1. Introduction
Type 1 diabetes (T1D) is one of the most common autoimmune diseases which is characterized by the destruction of insulin-producing β cells by autoreactive T cells. Patients require life-long management with daily glucose monitoring and multiple insulin injections. Clinical evidence and animal studies have demonstrated that CD4
+ T cells play a key role in the initiation and development of T1D [
1]. Specifically, autoimmune memory CD4
+ T cells become “stumbling blocks” that hinder most attempts to treat or heal T1D and other autoimmune diseases [
2,
3,
4]. Based on the different expressions of surface markers, such as the isoforms of leukocyte common antigens CD45RA or CD45RO and CC chemokine receptor type 7 (CCR7), human CD4
+ T cells were categorized with CD45RA
+CD45RO
−CCR7
+ naïve T cells, CD45RA
−CD45RO
+CCR7
+ central memory T cells (T
CM), and CD45RA
−CD45RO
+CCR7
− effector memory T cells (T
EM) [
5]. Due to the lack of CCR7 expression, CD4 T
EM cannot return to lymph/blood circulation and become the resident memory cells remaining in tissues [
3,
4,
6,
7]. These resident autoimmune memory CD4 T
EM cells may replicate quickly and evoke the immune system to destroy the newly-generated islet β cells. Therefore, these autoimmune memory T cells need to be eradicated in order to fundamentally correct the autoimmunity of T1D. Additionally, it will be essential to modulate CD4
+ T cells for the treatment of other autoimmune disease such as multiple sclerosis (MS) [
8] and infection diseases [
9], due to their dominant roles in orchestrate the innate and adaptive immune responses.
Recent clinical trials [
10,
11] have highlighted the limits of conventional immune therapy and underscored the need for novel approaches that not only overcome multiple immune dysfunctions, but also help to restore pancreatic islet β cells. To address these two key issues, we have developed a unique and novel process designated the Stem Cell Educator (SCE) therapy. With this SCE technology, a patient’s blood is circulated through a blood cell separator where the patient’s lymphocytes are co-cultured with adherent cord-blood-derived multipotent stem cells (CB-SC) in vitro. The “educated” lymphocytes are then returned to the patient’s circulation [
12]. Over the last 10 years, our unique technology has been evaluated through international multi-center clinical studies [
12,
13,
14,
15,
16] in the United States (NCT02624804), China (NCT01350219, NCT01415726, and NCT01673789), and Spain (NCT01350219), where we have demonstrated the clinical efficacy and safety of SCE Therapy. Notably, SCE therapy is the leading approach to date to safely and efficiently correct autoimmunity and restore β-cell function in T1D patients. Previous clinical trials have demonstrated that SCE therapy provided lasting reversal of autoimmune memory T cells evidenced by the up-regulation of the percentages of Naïve T cells and CD4
+ T
CM and the down-regulation of CD4
+ T
EM in Caucasian T1D subjects [
14]. During mechanistic studies, we found that platelets’ numbers of T1D patients were increased after receiving SCE therapy. Additionally, platelet-derived mitochondria displayed the immune tolerance-associated markers such as programmed death ligand-1 (PD-L1, CD274) and Herpes Virus Entry Mediator (CD270), stimulating the proliferation of human islet β cells [
16]. The effects of platelet-derived mitochondria on CD4
+ T cells remain unclear for the treatment of T1D.
Recently, Dache et al. and our lab discovered the existence of circulating mitochondria in human and animal blood [
17,
18]. This finding indicates that mitochondria act not only as organelles of “a cellular power plant” for ATP production, but also function as novel mediators contributing to the crosstalk among different cells, tissues, and organs, and homeostatic maintenance [
17]. Since red blood cells (RBC) and platelets comprise 99% of the cellular component of human peripheral blood [
19] and there are no mitochondria in mature RBC [
20], it is likely that these circulating mitochondria are primarily released by platelets [
16]. To further explore the immune modulation of platelet-derived mitochondria, the purified CD4
+ T cells were treated with platelet-derived mitochondria, respectively. The data established significant changes in CD4
+ T cells after the treatment with platelet-derived mitochondria.
3. Discussion
CD4+ T cells are the predominant cell population to orchestrate human innate and adaptive immune responses against infections, cancer formations, inflammations, and the developments of autoimmune diseases. Increasing clinical evidence demonstrated that dysfunctions of CD4+ T cells contributed to multiple chronic diseases such as type 1 diabetes and autoimmune diseases. The current study demonstrated that platelet-derived mitochondria act as novel immune modulators on the anti-CD3/CD28 bead-activated CD4+ T cells, which exhibited multiple changes including cell surface markers, functionality, cytokine productions, and gene expressions after mitochondrial treatment. The data established the direct immune modulation of platelet-derived mitochondria on CD4+ T cells through the SDF-1 and its ligand CXCR4.
