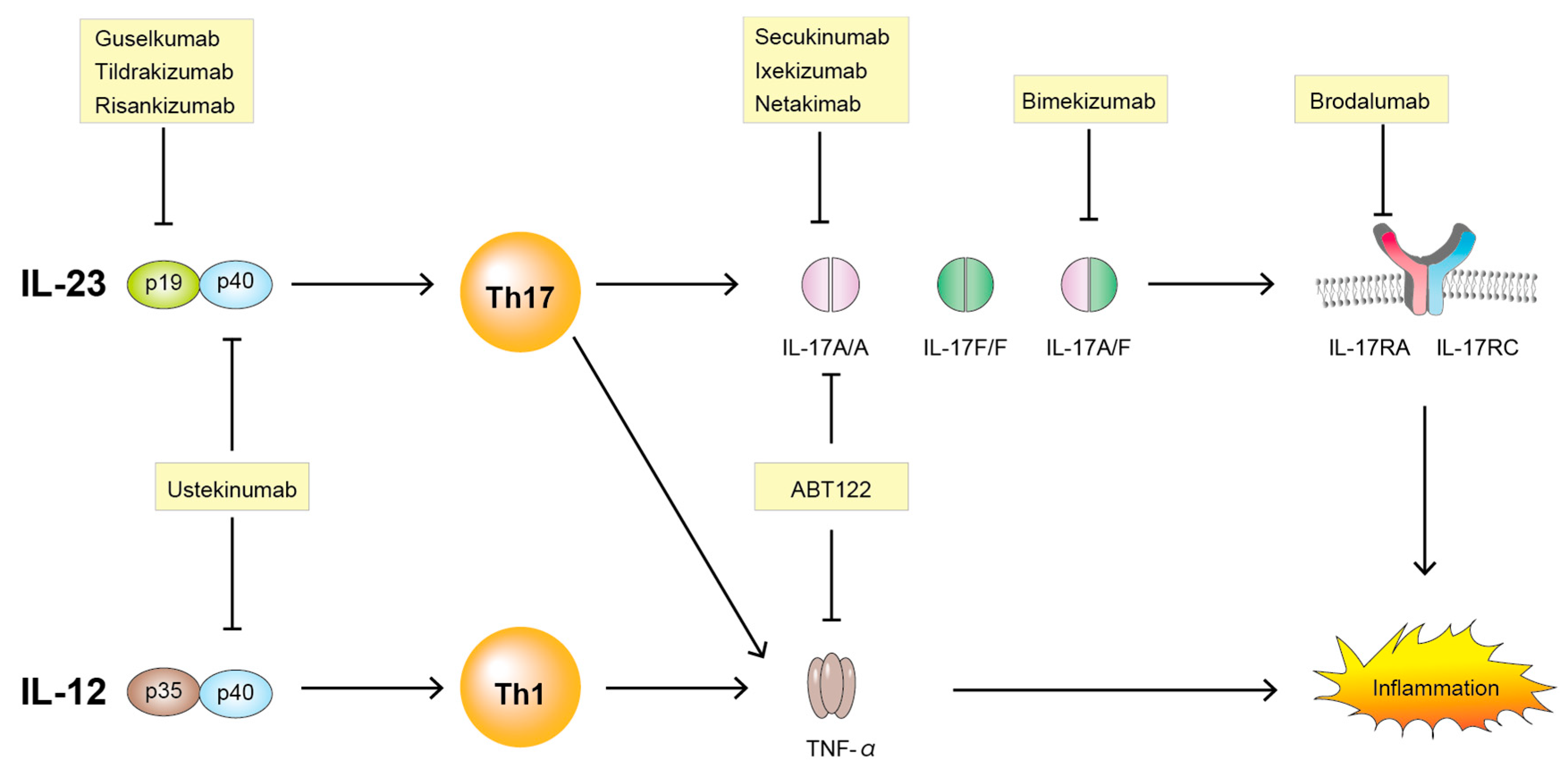
6. IL-23/IL-17 Axis-Targeting Therapies
6.1. Anti IL-23
6.1.1. Ustekinumab
6.1.2. Guselkumab
6.1.3. Tildrakizumab
6.1.4. Risankizumab
6.2. Anti IL-17
6.2.1. Secukinumab
6.2.2. Ixekizumab
6.2.3. Netakimab
6.2.4. Brodalumab
6.2.5. Bimekizumab
6.3. Bispecific TNF/IL-17A Inhibitor
ABT-122
7. Future Prospective
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SpA | Spondyloarthritis |
| AS | Ankylosing spondylitis |
| PsA | Psoriatic spondylitis |
| re-SpA | Reactive spondylitis |
| uSpA | Undifferentiated arthritis |
| IBD | Inflammatory bowel disease |
| Ps | Psoriasis |
| RA | Rheumatoid arthritis |
| IL-23 | Interleukin-23 |
| IL-17 | Interleukin-17 |
| IL-17R | Interleukin-17 receptor |
| Th1 | Type 1 helper T cell |
| Th2 | Type 2 helper T cell |
| Th17 | Type 17 helper T cell |
| TCR | T cell receptor |
| CTL | Cytotoxic T lymphocyte |
| HLA | Human leukocyte antigen |
| MHC | Major histocompatibility complex |
| ERAP1 | Endoplasmic reticulum aminopeptidase 1 |
| ER | Endoplasmic reticulum |
| SFB | Segmental filamentous bacteria |
| APC | Antigen presenting cells |
| DC | Dendric dell |
| RANKL | Receptor activator of NF-κB ligand |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| TNF-α | Tumor necrosis factor α |
| ILC3 | Type 3 innate lymphoid cells |
| MAIT | Mucosa-associated invariant T (MAIT) cells |
References
- Moll, J.M.H.; Haslock, I.; Macrae, I.F.; Wright, V. Associations between ankylosing spondylitis, psoriatic arthritis, Reiter’s disease, the intestinal arthropathies, and Behcet’s syndrome. Medicine 1974, 53, 343–364. [Google Scholar] [CrossRef] [PubMed]
- Kellgren, J.H. Epidemiology of Chronic Rheumatism. In Atlas of Standard Radiographs of Arthritis; Blackwell Science Ltd.: Oxford, UK, 1963. [Google Scholar]
- Van Der Linden, S.; Valkenburg, H.A.; Cats, A. Evaluation of Diagnostic Criteria for Ankylosing Spondylitis. Arthritis Rheum. 1984, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Amor, B.; Dougados, M.; Mijiyawa, M. Criteria of the classification of spondylarthropathies. Rev. Rhum. Mal. Osteoartic. 1990, 57, 85–89. [Google Scholar] [PubMed]
- Dougados, M.; Van Der Linden, S.; Juhlin, R.; Huitfeldt, B.; Amor, B.; Calin, A.; Cats, A.; Dijkmans, B.; Olivieri, I.; Pasero, G.; et al. The European Spondylarthropathy Study Group Preliminary Criteria for the Classification of Spondylarthropathy. Arthritis Rheum. 1991, 34, 1218–1227. [Google Scholar] [CrossRef]
- Rudwaleit, M.; Van Der Heijde, D.; Landewé, R.; Listing, J.; Akkoc, N.; Brandt, J.; Braun, J.; Chou, C.T.; Collantes-Estévez, E.; Dougados, M.; et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): Validation and final selection. Ann. Rheum. Dis. 2009, 68, 777–783. [Google Scholar] [CrossRef]
- Rudwaleit, M.; Van Der Heijde, D.; Landewe, R.; Akkoc, N.; Brandt, J.; Chou, C.T.; Dougados, M.; Huang, F.; Gu, J.; Kirazli, Y.; et al. The Assessment of SpondyloArthritis international Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann. Rheum. Dis. 2010, 70, 25–31. [Google Scholar] [CrossRef]
- Norment, A.M.; Salter, R.D.; Parham, P.; Engelhard, V.H.; Littman, D.R. Cell-cell adhesion mediated by CD8 and MHC class I molecules. Nature 1988, 336, 79–81. [Google Scholar] [CrossRef]
- Schlosstein, L.; Terasaki, P.I.; Bluestone, R.; Pearson, C.M. High Association of an HL-A Antigen, W27, with Ankylosing Spondylitis. N. Engl. J. Med. 1973, 288, 704–706. [Google Scholar] [CrossRef]
- Zhu, W.; He, X.; Cheng, K.; Zhang, L.; Chen, D.; Wang, X.; Qiu, G.; Cao, X.; Weng, X. Ankylosing spondylitis: Etiology, pathogenesis, and treatments. Bone Res. 2019, 7, 22. [Google Scholar] [CrossRef]
- Taurog, J.D.; Chhabra, A.; Colbert, R.A. Ankylosing Spondylitis and Axial Spondyloarthritis. N. Engl. J. Med. 2016, 374, 2563–2574. [Google Scholar] [CrossRef]
- Ruiz, D.G.; De Azevedo, M.N.L.; Lupi, O. HLA-B27 frequency in a group of patients with psoriatic arthritis. An. Bras. Dermatol. 2012, 87, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Baraliakos, X.; Coates, L.C.; Braun, J. The involvement of the spine in psoriatic arthritis. Clin. Exp. Rheumatol. 2015, 33 (Suppl. 93), S31–S35. [Google Scholar] [PubMed]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007, 39, 1329–1337. [Google Scholar] [PubMed]
