The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury


Abstract
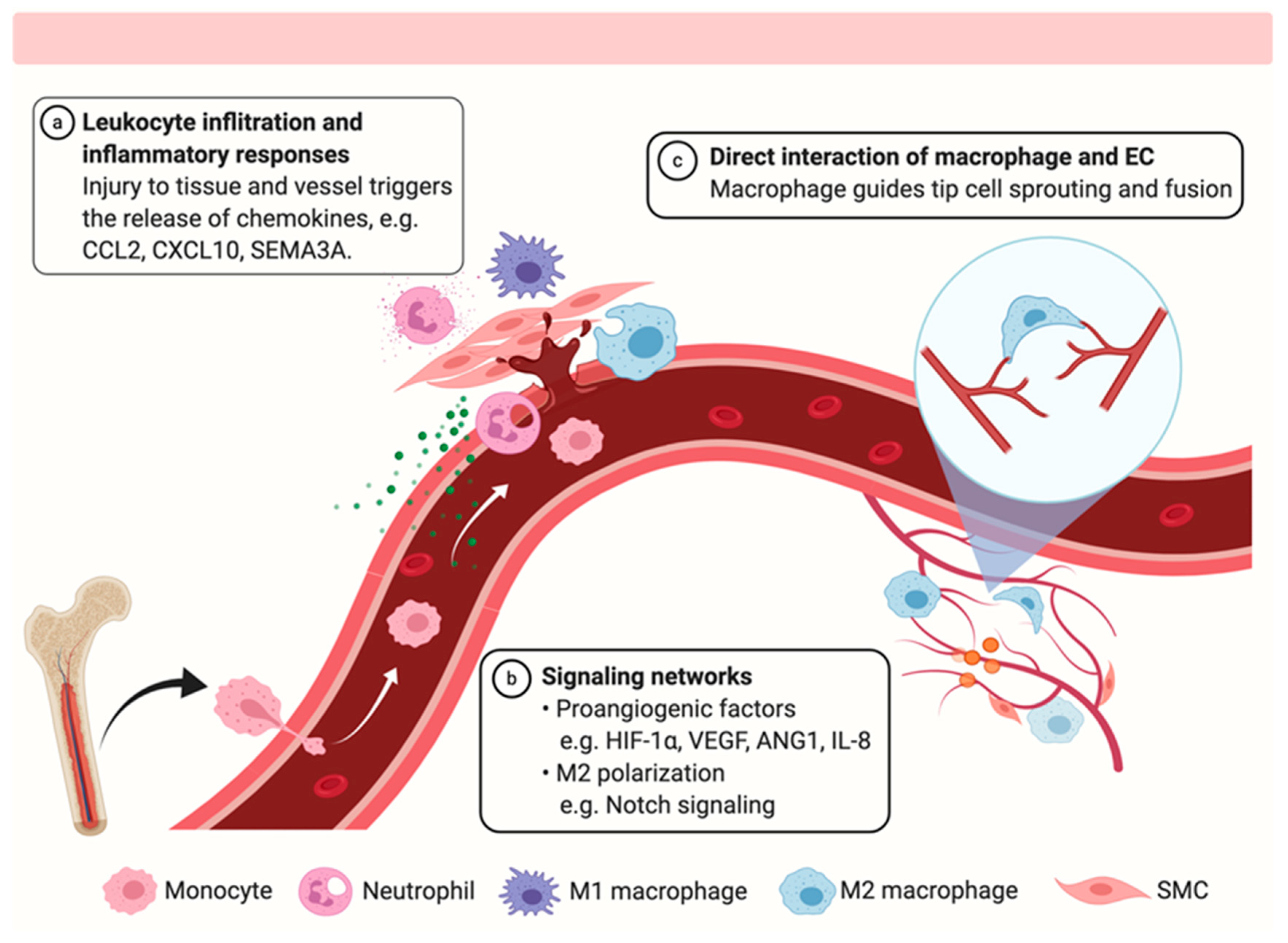
:1. Introduction
2. The Role of Macrophages in Vascular Repair and Regeneration
2.1. Influence of Macrophage Polarization on Vascular Inflammation and Repair
2.2. Chemotaxis and Recruitment of Various Cell Types during Vascular Repair
2.3. Phagocytic Macrophages in Vascular Repair
2.4. Pro-Angiogenesis Signaling Networks between Macrophages and Endothelial Cells
2.5. Physical Interaction of Macrophages and Endothelial Cells Post-Injury
3. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| ANG | Angiopoietin |
| CCR | CC chemokine receptor |
| CXCR | CXC chemokine receptor |
| EC | Endothelial cell |
| HIF | Hypoxia induced factor |
| IL | Interleukin |
| MMP | Matrix metalloproteinase |
| MoM | Monocyte derived macrophages |
| MSC | Mesenchymal stem cell |
| PAD | Peripheral artery diseases |
| PDGF | Platelet derived growth factor |
| PHD | Prolyl hydroxylase domain protein |
| TrM | Tissue resident macrophage |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ayadi, A.; Jay, J.W.; Prasai, A. Current Approaches Targeting the Wound Healing Phases to Attenuate Fibrosis and Scarring. Int. J. Mol. Sci. 2020, 21, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. Mech. Dis. 2012, 8, 241–276. [Google Scholar] [CrossRef] [Green Version]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- La Sala, A.; Pontecorvo, L.; Agresta, A.; Rosano, G.; Stabile, E. Regulation of collateral blood vessel development by the innate and adaptive immune system. Trends Mol. Med. 2012, 18, 494–501. [Google Scholar] [CrossRef]
- López-Díez, R.; Shen, X.; Daffu, G.; Khursheed, M.; Hu, J.; Song, F.; Rosario, R.; Xu, Y.; Li, Q.; Xi, X.; et al. Ager Deletion Enhances Ischemic Muscle Inflammation, Angiogenesis, and Blood Flow Recovery in Diabetic Mice. Arter. Thromb. Vasc. Boil. 2017, 37, 1536–1547. [Google Scholar] [CrossRef] [Green Version]
- Heil, M.; Ziegelhoeffer, T.; Wagner, S.; Fernández, B.; Helisch, A.; Martín, S.; Tribulova, S.; Kuziel, W.A.; Bachmann, G.; Schaper, W. Collateral Artery Growth (Arteriogenesis) After Experimental Arterial Occlusion Is Impaired in Mice Lacking CC-Chemokine Receptor-2. Circ. Res. 2004, 94, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Willenborg, S.; Lucas, T.; Van Loo, G.; Knipper, J.A.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Waeckel, L.; Mallat, Z.; Potteaux, S.; Combadière, C.; Clergue, M.; Duriez, M.; Bao, L.; Gerard, C.; Rollins, B.J.; Tedgui, A.; et al. Impairment in Postischemic Neovascularization in Mice Lacking the CXC Chemokine Receptor 3. Circ. Res. 2005, 96, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, G.C.; Raghuram, S.; Jang, C.; Nagy, J.A.; Patten, I.S.; Goyal, A.; Chan, M.C.; Liu, L.X.; Jiang, A.; Spokes, K.C.; et al. PGC-1α Induces SPP1 to Activate Macrophages and Orchestrate Functional Angiogenesis in Skeletal Muscle. Circ. Res. 2014, 115, 504–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejda, A.; Mawambo, G.; Cerani, A.; Miloudi, K.; Shao, Z.; Daudelin, J.