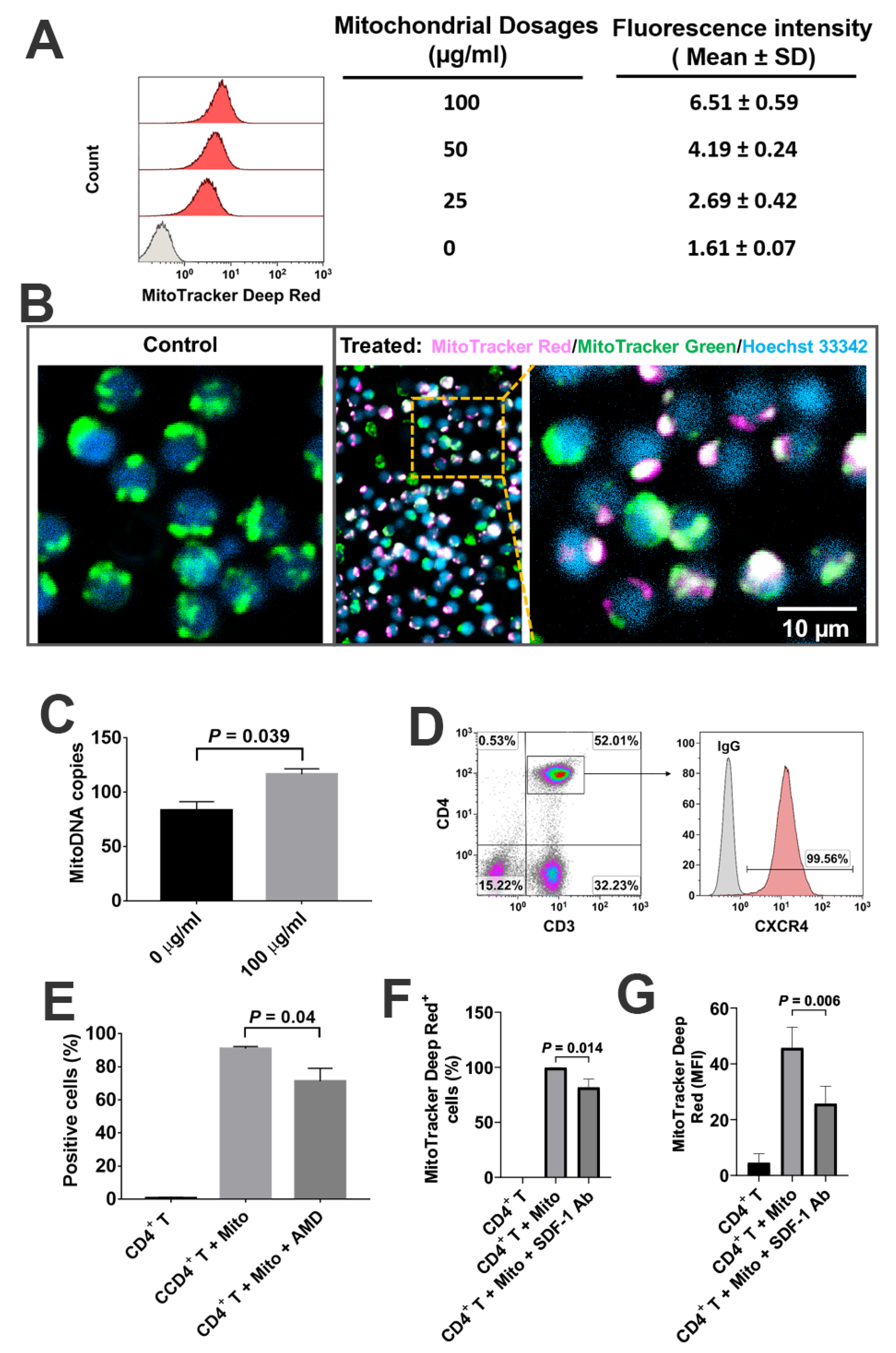
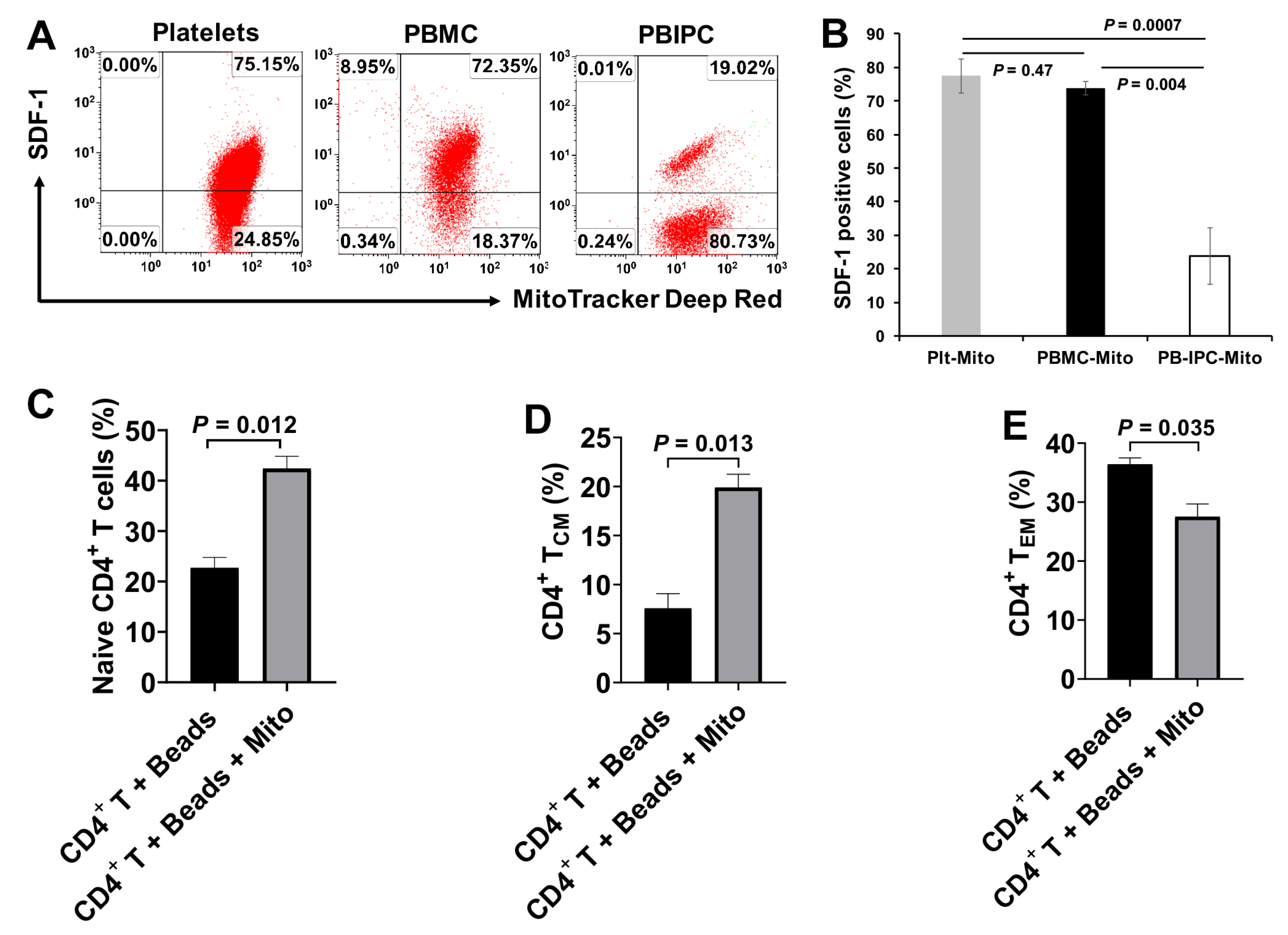
Based on the co-localization of MitoTracker Deep Red-positive platelet-derived mitochondria and MitoTracker Green-positive intrinsic mitochondria in CD4
+ T cells, together with the significant up-regulation of mtDNA-specific gene markers ND1 and ND5 in CD4
+ T cells, these data suggest that platelet-derived mitochondria may enter into CD4
+ T cells. This was similar to our previous work on the penetration of platelet-derived mitochondria into human peripheral blood-derived insulin-producing cells (PB-IPC) [
21]. There is a high expression of CXCR4 on CD4
+ T cells, contributing to the migration of CD4
+ T cells to pancreatic islets and destruction of β cells in T1D [
25,
26] and other autoimmune diseases [
27]. Due to the expression of CXCR12 (SDF-1) on platelet-derived mitochondria [
21], they may target these pathogenic CD4
+ T cells through the interaction of CXCR4/CXCRL12. Importantly, platelet-derived mitochondria display the immune tolerance-associated markers including programmed death ligand-1 (PD-L1) and CD270 [
16], while CD4
+ T cells present their ligands PD-1 and B- and T- lymphocyte attenuator (BTLA), respectively, mitochondria may modulate CD4