- Strange, A.; Capon, F.; Spencer, C.C.A.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.; Bellenguez, C.; Bergboer, J.G.M.; et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [Google Scholar]
- York, I.A.; Chang, S.-C.; Saric, T.; Keys, J.A.; Favreau, J.M.; Goldberg, A.L.; Rock, K.L. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat. Immunol. 2002, 3, 1177–1184. [Google Scholar] [CrossRef]
- DeLay, M.L.; Turner, M.J.; Klenk, E.I.; Smith, J.A.; Sowders, D.P.; Colbert, R.A. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009, 60, 2633–2643. [Google Scholar] [CrossRef]
- Cargill, M.; Schrodi, S.J.; Chang, M.; Garcia, V.E.; Brandon, R.; Callis, K.P.; Matsunami, N.; Ardlie, K.G.; Civello, D.; Catanese, J.J.; et al. A Large-Scale Genetic Association Study Confirms IL12B and Leads to the Identification of IL23R as Psoriasis-Risk Genes. Am. J. Hum. Genet. 2007, 80, 273–290. [Google Scholar] [CrossRef]
- Cenit, M.C.; Ortego-Centeno, N.; Raya, E.; Callejas, J.L.; García-Hernández, F.J.; Castillo-Palma, M.J.; Fernández-Sueiro, J.; Magro, C.; Solans, R.; Castañeda, S.; et al. Influence of the STAT3 genetic variants in the susceptibility to psoriatic arthritis and Behcet’s disease. Hum. Immunol. 2013, 74, 230–233. [Google Scholar] [CrossRef]
- Ellinghaus, D.; The International IBD Genetics Consortium (IIBDGC); Jostins, L.; Spain, S.L.; Cortes, A.; Bethune, J.; Han, B.; Park, Y.R.; Raychaudhuri, S.; Pouget, J.G.; et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 2016, 48, 510–518. [Google Scholar] [CrossRef]
- Jacques, P.; McGonagle, D. The role of mechanical stress in the pathogenesis of spondyloarthritis and how to combat it. Best Pract. Res. Clin. Rheumatol. 2014, 28, 703–710. [Google Scholar] [CrossRef]
- Schett, G.; Lories, R.J.; D’Agostino, M.-A.; Elewaut, D.; Kirkham, B.; Soriano, E.R.; McGonagle, D. Enthesitis: From pathophysiology to treatment. Nat. Rev. Rheumatol. 2017, 13, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Ruff, C.B. Mechanical determinants of bone form: Insights from skeletal remains. J. Musculoskelet. Neuronal Interact. 2005, 5, 202. [Google Scholar] [PubMed]
- Jacques, P.; Lambrecht, S.; Verheugen, E.; Pauwels, E.; Kollias, G.; Armaka, M.; Verhoye, M.; Van Der Linden, A.M.; Achten, R.; Lories, R.J.; et al. Proof of concept: Enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann. Rheum. Dis. 2013, 73, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef]
- Scher, J.U.; Littman, D.R.; Abramson, S.B. Microbiome in Inflammatory Arthritis and Human Rheumatic Diseases. Arthritis Rheumatol. 2016, 68, 35–45. [Google Scholar] [CrossRef]
- Taurog, J.D.; Richardson, J.A.; Croft, J.T.; Simmons, W.A.; Zhou, M.; Fernández-Sueiro, J.L.; Balish, E.; Hammer, R.E. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 1994, 180, 2359–2364. [Google Scholar] [CrossRef]
- Cua, D.J.; Sherlock, J.P. Autoimmunity’s collateral damage: Gut microbiota strikes “back”. Nat. Med. 2011, 17, 1055–1056. [Google Scholar] [CrossRef]
- Van Praet, L.; Bosch, F.E.V.D.; Jacques, P.; Carron, P.; Jans, L.; Colman, R.; Glorieus, E.; Peeters, H.; Mielants, H.; De Vos, M.; et al. Microscopic gut inflammation in axial spondyloarthritis: A multiparametric predictive model. Ann. Rheum. Dis. 2012, 72, 414–417. [Google Scholar] [CrossRef]
- Van Praet, L.; Jans, L.; Carron, P.; Jacques, P.; Glorieus, E.; Colman, R.; Cypers, H.; Mielants, H.; De Vos, M.; Cuvelier, C.; et al. Degree of bone marrow oedema in sacroiliac joints of patients with axial spondyloarthritis is linked to gut inflammation and male sex: Results from the GIANT cohort. Ann. Rheum. Dis. 2013, 73, 1186–1189. [Google Scholar] [CrossRef]
- Scher, J.U.; Ubeda, C.; Artacho, A.; Attur, M.; Isaac, S.; Reddy, S.M.; Marmon, S.; Neimann, A.; Brusca, S.B.; Patel, T.; et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015, 67, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.-E.; Ciccia, F.; Willner, D.; Warrington, N.M.; Robinson, P.C.; Gardiner, B.; Marshall, M.; Kenna, T.J.; Triolo, G.; Brown, M.A. Brief Report: Intestinal Dysbiosis in Ankylosing Spondylitis. Arthritis Rheumatol. 2015, 67, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunology 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; De Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 2002, 278, 1910–1914. [Google Scholar] [CrossRef]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef]