-F.; Boulet, S.; Oubaha, M.; Beaudoin, F.; Akla, N.; et al. Neuropilin-1 mediates myeloid cell chemoattraction and influences retinal neuroimmune crosstalk. J. Clin. Investig. 2014, 124, 4807–4822. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Shiraishi, Y.; Cohen, R.A.; Tong, X. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase C674 promotes ischemia- and hypoxia-induced angiogenesis via coordinated endothelial cell and macrophage function. J. Mol. Cell. Cardiol. 2014, 76, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laban, H.; Weigert, A.; Zink, J.; Elgheznawy, A.; Schürmann, C.; Günther, L.; Malik, R.A.; Bothur, S.; Wingert, S.; Bremer, R.; et al. VASP regulates leukocyte infiltration, polarization, and vascular repair after ischemia. J. Cell Boil. 2018, 217, 1503–1519. [Google Scholar] [CrossRef] [Green Version]
- Hamm, A.; Veschini, L.; Takeda, Y.; Costa, S.; Delamarre, E.; Squadrito, M.L.; Henze, A.-T.; Wenes, M.; Serneels, J.; Pucci, F.; et al. PHD2 regulates arteriogenic macrophages through TIE2 signalling. EMBO Mol. Med. 2013, 5, 843–857. [Google Scholar] [CrossRef]
- Patel, A.S.; Smith, A.; Nucera, S.; Biziato, D.; Saha, P.; Attia, R.Q.; Humphries, J.; Mattock, K.; Grover, S.P.; Lyons, O.; et al. TIE2-expressing monocytes/macrophages regulate revascularization of the ischemic limb. EMBO Mol. Med. 2013, 5, 858–869. [Google Scholar] [CrossRef]
- Takeda, Y.; Costa, S.; Delamarre, E.; Roncal, C.; De Oliveira, R.L.; Squadrito, M.L.; Finisguerra, V.; Deschoemaeker, S.; Bruyère, F.; Wenes, M.; et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 2011, 479, 122–126. [Google Scholar] [CrossRef]
- Krishnasamy, K.; Limbourg, A.; Kapanadze, T.; Gamrekelashvili, J.; Beger, C.; Häger, C.; Lozanovski, V.J.; Falk, C.S.; Napp, L.C.; Bauersachs, J.; et al. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun. 2017, 8, 952. [Google Scholar] [CrossRef]
- Limbourg, A.; Ploom, M.; Elligsen, D.; Sörensen, I.; Ziegelhoeffer, T.; Gossler, A.; Drexler, H.; Limbourg, F.P. Notch Ligand Delta-Like 1 Is Essential for Postnatal Arteriogenesis. Circ. Res. 2007, 100, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, T.; Kurobe, H.; Sugasawa, N.; Kinoshita, H.; Higashida, M.; Matsuoka, Y.; Yoshida, Y.; Hirata, Y.; Sakata, M.; Maxfield, M.W.; et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1α as a mediator of vascular remodelling. Cardiovasc. Res. 2013, 99, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, R.J.; O’Neill, C.L.; O’Doherty, T.M.; Knott, H.; Guduric-Fuchs, J.; Gardiner, T.A.; Stitt, A.W. Myeloid Angiogenic Cells Act as Alternative M2 Macrophages and Modulate Angiogenesis through Interleukin-8. Mol. Med. 2011, 17, 1045–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petreaca, M.L.; Yao, M.; Liu, Y.; Defea, K.; Martins-Green, M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol. Biol. Cell 2007, 18, 5014–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganta, V.C.; Choi, M.H.; Kutateladze, A.; Fox, T.E.; Farber, C.R.; Annex, B.H. A MicroRNA93-Interferon Regulatory Factor-9-Immunoresponsive Gene-1-Itaconic Acid Pathway Modulates M2-Like Macrophage Polarization to Revascularize Ischemic Muscle. Circulation 2017, 135, 2403–2425. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Sung, H.-J.; Lessner, S.M.; Fini, M.E.; Galis, Z.S. Matrix Metalloproteinase-9 Is Required for Adequate Angiogenic Revascularization of Ischemic Tissues. Circ. Res. 2004, 94, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Post, M.; Volk, R.; Gao, Y.; Li, M.; Metais, C.; Sato, K.; Tsai, J.; Aird, W.; Rosenberg, R.D.; et al. PR39, a peptide regulator of angiogenesis. Nat. Med. 2000, 6, 49–55. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling ? in control of vascular function. Nat. Rev. Mol. Cell. Boil. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Tidball, J. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 2017, 17, 165–178. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate immune responses to trauma. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2013, 17, 109–118. [Google Scholar] [CrossRef]
- Jetten, N.; Donners, M.M.P.C.; Wagenaar, A.; Cleutjens, J.P.M.; Van Rooijen, N.; De Winther, M.P.J.; Post, M.J. Local Delivery of Polarized Macrophages Improves Reperfusion Recovery in a Mouse Hind Limb Ischemia Model. PLoS ONE 2013, 8, e68811. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.; Devi, T.D.; Mäkinen, P.; Kaikkonen, M.U.; Lesch, H.P.; Junttila, S.; Laiho, A.; Ghimire, B.; Gyenesei, A.; Ylä-Herttuala, S. Differential Promoter Methylation of Macrophage Genes Is Associated with Impaired Vascular Growth in Ischemic Muscles of Hyperlipidemic and Type 2 Diabetic Mice. Circ. Res. 2015, 117, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.W.; Vunjak-Novakovic, G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014, 35, 4477–4488. [Google Scholar] [CrossRef] [Green Version]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graney, P.L.; Ben-Shaul, S.; Landau, S.; Bajpai, A.; Singh, B.; Eager, J.; Cohen, A.; Levenberg, S.; Spiller, K.L. Macrophages of diverse phenotypes drive vascularization of engineered tissues. Sci. Adv. 2020, 6, eaay6391. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Fouda, A.Y.; Xu, Z.; Shosha, E.; Lemtalsi, T.; Chen, J.; Toque, H.A.; Tritz, R.; Cui, X.; Stansfield, B.K.; Huo, Y.; et al. Arginase 1 promotes retinal neurovascular protection from ischemia through suppression of macrophage inflammatory responses. Cell. Death. Dis. 2018, 9, 1001. [Google Scholar] [CrossRef]