+ T cells through the interactions of PD-1/PD-L1 and BTLA/CD270, respectively. This highlights the translational potentials of platelet-derived mitochondria for the treatment of autoimmune diseases in clinics. Since the platelet numbers were increased in T1D subjects after receiving SCE therapy [
16], which may release more mitochondria into blood circulation [
17], the immunomodulation of platelet-derived mitochondria provides additional mechanisms underlying the SCE therapy for the treatment of T1D and other autoimmune diseases.
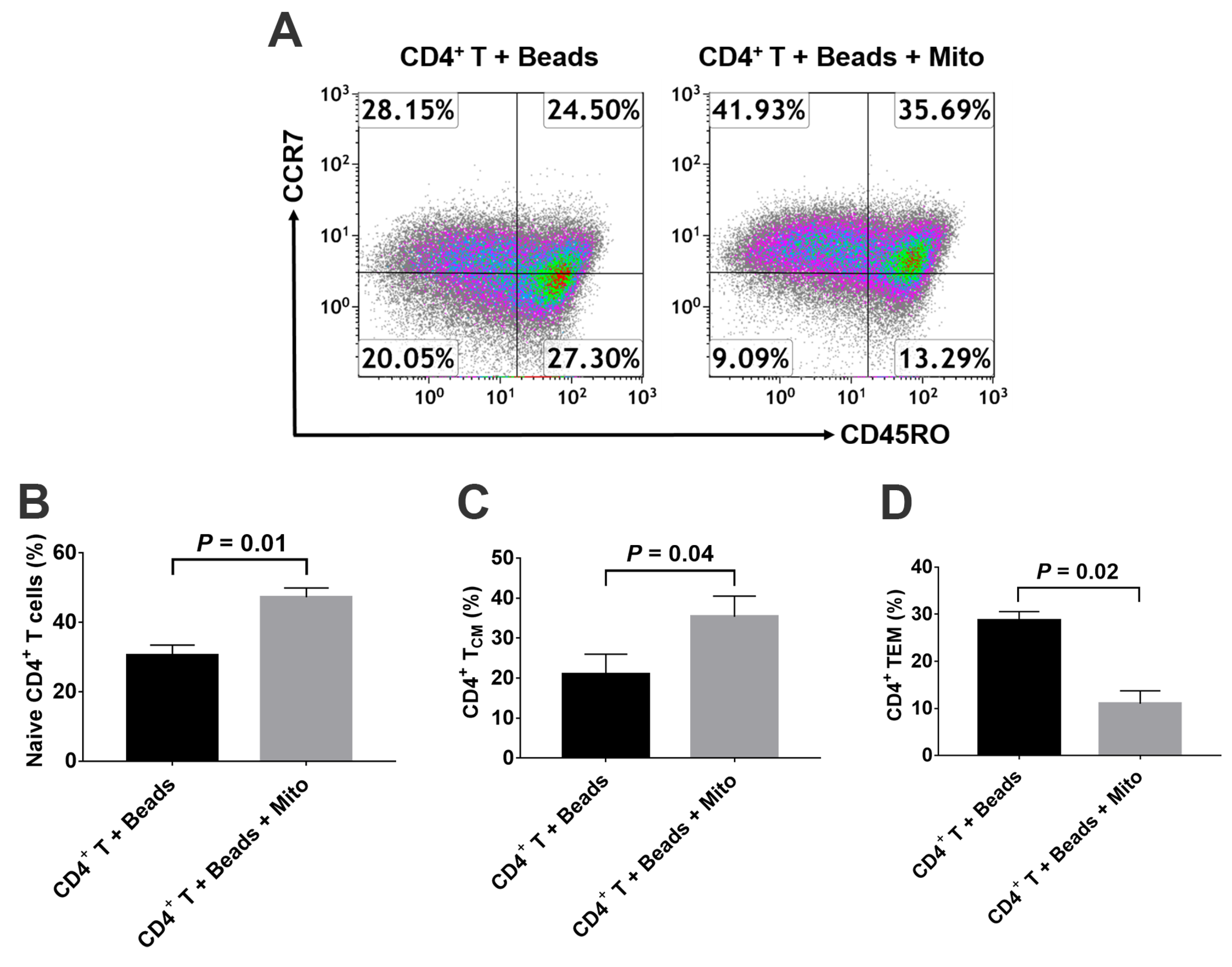
Based on different surface markers (e.g., CCR7 and CD45RA or CD45RO), CD4
+ T cells are sub-divided into Naïve T cells, T
CM, T
EM, and regulatory T cells (Tregs) [
5]. Naïve T cells are the pool of precursors that have the capability to give rise to effector and memory T cells upon receiving the antigen signals from antigen-presenting cells (APC). Spainer et al. reported the increased insulin-specific CD4
+ T
EM cells in recent onset T1D patients [
2]. Matteucci et al. reported that both percentages and absolute numbers of naïve and T
CM cells were reduced, while the terminally differentiated effector memory T cells were markedly increased in T1D patients comparing with the healthy control [