- Lubberts, E. The IL-23–IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef]
- Duvallet, E.; Semerano, L.; Assier, E.; Falgarone, G.; Boissier, M.-C. Interleukin-23: A key cytokine in inflammatory diseases. Ann. Med. 2011, 43, 503–511. [Google Scholar] [CrossRef]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef]
- Teng, M.W.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.-L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1090–1100. [Google Scholar] [CrossRef]
- Wang, X.-W.; Lin, Z.; Wei, Q.; Jiang, Y.; Gu, J. Expression of IL-23 and IL-17 and effect of IL-23 on IL-17 production in ankylosing spondylitis. Rheumatol. Int. 2009, 29, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Pan, F.; Gao, J.; Ge, R.; Duan, Z.; Zeng, Z.; Liao, F.; Xia, G.; Wang, S.; Xu, S.; et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin. Rheumatol. 2010, 30, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased Expression of Interleukin 23 p19 and p40 in Lesional Skin of Patients with Psoriasis Vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Celis, R.; Planell, N.; Fernandez-Sueiro, J.L.; Sanmartí, R.; Ramirez, J.; González-Álvaro, I.; Pablos, J.L.; Cañete, J.D. Synovial cytokine expression in psoriatic arthritis and associations with lymphoid neogenesis and clinical features. Arthritis Res. Ther. 2012, 14, R93. [Google Scholar] [CrossRef] [PubMed]
- Melis, L.; Vandooren, B.; Kruithof, E.; Jacques, P.; De Vos, M.; Mielants, H.; Verbruggen, G.; De Keyser, F.; Elewaut, D. Systemic levels of IL-23 are strongly associated with disease activity in rheumatoid arthritis but not spondyloarthritis. Ann. Rheum. Dis. 2010, 69, 618–623. [Google Scholar]
- Ciccia, F.; Bombardieri, M.; Principato, A.; Giardina, A.; Tripodo, C.; Porcasi, R.; Peralta, S.; Franco, V.; Giardina, E.; Craxi, A.; et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009, 60, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Saieva, L.; Peralta, S.; Giardina, A.; Cannizzaro, A.; Sireci, G.; De Leo, G.; Alessandro, R.; et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann. Rheum. Dis. 2015, 74, 1739–1747. [Google Scholar] [CrossRef]
- Rouvier, E.; Luciani, M.F.; Mattéi, M.G.; Denizot, F.; Golstein, P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 1993, 150, 5445–5456. [Google Scholar]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef]
- Ye, P.; Rodriguez, F.H.; Kanaly, S.; Stocking, K.L.; Schurr, J.; Schwarzenberger, P.; Oliver, P.; Huang, W.; Zhang, P.; Zhang, J.; et al. Requirement of Interleukin 17 Receptor Signaling for Lung Cxc Chemokine and Granulocyte Colony-Stimulating Factor Expression, Neutrophil Recruitment, and Host Defense. J. Exp. Med. 2001, 194, 519–528. [Google Scholar] [CrossRef]
- Huang, W.; Na, L.; Fidel, P.L.; Schwarzenberger, P. Requirement of Interleukin-17A for Systemic Anti–Candida albicans Host Defense in Mice. J. Infect. Dis. 2004, 190, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Taams, L.S.; Steel, K.J.A.; Srenathan, U.; Burns, L.A.; Kirkham, B.W. IL-17 in the immunopathogenesis of spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 453–466. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Schett, G. Effects of the IL-23–IL-17 pathway on bone in spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 365–370. [Google Scholar] [CrossRef]
- Romero-Sanchez, C.; Jaimes, D.A.; Londoño, J.; De Avila, J.; Castellanos, J.E.; Bello, J.M.; Bautista, W.; Valle-Oñate, R. Association between Th-17 cytokine profile and clinical features in patients with spondyloarthritis. Clin. Exp. Rheumatol. 2011, 29, 828–834. [Google Scholar]
- Wendling, D.; Cedoz, J.-P.; Racadot, E.; Dumoulin, G. Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Jt. Bone Spine 2007, 74, 304–305. [Google Scholar] [CrossRef]
- Chen, W.-S.; Chang, Y.-S.; Lin, K.-C.; Lai, C.-C.; Wang, S.-H.; Hsiao, K.-H.; Lee, H.-T.; Chen, M.-H.; Tsai, C.-Y.; Chou, C.-T. Association of serum interleukin-17 and interleukin-23 levels with disease activity in Chinese patients with ankylosing spondylitis. J. Chin. Med Assoc. 2012, 75, 303–308. [Google Scholar] [CrossRef]
- Xueyi, L.; Lina, C.; Zhenbiao, W.; Qing, H.; Qiang, L.; Zhu, P. Levels of Circulating Th17 Cells and Regulatory T Cells in Ankylosing Spondylitis Patients with an Inadequate Response to Anti−TNF-α Therapy. J. Clin. Immunol. 2012, 33, 151–161. [Google Scholar] [CrossRef]