- He, S.; Gleason, J.; Fik-Rymarkiewicz, E.; DiFiglia, A.; Bharathan, M.; Morschauser, A.; Djuretic, I.; Xu, Y.; Krakovsky, M.; Jankovic, V.; et al. Human Placenta-Derived Mesenchymal Stromal-Like Cells Enhance Angiogenesis via T Cell-Dependent Reprogramming of Macrophage Differentiation. Stem Cells 2017, 35, 1603–1613. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Johnson, T.K.; Wang, Y.; Thomas, M.; Huynh, K.; Yang, Q.; Bond, V.C.; Chen, Y.E.; Liu, D. Macrophage M2 polarization induced by exosomes from adipose-derived stem cells contributes to the exosomal proangiogenic effect on mouse ischemic hindlimb. Stem Cell Res. Ther. 2020, 11, 162. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, G.; Nishinakamura, H.; Kojima, D.; Tashiro, T.; Kodama, S. GM-CSF treated F4/80+ BMCs improve murine hind limb ischemia similar to M-CSF differentiated macrophages. PLoS ONE 2014, 9, e106987. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9, 585. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Mehina, E.; White, E.; Reeson, P.; Yongblah, K.; Doyle, K.; Brown, C.E. Suppressing Interferon-γ Stimulates Microglial Responses and Repair of Microbleeds in the Diabetic Brain. J. Neurosci. 2018, 38, 8707–8722. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Theret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 Regulates Macrophage Skewing at the Time of Resolution of Inflammation during Skeletal Muscle Regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef] [Green Version]
- Perdiguero, E.; Sousa-Victor, P.; Ruiz-Bonilla, V.; Jardí, M.; Caelles, C.; Serrano, A.L.; Muñoz-Cánoves, P. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell. Biol. 2011, 195, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Gerri, C.; Marín-Juez, R.; Marass, M.; Marks, A.; Maischein, H.-M.; Stainier, D.Y.R. Hif-1α regulates macrophage-endothelial interactions during blood vessel development in zebrafish. Nat. Commun. 2017, 8, 15492. [Google Scholar] [CrossRef] [Green Version]
- Cattin, A.-L.; Burden, J.J.; Van Emmenis, L.; MacKenzie, F.E.; Hoving, J.J.; Calavia, N.G.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Ahn, G.-O.; Seita, J.; Hong, B.-J.; Kim, Y.-E.; Bok, S.; Lee, C.-J.; Kim, K.S.; Lee, J.C.; Leeper, N.J.; Cooke, J.P.; et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc. Natl. Acad. Sci. USA 2014, 111, 2698–2703. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular Endothelial Growth Factor Signaling in Hypoxia and Inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, M.; Leppänen, V.-M.; Saharinen, P.; Alitalo, K. Receptor Tyrosine Kinase-Mediated Angiogenesis. Cold Spring Harb. Perspect. Boil. 2013, 5, a009183. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Nurmi, H.; Jakobsson, L.; Heinolainen, K.; Tvorogov, D.; Zheng, W.; Franco, C.-A.; Murtomäki, A.; Aranda, E.; et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nature 2011, 13, 1202–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkinen, K.; Hedman, M.; Matsi, P.; Mussalo, H.; Alhava, E.; Ylä-Herttuala, S.; Auml, K. Increased Vascularity Detected by Digital Subtraction Angiography after VEGF Gene Transfer to Human Lower Limb Artery: A Randomized, Placebo-Controlled, Double-Blinded Phase II Study. Mol. Ther. 2002, 6, 127–133. [Google Scholar] [CrossRef]