28]. The persistence of autoreactive memory T cells leads to the difficulty in rescuing the residual islet β cells in new-onset T1D and improving the therapeutic potentials of islet transplants [
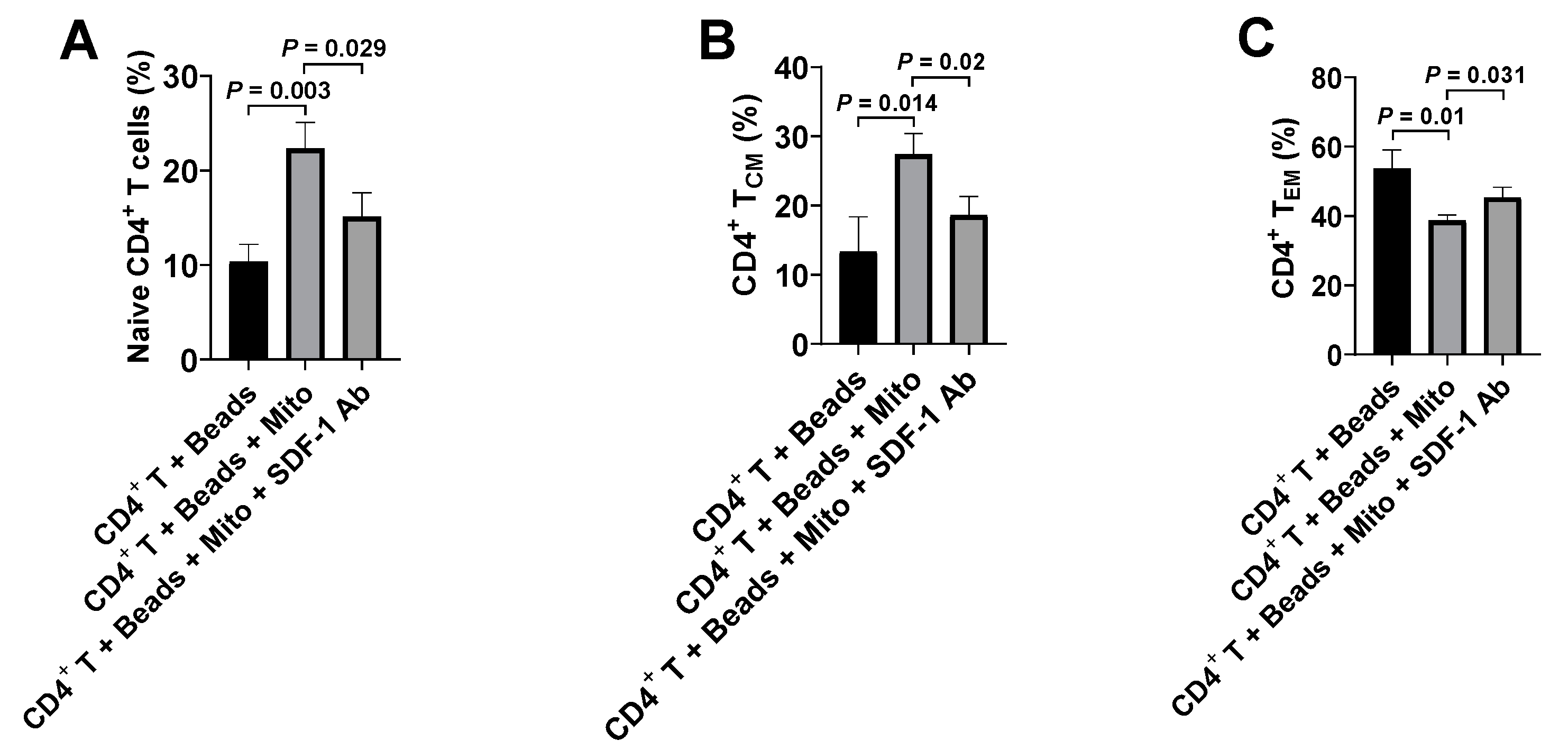
4]. Therefore, it will be essential to eliminate these autoimmune memory T cells and avoid the persistent destructions of the transplanted islet β cells and/or newly-generated insulin-producing cells. The current study demonstrated the up-regulated percentages of CD4
+ Naïve T and CD4
+ T
CM cells, and the reduced percentage of CD4
+ T
EM cells in the anti-CD3/CD28 bead-activated CD4
+ T cells after the treatment with platelet-derived mitochondria. Current ex vivo data were similar to our previous clinical studies in T1D patients after receiving Stem Cell Educator therapy [
14]. Additional data demonstrated the direct immune modulation of platelet-derived mitochondria in the anti-CD3/CD28 bead-activated CD4
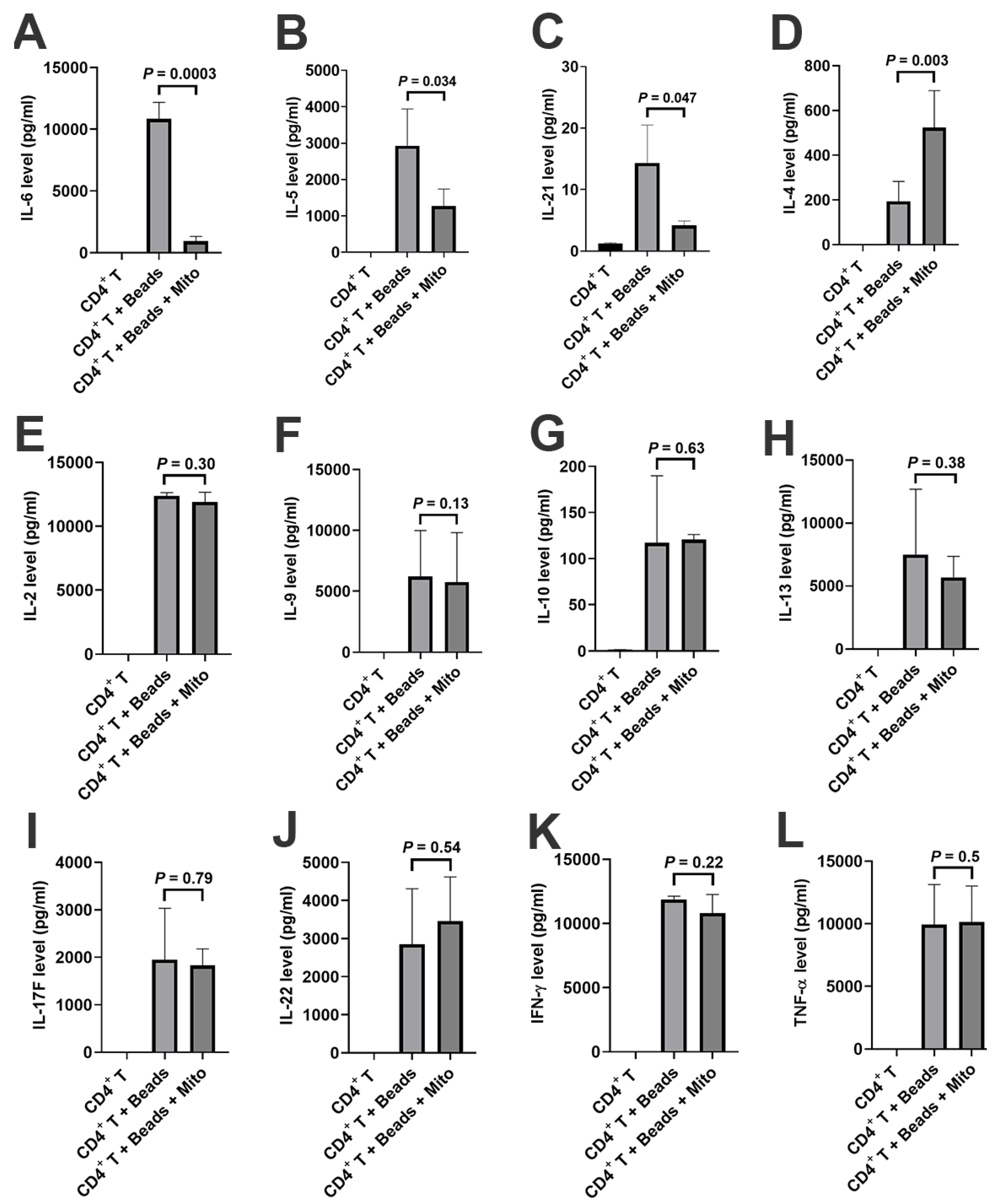
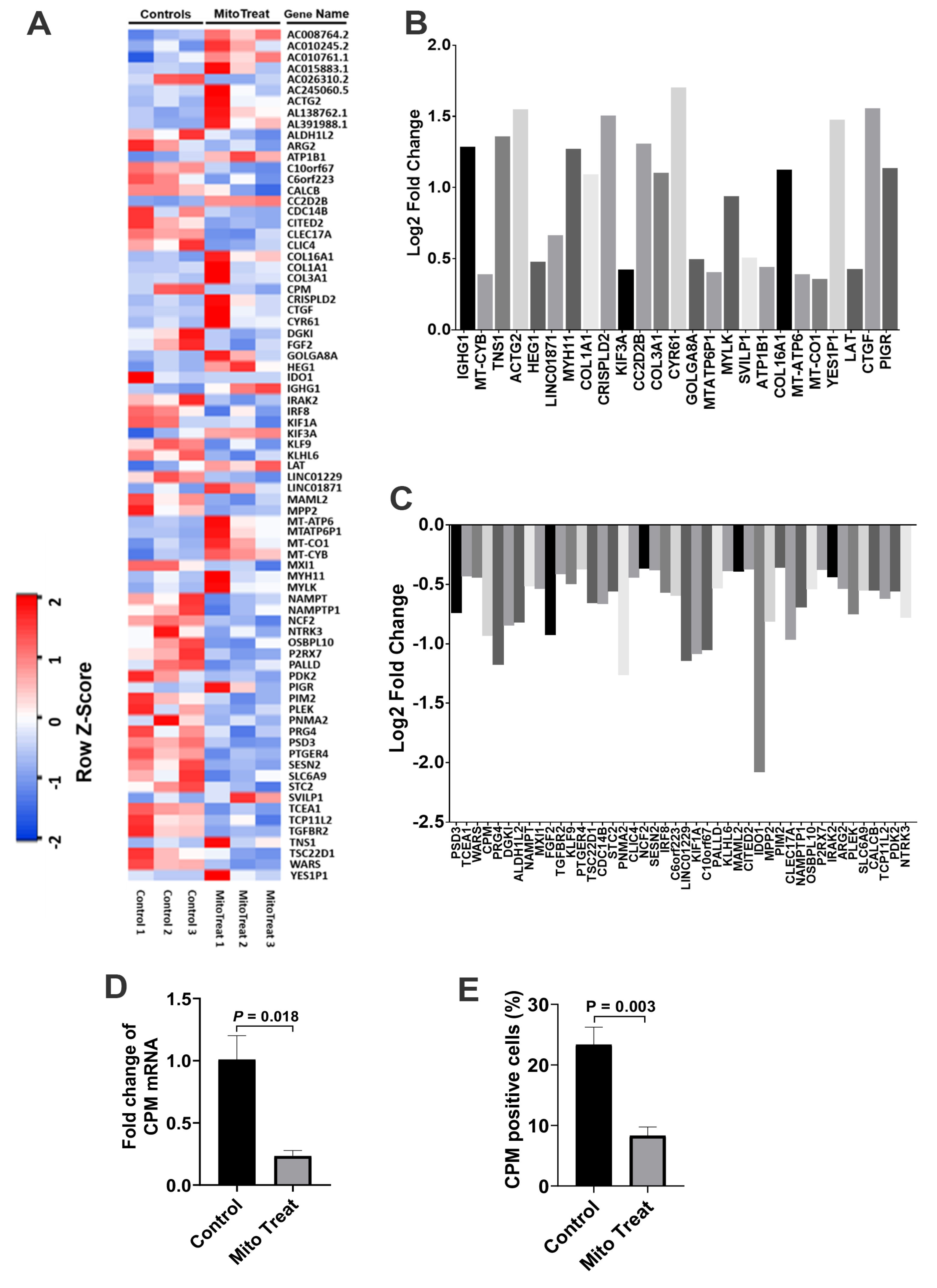
+ T cells such as down-regulation of inflammation-associated cytokines (e.g., IL-6 and IL-21), up-regulation of Th2-associated cytokine IL-4 but down-regulation of IL-5, and changes in gene profiling. For instance, carboxypeptidase M (CPM) was highly expressed on the membrane of activated CD4
+ T cells [
23], contributing to the cleavage of C-terminal arginine (Arg) or lysine (Lys) of growth factors or cytokine for releasing [