- Singh, R.; Aggarwal, A.; Misra, R. Th1/Th17 cytokine profiles in patients with reactive arthritis/undifferentiated spondyloarthropathy. J. Rheumatol. 2007, 34, 2285–2290. [Google Scholar] [PubMed]
- Leipe, J.; Grunke, M.; DeChant, C.; Reindl, C.; Kerzendorf, U.; Skapenko, A.; Schulze-Koops, H. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010, 62, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.P.; Raychaudhuri, S.K.; Genovese, M.C. IL-17 receptor and its functional significance in psoriatic arthritis. Mol. Cell. Biochem. 2011, 359, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the TH17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef]
- Jandus, C.; Bioley, G.; Rivals, J.-P.; Dudler, J.; Speiser, D.E.; Romero, P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum. 2008, 58, 2307–2317. [Google Scholar] [CrossRef]
- Shen, H.; Goodall, J.C.; Gaston, H. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. 2009, 60, 1647–1656. [Google Scholar] [CrossRef]
- Menon, B.; Gullick, N.J.; Walter, G.J.; Rajasekhar, M.; Garrood, T.; Evans, H.G.; Taams, L.S.; Kirkham, B.W. Interleukin-17+CD8+ T Cells Are Enriched in the Joints of Patients With Psoriatic Arthritis and Correlate With Disease Activity and Joint Damage Progression. Arthritis Rheumatol. 2014, 66, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Goodall, J.C.; Gaston, J.H. Frequency and Phenotype of T Helper 17 Cells in Peripheral Blood and Synovial Fluid of Patients with Reactive Arthritis. J. Rheumatol. 2010, 37, 2096–2099. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Norris, P.; Goodall, J.C.; Wechalekar, M.D.; Fitzgerald, O.; Szenpetery, A.; Smith, M.; Thomas, R.; Gaston, H. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res. Ther. 2013, 15, R136. [Google Scholar] [CrossRef] [PubMed]
- Appel, H.; Heydrich, R.; Wu, P.; Scheer, R.; Hempfing, A.; Kayser, R.; Thiel, A.; Radbruch, A.; Loddenkemper, C.; Sieper, J. Analysis of IL-17+ cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res. Ther. 2011, 13, R95. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Mielke, L.A.; Mills, K.H.G. IL-17-producing γδ T cells and innate lymphoid cells. Eur. J. Immunol. 2012, 42, 2221–2231. [Google Scholar] [CrossRef]
- Hamada, S.; Umemura, M.; Shiono, T.; Tanaka, K.; Yahagi, A.; Begum, M.D.; Oshiro, K.; Okamoto, Y.; Watanabe, H.; Kawakami, K.; et al. IL-17A Produced by γδ T Cells Plays a Critical Role in Innate Immunity against Listeria monocytogenes Infection in the Liver. J. Immunol. 2008, 181, 3456–3463. [Google Scholar] [CrossRef]
- Nielsen, M.M.; Witherden, D.A.; Havran, W.L. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol. 2017, 17, 733–745. [Google Scholar] [CrossRef]
- Roark, C.L.; Simonian, P.L.; Fontenot, A.P.; Born, W.K.; O’Brien, R.L. γδ T cells: An important source of IL-17. Curr. Opin. Immunol. 2008, 20, 353–357. [Google Scholar] [CrossRef]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H.G. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.G.; Wang, T.; Zheng, J.; et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef]
- Kenna, T.J.; Davidson, S.I.; Duan, R.; Bradbury, L.A.; McFarlane, J.; Smith, M.; Weedon, H.; Street, S.; Thomas, R.; Thomas, G.P.; et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012, 64, 1420–1429. [Google Scholar] [CrossRef]
- Guggino, G.; Ciccia, F.; Di Liberto, D.; Lo Pizzo, M.; Ruscitti, P.; Cipriani, P.; Ferrante, A.; Sireci, G.; Dieli, F.; Fourniè, J.J. Interleukin (IL)-9/IL-9R axis drives γδ T cells activation in psoriatic arthritis patients. Clin. Exp. Immunol. 2016, 186, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, R.J.; Watad, A.; Fragkakis, E.M.; Dunsmuir, R.; Loughenbury, P.; Khan, A.; Millner, P.A.; Davison, A.; Marzo-Ortega, H.; Newton, D.; et al. Evidence that tissue resident human enthesis γδT-cells can produce IL-17A independently of IL-23R transcript expression. Ann. Rheum. Dis. 2019, 78, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Treiner, E.; Duban, L.; Guerri, L.; Laude, H.; Toly, C.; Prémel, V.; Devys, A.; Moura, I.C.; Tilloy, F.; et al. Stepwise development of MAIT cells in mouse and human. PLoS Biol. 2009, 7, e54. [Google Scholar] [CrossRef]