- Rasmussen, H.S.; Rasmussen, C.S.; Macko, J. VEGF gene therapy for coronary artery disease and peripheral vascular disease. Cardiovasc. Radiat. Med. 2002, 3, 114–117. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Mohler, E.R.; Lederman, R.J.; Mendelsohn, F.O.; Saucedo, J.F.; Goldman, C.K.; Blebea, J.; Macko, J.; Kessler, P.D.; Rasmussen, H.S.; et al. Regional Angiogenesis With Vascular Endothelial Growth Factor in Peripheral Arterial Disease. Circulation 2003, 108, 1933–1938. [Google Scholar] [CrossRef]
- Ganta, V.C.; Choi, M.; Kutateladze, A.; Annex, B.H. VEGF165b Modulates Endothelial VEGFR1-STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease. Circ. Res. 2017, 120, 282–295. [Google Scholar] [CrossRef] [Green Version]
- Ganta, V.C.; Choi, M.; Farber, C.R.; Annex, B.H. Antiangiogenic VEGF(165)b Regulates Macrophage Polarization via S100A8/S100A9 in Peripheral Artery Disease. Circulation 2019, 139, 226–242. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Iruela-Arispe, M.L. Notch Signaling in Blood Vessels. Circ. Res. 2007, 100, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Suchting, S.; Freitas, C.; Le Noble, F.; Benedito, R.; Bréant, C.; Duarte, A.; Eichmann, A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. USA 2007, 104, 3225–3230. [Google Scholar] [CrossRef] [Green Version]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin–Tie system. Nat. Rev. Mol. Cell Boil. 2009, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Kränkel, N. You can teach an old dog new tricks: Angiopoietin-1 instructs Tie2 pos myeloid cells to promote neovascularization in ischemic limbs. EMBO Mol. Med. 2013, 5, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Khmelewski, E.; Becker, A.; Meinertz, T.; Ito, W.D. Tissue Resident Cells Play a Dominant Role in Arteriogenesis and Concomitant Macrophage Accumulation. Circ. Res. 2004, 95, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wu, C.; Yang, Q.; Gao, J.; Li, L.; Yang, D.; Luo, L. Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity 2016, 44, 1162–1176. [Google Scholar] [CrossRef] [Green Version]
- Melgar-Lesmes, P.; Edelman, E.R. Monocyte-endothelial cell interactions in the regulation of vascular sprouting and liver regeneration in mouse. J. Hepatol. 2015, 63, 917–925. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Functions | Signaling Molecules | Effects on Macrophages | Phenotypes upon Injury | Ref. |
|---|---|---|---|---|
| Chemotaxis and cell recruitment | AGE-RAGE | Reduce macrophage infiltration and interaction with EC | RAGE KO or overexpression of reduced AGE enhance vascular repair in diabetic mice upon HLI | [8] |
| CCL2-CCR2 | Recruitment of proangiogenic monocytes/macrophages | CCR2 KO impairs recovery of blood flow recovery, vessel size, and active foot movement in HLI mice | [9,10] | |