24]. Importantly, both gene and protein expressions of CPM were markedly down-regulated in the presence of platelet-derived mitochondria, which were consistent with the suppression of CD4
+ T cells after the treatment with platelet-derived mitochondria. The data provides new molecular mechanisms about mitochondrial immune modulation involved in the SCE therapy.
Similar to most autoimmune diseases, the autoimmunity of T1D is complicated and involves different compartments of the immune system including CD4
+, CD8
+ T cells, Tregs, B cells, DCs, monocyte/macrophages (Mo/Mϕs), and natural killer T cells (NKTs). Consequently, most efforts to develop successful treatments or a cure for T1D have been hindered over the last 40 years. Mitochondria circulate in human and animal peripheral blood [
17]. Flow cytometry proved that they may directly interact with monocytes, DC, B cells and other cell compartments, except CD4
+ T cells. As novel immune modulators, circulating mitochondria may play a central role in the induction of immune tolerance and balance. Additionally, platelets are the second largest cell population in human blood, without a nucleus. The highly-purified mitochondria derived from platelets offer a promising research tool to explore their immunomodulatory effects and translational potentials for the clinical treatment of human diseases.
4. Materials and Methods
4.1. PBMC and CD4+ T Cells Collection
Human buffy coat blood units (
n = 26; mean age of 44 ± 16.8; age range from 16 to 66 years old; 17 males and 9 females) were purchased from the New York Blood Center (New York, NY, USA,
http://nybloodcenter.org/). Human buffy coats were added to 40 mL of chemical-defined serum-free culture X-VIVO 15
TM medium (Lonza, Walkersville, MD, USA) and mixed with 10 mL pipette. Next, they were used for isolation of peripheral blood-derived mononuclear cells (PBMC). Mononuclear cells were isolated from buffy coats blood by Ficoll-Paque
TM PLUS (
γ = 1.007, GE Healthcare, Chicago, IL, USA), followed by the removal of the red blood cells using Red Blood Cell Lysis buffer (eBioscience, San Diego, CA, USA). After three washes with saline, the whole PBMC were seeded in chemical-defined serum-free culture X-VIVO 15
TM medium (Lonza, Walkersville, MD, USA), without adding any other growth factors, and incubated at 37 °C in 8% CO
2 conditions.
To get the purified CD4+ T cells, PBMC were stained with CD4-FITC antibody for 30 min and purified with Anti-FITC Magnetic Beads (Miltenyi Biotech, Gladbach, Germany) according to the manufacturer’s instructions.