- Teunissen, M.B.M.; Yeremenko, N.; Baeten, D.L.P.; Chielie, S.; Spuls, P.I.; De Rie, M.A.; Lantz, O.; Res, P.C. The IL-17A-Producing CD8 + T-Cell Population in Psoriatic Lesional Skin Comprises Mucosa-Associated Invariant T Cells and Conventional T Cells. J. Investig. Dermatol. 2014, 134, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- Gracey, E.; Qaiyum, Z.; Almaghlouth, I.; Lawson, D.O.; Karki, S.; Avvaru, N.; Zhang, Z.; Yao, Y.; Ranganathan, V.; Baglaenko, Y.; et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann. Rheum. Dis. 2016, 75, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, E.; Chiba, A.; Tada, K.; Haga, K.; Kitagaichi, M.; Nakajima, S.; Kusaoi, M.; Sekiya, F.; Ogasawara, M.; Yamaji, K.; et al. Involvement of Mucosal-associated Invariant T cells in Ankylosing Spondylitis. J. Rheumatol. 2016, 43, 1695–1703. [Google Scholar] [CrossRef]
- Toussirot, É.; Laheurte, C.; Gaugler, B.; Gabriel, D.; Saas, P. Increased IL-22- and IL-17A-Producing Mucosal-Associated Invariant T Cells in the Peripheral Blood of Patients With Ankylosing Spondylitis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Serafini, N.; Vosshenrich, C.A.J.; Di Santo, J.P. Transcriptional regulation of innate lymphoid cell fate. Nat. Rev. Immunol. 2015, 15, 415–428. [Google Scholar] [CrossRef]
- Soare, A.; Weber, S.; Maul, L.; Rauber, S.; Gheorghiu, A.M.; Luber, M.; Houssni, I.; Kleyer, A.; Von Pickardt, G.; Gado, M.; et al. Cutting Edge: Homeostasis of Innate Lymphoid Cells Is Imbalanced in Psoriatic Arthritis. J. Immunol. 2018, 200, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, R.J.R.J.; Fragkakis, E.M.; Dunsmuir, R.; Li, Z.; Coles, M.; Marzo-Ortega, H.; Giannoudis, P.V.; Jones, E.; El-Sherbiny, Y.; McGonagle, D. Brief Report: Group 3 Innate Lymphoid Cells in Human Enthesis. Arthritis Rheumatol. 2017, 69, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liao, Q.; Hu, Y.; Zhong, D. T lymphocyte subset imbalances in patients contribute to ankylosing spondylitis. Exp. Ther. Med. 2014, 9, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Al-Mossawi, M.H.; Chen, L.; Fang, H.; Ridley, A.; De Wit, J.; Yager, N.; Hammitzsch, A.; Pulyakhina, I.; Fairfax, B.P.; Simone, D.; et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat. Commun. 2017, 8, 1510. [Google Scholar] [CrossRef]
- Chowdhury, A.C.; Chaurasia, S.; Mishra, S.K.; Aggarwal, A.; Misra, R. IL-17 and IFN-γ producing NK and γδ-T cells are preferentially expanded in synovial fluid of patients with reactive arthritis and undifferentiated spondyloarthritis. Clin. Immunol. 2017, 183, 207–212. [Google Scholar] [CrossRef]
- Noordenbos, T.; Yeremenko, N.; Gofita, I.; Van De Sande, M.; Tak, P.P.; Cañete, J.D.; Baeten, D. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis Rheum. 2011, 64, 99–109. [Google Scholar] [CrossRef]
- Steel, K.; Wu, S.-Y.; Srenathan, U.; Chan, E.; Kirkham, B.; Taams, L. O016 Synovial IL-17+ CD8+ T cells are a pro-inflammatory tissue resident population enriched in spondyloarthritis. Ann. Rheum. Dis. 2018, 77 (Suppl. 1), A8–A9. [Google Scholar]
- Gottlieb, A.B.; Menter, A.; Mendelsohn, A.; Shen, Y.-K.; Li, S.; Guzzo, C.; Fretzin, S.; Kunynetz, R.; Kavanaugh, A. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: Randomised, double-blind, placebo-controlled, crossover trial. Lancet 2009, 373, 633–640. [Google Scholar] [CrossRef]
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 2013, 382, 780–789. [Google Scholar] [CrossRef]
- Helliwell, P.S.; Gladman, D.D.; Chakravarty, S.D.; Kafka, S.; Karyekar, C.S.; You, Y.; Campbell, K.; Sweet, K.; Kavanaugh, A.; Gensler, L.S. Effects of ustekinumab on spondylitis-associated endpoints in TNFi-naïve active psoriatic arthritis patients with physician-reported spondylitis: Pooled results from two phase 3, randomised, controlled trials. RMD Open 2020, 6, e001149. [Google Scholar] [CrossRef]
- Deodhar, A.; Gensler, L.S.; Sieper, J.; Clark, M.; Calderon, C.; Wang, Y.; Zhou, Y.; Leu, J.H.; Campbell, K.; Sweet, K.; et al. Three Multicenter, Randomized, Double-Blind, Placebo-Controlled Studies Evaluating the Efficacy and Safety of Ustekinumab in Axial Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, A.; Helliwell, P.S.; Boehncke, W.-H.; Kollmeier, A.P.; Hsia, E.C.; Subramanian, R.A.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B.; et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2020, 395, 1115–1125. [Google Scholar] [CrossRef]