| CXCL10-CXCR3 | Regulate leucocyte infiltration | CXCR3 KO reduces VEGF production, angiogenesis, blood perfusion, and capillary density | [11] | |
| PGC-1α-SPP1 | Recruit macrophage and upregulate CCL2 production | PGC-1α overexpression improves angiogenesis and blood flow recovery in adult, aged, diabetic mice; SPP1 KO induces immature capillarization and blunted arterialization | [12] | |
| SEMA3A/VEGF-NRP-1 | Recruit NRP-1+ macrophage | NRP-1 deficient macrophage fail to enter retinal and reduce neovascularization in OIR mice | [13] | |
| SERCA 2 | Regulated VEGF production and adhesion to EC | Mediated immune cells infiltration and adhesion via ERO1 and VCAM-1 expression in EC | [14] | |
| VASP | Form complex with CCR2, suppress macrophage differentiation via STAT signaling | KO increase blood flow recovery, angiogenesis, arteriogenesis, and leukocyte infiltration upon HLI | [15] | |
| Angiogenesis | ANG/TIE2 | Upregulate HIF signaling via repressing Phd2 and M2 polarization | ANG or TIE2 overexpression increases vessel density, reduced ischemic necrosis in HLI mice | [16,17,18] |
| DLL1-NOTCH | Promote differentiation from Ly6Chi monocyte, enhanced phagocytic capacity and anti-inflammatory phenotype | Heterozygous Dll1 mutant prevents arteriogenesis, blood perfusion, and tissue recovery in HLI mice | [19,20] | |
| HIF | HIF-1α KO reduced macrophage migration and suppressed pro-inflammatory phenotype | KO impairs ruptured vessel repair, angiogenesis, and tissue repair | [21] | |
| IL-8 | M2 polarization | Blockade of IL-8 suppresses angiogenesis | [22,23] | |
| MiR93/IRF9/IRG1/itaconic acid | MiR93-mediated suppression of IRF9/IRG1/itaconic acid induces M2 polarization | MiR93 overexpression promotes angiogenesis, arteriogenesis, and blood perfusion | [24] | |
| MMP-9 | Secreted by M2 macrophage | KO reduces capillary branching | [25] | |
| PR39 peptide | Inhibited the degradation of HIF-1 α | Promote angiogenesis | [26] | |
| VEGF-VEGFR | Activate NOTCH signaling, induce maturation and M2 polarization | Promote EC migration, proliferation and angiogenesis | [19,27] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, H.; Tian, X.Y. The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury. Int. J. Mol. Sci. 2020, 21, 6328. https://doi.org/10.3390/ijms21176328
Hong H, Tian XY. The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury. International Journal of Molecular Sciences. 2020; 21(17):6328. https://doi.org/10.3390/ijms21176328
Chicago/Turabian StyleHong, Huiling, and Xiao Yu Tian. 2020. "The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury" International Journal of Molecular Sciences 21, no. 17: 6328. https://doi.org/10.3390/ijms21176328
APA StyleHong, H., & Tian, X. Y. (2020). The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury. International Journal of Molecular Sciences, 21(17), 6328. https://doi.org/10.3390/ijms21176328