4.2. Isolation of Mitochondria from Platelets and Other Cells
Human platelets samples (
n = 15) were separated from adult volunteer donors and purchased at New York Blood Centers (New York, NY, USA,
http://nybloodcenter.org/). The mitochondria were isolated from PB-platelets by a Mitochondria Isolation kit (Thermo scientific, Rockford, IL, USA, Prod: 89874) according to the manufacturer’s recommended protocol [
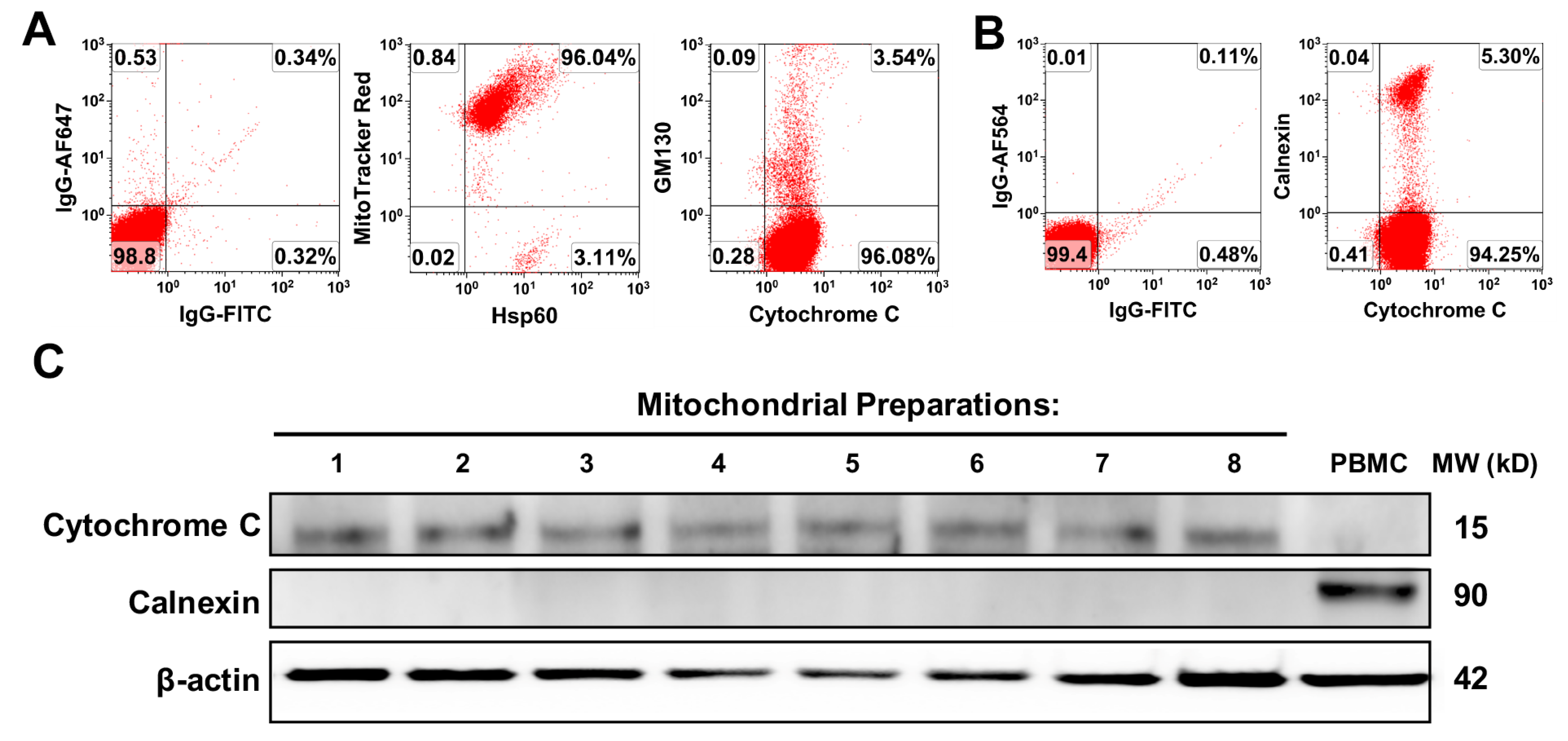
16]. The protein concentration of mitochondria was measured by a NanoDrop 2000 Spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA). The isolated mitochondria were aliquoted and kept in a −80 °C freezer.
Platelets-derived mitochondria were stained with MitoTracker Deep Red FM (100 nM) (Thermo Fisher Scientific, Waltham, MA, USA), at 37 °C for 15 min according to the manufacturer’s protocol, followed by three washes with PBS at 12,000 rpm × 15 min at 4 °C. CD4+ T cells were stained with MitoTracker Green FM (100 nM) (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C for 15 min according to the manufacturer’s protocol, followed by three washes with PBS at 300× g at 4 °C for 10 min.
To compare the phenotypic differences of platelet-derived mitochondria with other cells, mitochondria were isolated from PBMC and peripheral blood-derived insulin-producing cells (PB-IPC) by using the same Mitochondria Isolation kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s recommended protocol. Culture of PB-IPC from adult peripheral blood was performed as previously described [
21,
22]. Briefly, the whole PBMC were seeded in 150 × 15 mm Petri dishes (BD Falcon, NC, USA) at 1 × 10
6 cells/mL, 25mL/dish in chemical-defined serum-free culture X-VIVO 15
TM medium (Lonza, Walkersville, MD, USA) without adding any other growth factors and incubated at 37 °C in 8% CO
2 [
29]. Seven days later, PB-IPC were growing and expanded by adhering to the hydrophobic bottom of Petri dishes for experiments.