- Mease, P.J.; Rahman, P.; Gottlieb, A.B.; Kollmeier, A.P.; Hsia, E.C.; Xu, X.L.; Sheng, S.; Agarwal, P.; Zhou, B.; Zhuang, Y.; et al. Guselkumab in biologic-naive patients with active psoriatic arthritis (DISCOVER-2): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2020, 395, 1126–1136. [Google Scholar] [CrossRef]
- McHugh, J. IL-23 inhibitor guselkumab shows promise for PsA. Nat. Rev. Rheumatol. 2020, 16, 247. [Google Scholar] [CrossRef]
- Banaszczyk, K. Tildrakizumab in the treatment of psoriasis—literature review. Reumatologia 2019, 57, 234–238. [Google Scholar] [CrossRef]
- Mease, P.J.; Chohan, S.; Fructuoso, F.J.G.; Chou, R.C.; Nograles, K.E.; Mendelsohn, A.M.; Luggen, M.E. Lb0002 Randomised, Double-blind, Placebo-controlled, Multiple-dose, Phase 2b Study to Demonstrate the Safety and Efficacy Of Tildrakizumab, a High-affinity Anti-interleukin-23p19 Monoclonal Antibody, un Patients with Active Psoriatic Arthritis. Ann. Rheum. Dis. 2019, 78 (Suppl. 2), 78–79. [Google Scholar]
- Krueger, J.G.; Ferris, L.K.; Menter, A.; Wagner, F.; White, A.; Visvanathan, S.; Lalovic, B.; Aslanyan, S.; Wang, E.E.; Hall, D.; et al. Anti–IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: Safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J. Allergy Clin. Immunol. 2015, 136, 116–124. [Google Scholar] [CrossRef]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef]
- Mease, P.J.; Kellner, H.; Morita, A.; Kivitz, A.; Papp, K.; Aslanyan, S.; Berner, B.; Chen, S.; Eldred, A.; Behrens, F. OP0307 Efficacy and safety of risankizumab, a selective il-23p19 inhibitor, in patients with active psoriatic arthritis over 24 weeks: Results from a phase 2 trial. Ann. Rheum. Dis. 2018, 77 (Suppl. 2), 200–201. [Google Scholar]
- Baeten, D.; Østergaard, M.; Wei, J.C.-C.; Sieper, J.; Järvinen, P.; Tam, L.-S.; Salvarani, C.; Kim, T.-H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: Results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann. Rheum. Dis. 2018, 77, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; McInnes, I.B.; Kirkham, B.; Kavanaugh, A.; Rahman, P.; Van Der Heijde, D.; Landewé, R.; Nash, P.; Pricop, L.; Yuan, J.; et al. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N. Engl. J. Med. 2015, 373, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; Van Der Heijde, D.; Landewé, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef]
- Braun, J.; Baraliakos, X.; Deodhar, A.; Baeten, M.; Sieper, J.; Emery, P.; Readie, A.; Martin, R.; Mpofu, S.; Richards, H.B. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann. Rheum. Dis. 2016, 76, 1070–1077. [Google Scholar] [CrossRef]
- Pavelka, K.; Kivitz, A.; Dokoupilova, E.; Blanco, R.; Maradiaga, M.; Tahir, H.; Pricop, L.; Andersson, M.; Readie, A.; Porter, B. Efficacy, safety, and tolerability of secukinumab in patients with active ankylosing spondylitis: A randomized, double-blind phase 3 study, MEASURE 3. Arthritis Res. Ther. 2017, 19, 285. [Google Scholar] [CrossRef]
- Baraliakos, X.; Kivitz, A.J.; Deodhar, A.A.; Braun, J.; Wei, J.C.; Delicha, E.M.; Tolloczy, Z.; Porter, B. Long-term effects of interleukin-17A inhibition with secukinumab in active ankylosing spondylitis: 3-year efficacy and safety results from an extension of the Phase 3 MEASURE 1 trial. Clin. Exp. Rheumatol. 2018, 36, 50–55. [Google Scholar]
- Coates, L.C.; Gladman, D.D.; Nash, P.; Fitzgerald, O.; Kavanaugh, A.; Kvien, T.K.; Gossec, L.; Strand, V.; Rasouliyan, L.; Pricop, L.; et al. Secukinumab provides sustained PASDAS-defined remission in psoriatic arthritis and improves health-related quality of life in patients achieving remission: 2-year results from the phase III FUTURE 2 study. Arthritis Res. 2018, 20, 272. [Google Scholar] [CrossRef]
- McInnes, I.B.; Mease, P.J.; Kivitz, A.J.; Nash, P.; Rahman, P.; Rech, J.; Conaghan, P.G.; Kirkham, B.; Navarra, S.; Belsare, A.D.; et al. Long-term efficacy and safety of secukinumab in patients with psoriatic arthritis: 5-year (end-of-study) results from the phase 3 FUTURE 2 study. Lancet Rheumatol. 2020, 2, e227–e235. [Google Scholar] [CrossRef]
- McInnes, I.B.; Behrens, F.; Mease, P.J.; Kavanaugh, A.; Ritchlin, C.; Nash, P.; Masmitja, J.G.; Goupille, P.; Korotaeva, T.; Gottlieb, A.B.; et al. Secukinumab versus adalimumab for treatment of active psoriatic arthritis (EXCEED): A double-blind, parallel-group, randomised, active-controlled, phase 3b trial. Lancet 2020, 395, 1496–1505. [Google Scholar] [CrossRef]
- Liu, L.; Lu, J.; Allan, B.W.; Tang, Y.; Tetreault, J.; Chow, C.-K.; Barmettler, B.; Nelson, J.; Bina, H.; Huang, L.; et al. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin-17A. J. Inflamm. Res. 2016, 9, 39–50. [Google Scholar] [CrossRef]
- Paul, C. Ixekizumab or secukinumab in psoriasis: What difference does it make? Br. J. Dermatol. 2018, 178, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.; Brnabic, A.; Saure, D.; Langley, R.G.; See, K.; Wu, J.J.; Schacht, A.; Mallbris, L.; Nast, A. Matching-adjusted indirect comparison of efficacy in patients with moderate-to-severe plaque psoriasis treated with ixekizumab vs. secukinumab. Br. J. Dermatol. 2018, 178, 1064–1071. [Google Scholar] [CrossRef]
- Mease, P.J.; Van Der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.-Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D.; et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann. Rheum. Dis. 2016, 76, 79–87. [Google Scholar] [PubMed]
- Chandran, V.; Van Der Heijde, D.; Fleischmann, R.M.; Lespessailles, E.; Helliwell, P.S.; Kameda, H.; Burgos-Vargas, R.; Erickson, J.S.; Rathmann, S.S.; Sprabery, A.T.; et al. Ixekizumab treatment of biologic-naïve patients with active psoriatic arthritis: 3-year results from a phase III clinical trial (SPIRIT-P1). Rheumatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijde, D.; Wei, J.C.-C.; Dougados, M.; Mease, P.J.; Deodhar, A.; Maksymowych, W.P.; Bosch, F.V.D.; Sieper, J.; Tomita, T.; Landewé, R.; et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet 2018, 392, 2441–2451. [Google Scholar] [PubMed]
- Mease, P.J.; Smolen, J.S.; Behrens, F.; Nash, P.; Leage, S.L.; Li, L.; Tahir, H.; Gooderham, M.; Krishnan, E.; Liu-Seifert, H.; et al. A head-to-head comparison of the efficacy and safety of ixekizumab and adalimumab in biological-naïve patients with active psoriatic arthritis: 24-week results of a randomised, open-label, blinded-assessor trial. Ann. Rheum. Dis. 2019, 79, 123–131. [Google Scholar] [CrossRef]
- Kostareva, O.S.; Kolyadenko, I.; Ulitin, A.; Ekimova, V.; Evdokimov, S.; Garber, M.; Tishchenko, S.V.; Gabdoulkhakov, A. Fab Fragment of VHH-Based Antibody Netakimab: Crystal Structure and Modeling Interaction with Cytokine IL-17A. Crystals 2019, 9, 177. [Google Scholar] [CrossRef]
- Erdes, S.; Nasonov, E.; Kunder, E.; Pristrom, A.; Soroka, N.; Shesternya, P.; Dubinina, T.; Smakotina, S.; Raskina, T.; Krechikova, D.; et al. Primary efficacy of netakimab, a novel interleukin-17 inhibitor, in the treatment of active ankylosing spondylitis in adults. Clin. Exp. Rheumatol. 2019, 38, 27–34. [Google Scholar]
- Korotaeva, T.; Gaydukova, I.; Mazurov, V.; Samtsov, A.; Khayrutdinov, V.; Bakulev, A.; Kokhan, M.; Kundzer, A.; Soroka, N.; Dokukina, E.; et al. AB0791 Netakimab Reduces Skin Manifestations of Psoriatic Arthritis: Results of Subanalysis From a Double-blind Randomized Phase 3 Study (Patera). Ann. Rheum. Dis. 2020, 79 (Suppl. 1), 1690–1691. [Google Scholar]
- Gaydukova, I.; Mazurov, V.; Erdes, S.; Dubinina, T.; Nesmeyanova, O.; Ilivanova, E.; Kundzer, A.; Soroka, N.; Kastanayan, A.; Povarova, T.; et al. OP0232 Netakimab Reduces the Disease Activity of Radiographic Axial Spondyloarthritis. Results of Astera Study. Ann. Rheum. Dis. 2019, 78 (Suppl. 2), 193–194. [Google Scholar]
- Papp, K.A.; Reid, C.; Foley, P.; Sinclair, R.; Salinger, D.H.; Williams, G.; Dong, H.; Krueger, J.G.; Russell, C.B.; Martin, D.A.; et al. Anti-IL-17 Receptor Antibody AMG 827 Leads to Rapid Clinical Response in Subjects with Moderate to Severe Psoriasis: Results from a Phase I, Randomized, Placebo-Controlled Trial. J. Investig. Dermatol. 2012, 132, 2466–2469. [Google Scholar] [CrossRef]
- Mease, P.J.; Genovese, M.C.; Greenwald, M.W.; Ritchlin, C.T.; Beaulieu, A.D.; Deodhar, A.; Newmark, R.; Feng, J.; Erondu, N.; Nirula, A. Brodalumab, an Anti-IL17RA Monoclonal Antibody, in Psoriatic Arthritis. N. Engl. J. Med. 2014, 370, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Verhoeven, F.; Prati, C. Anti-IL-17 monoclonal antibodies for the treatment of ankylosing spondylitis. Expert Opin. Boil. Ther. 2018, 19, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.G.; Papp, K.A.; Marangell, L.B.; Koo, J.; Blauvelt, A.; Gooderham, M.; Wu, J.J.; Rastogi, S.; Harris, S.; Pillai, R.; et al. Psychiatric adverse events during treatment with brodalumab: Analysis of psoriasis clinical trials. J. Am. Acad. Dermatol. 2018, 78, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Nash, P. Inhibition of interleukins 17A and 17F in psoriatic arthritis. Lancet 2020, 395, 395–396. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Kavanaugh, A.; Merola, J.F.; Schett, G.; Scher, J.U.; Warren, R.B.; Gottlieb, A.B.; Assudani, D.; Bedford-Rice, K.; Coarse, J.; et al. Bimekizumab in patients with active psoriatic arthritis: Results from a 48-week, randomised, double-blind, placebo-controlled, dose-ranging phase 2b trial. Lancet 2020, 395, 427–440. [Google Scholar] [CrossRef]
- Van Der Heijde, D.; Gensler, L.S.; Deodhar, A.; Baraliakos, X.; Poddubnyy, D.; Kivitz, A.; Farmer, M.K.; Baeten, D.; Goldammer, N.; Coarse, J.; et al. Dual neutralisation of interleukin-17A and interleukin-17F with bimekizumab in patients with active ankylosing spondylitis: Results from a 48-week phase IIb, randomised, double-blind, placebo-controlled, dose-ranging study. Ann. Rheum. Dis. 2020, 79, 595–604. [Google Scholar] [CrossRef]
- Hsieh, C.-M.; Cuff, C.; Tarcsa, E.; Hugunin, M. FRI0303 Discovery and Characterization of Abt-122, an Anti-TNF/IL-17 Dvd-Ig™ Molecule as A Potential Therapeutic Candidate for Rheumatoid Arthritis. Ann. Rheum. Dis. 2014, 73, 495. [Google Scholar] [CrossRef]
- Genovese, M.C.; Weinblatt, M.E.; Mease, P.J.; Aelion, J.A.; Peloso, P.M.; Chen, K.; Li, Y.; Liu, J.; Othman, A.A.; Khatri, A.; et al. Dual inhibition of tumour necrosis factor and interleukin-17A with ABT-122: Open-label long-term extension studies in rheumatoid arthritis or psoriatic arthritis. Rheumatology 2018, 57, 1972–1981. [Google Scholar] [CrossRef]
- Mease, P.J.; Genovese, M.C.; Weinblatt, M.E.; Peloso, P.M.; Chen, K.; Othman, A.A.; Li, Y.; Mansikka, H.T.; Khatri, A.; Wishart, N.; et al. Phase II Study of ABT-122, a Tumor Necrosis Factor- and Interleukin-17A-Targeted Dual Variable Domain Immunoglobulin, in Patients With Psoriatic Arthritis With an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2018, 70, 1778–1789. [Google Scholar] [CrossRef]
- Khatri, A.; Klünder, B.; Peloso, P.M.; Othman, A.A. Exposure-response analyses demonstrate no evidence of interleukin 17A contribution to efficacy of ABT-122 in rheumatoid or psoriatic arthritis. Rheumatology 2019, 58, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Gasink, C.; Gao, L.-L.; Blank, M.A.; Johanns, J.; Guzzo, C.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Rutgeerts, P.; et al. Ustekinumab Induction and Maintenance Therapy in Refractory Crohn’s Disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Roblin, X.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name | Target Cytokine | AS | PsA | Other Autoinflammatory Diseases |
|---|---|---|---|---|
| Ustekinumab | IL-12-p40 and IL-23-p40 | Phase 3 | Marketed | CD,UC (Marketed) |
| Guselkumab | IL-23-p19 | - | Marketed | CD,UC (Phase 3) |
| Tildrakizumab | IL-23-p19 | Phase 2 | Marketed | - |
| Risankizumab | IL-23-p19 | Phase 2 | Marketed | CD,UC (Phase 3) |
| Secukinumab | IL-17A | Marketed | Marketed | - |
| Ixekizumab | IL-17A | Marketed | Marketed | - |
| Netakimab | IL-17A | Phase 3 | Phase 3 | - |
| Brodalumab | IL-17RA | Phase 3 | Phase 3 | CD (Phase 2) |
| Bimekizumab | IL-17A/F | Phase 2 | Phase 3 | - |
| ABT-122 | IL-17A and TNF-α | - | Phase 2 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsukazaki, H.; Kaito, T. The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis. Int. J. Mol. Sci. 2020, 21, 6401. https://doi.org/10.3390/ijms21176401
Tsukazaki H, Kaito T. The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis. International Journal of Molecular Sciences. 2020; 21(17):6401. https://doi.org/10.3390/ijms21176401
Chicago/Turabian StyleTsukazaki, Hiroyuki, and Takashi Kaito. 2020. "The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis" International Journal of Molecular Sciences 21, no. 17: 6401. https://doi.org/10.3390/ijms21176401
APA StyleTsukazaki, H., & Kaito, T. (2020). The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis. International Journal of Molecular Sciences, 21(17), 6401. https://doi.org/10.3390/ijms21176401