4.3. PBMC Proliferation Assay
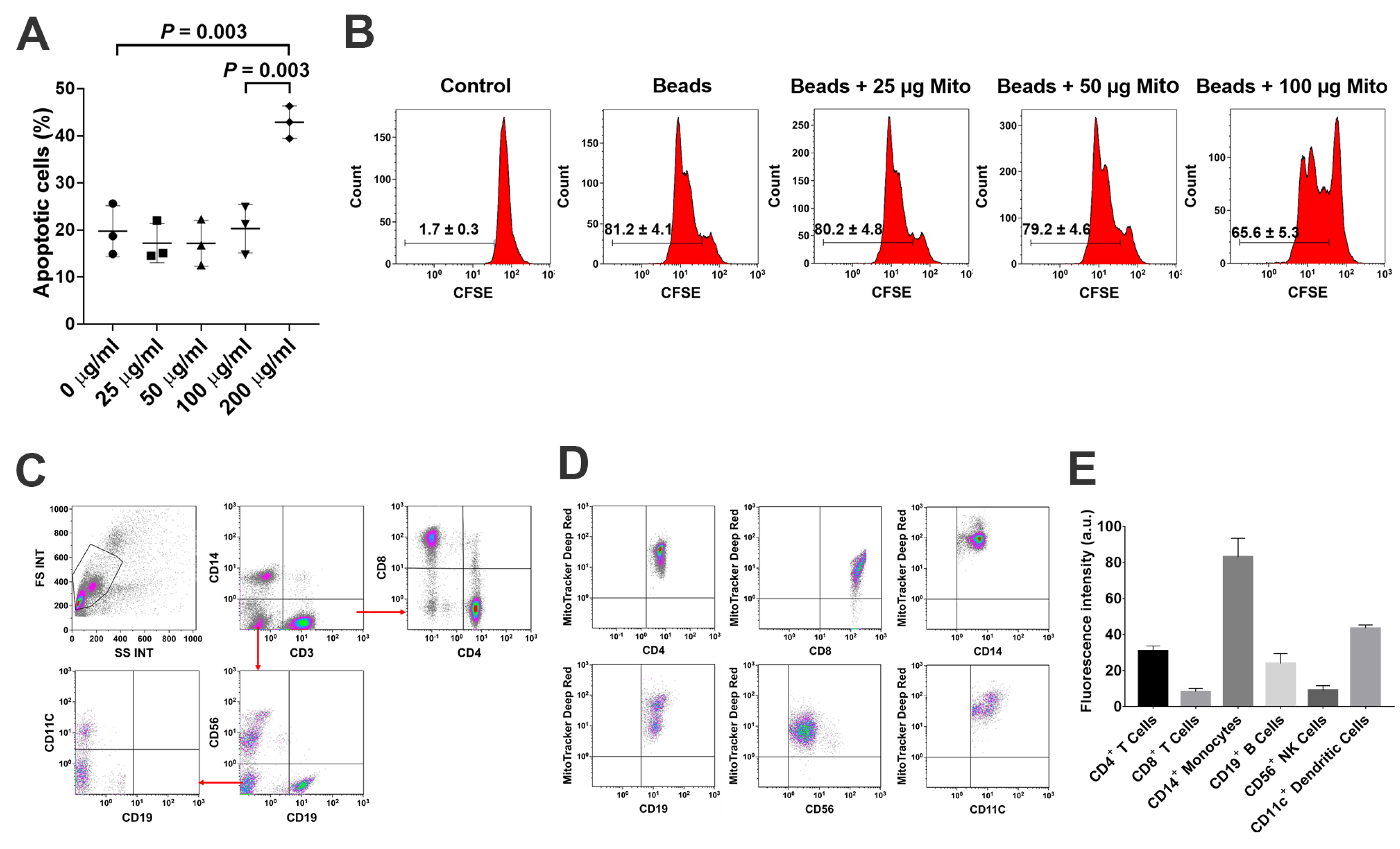
PBMC were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions [
30]. Next, they were cocultured with Dynabeads coupled with anti-CD3 and anti-CD28 antibodies (Thermo Fisher Scientific, Waltham, MA, USA) for 72 h in the presence of mitochondria at 25 μg/mL, 50 μg/mL, 100 μg/mL and 200 μg/mL, respectively. Lastly, they were incubated at 37 °C in 5% CO
2. Mitochondria-untreated cells served as control.
4.4. Flow Cytometry
For surface staining, samples were pre-incubated with human BD Fc Block (BD Pharmingen, Franklin Lakes, NJ, USA) for 15 min at room temperature and then directly aliquoted for different antibody staining [
16]. Cells were stained with antibody for 30 min at room temperature and then washed with PBS at 300×
g for 10 min before flow analysis. Cells were next incubated with different mouse anti-human mAb from Beckman Coulter (Brea, CA, USA), including PE-conjugated anti-CD56 and anti-CCR7, PE-Cy5-conjugated anti-CD19, PE-Cy7-conjugated anti-CD11c and anti-CD45RO, APC-Alexa Fluor 750-conjugated anti-CD8, and Krome Orange-conjugated anti-CD14. Anti-human mAb from BD Biosciences (BD Biosciences, San Jose, CA, USA) included the following: APC-conjugated anti-human CD4 antibody and propidium iodide (PI). Pacific Blue (PB)-conjugated anti-human CD3 Ab, FITC-conjugated anti-human Hsp60, and AlexaFluor-488-conjugated anti-human Cytochrome C were from BioLegend (San Diego, CA, USA). Anti-SDF-1 polyclonal mAB was purchased from Abcam (Cambridge, MA, USA). Anti-carboxypeptidase M (CPM) monoclonal mAB was purchased from Novocastra (Newcastle, UK). The FITC-conjugated AffiniPure donkey anti-mouse 2nd Ab was from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). AlexaFluor-546-conjugated Calnexin and isotype-matched IgG and AlexaFluor-647-conjugated GM130 were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). For intra-cellular and intra-mitochondrial staining, cells or mitochondria were fixed and permeabilized according to the PerFix-nc kit (Beckman Coulter, Brea, CA, USA) manufacturer’s recommended protocol. Isotype-matched mouse anti-human IgG antibodies (Beckman Coulter, Brea, CA, USA) served as a negative control. After staining, cells were collected and analyzed by a Gallios Flow Cytometer (Beckman Coulter, Brea, CA, USA) equipped with three lasers (488 nm blue, 638 red, and 405 violet lasers) for the concurrent reading of up to 10 colors. The final data were analyzed using the Kaluza Flow Cytometry Analysis Software version 2.1 (Beckman Coulter, Brea, CA, USA).
To determine the direct modulation of mitochondria on CD4+ T cells, the purified CD4+ T cells (1 × 105 cells/well) from PBMC of healthy donors (n = 6) were activated with T-cell activator anti-CD3/CD28 Dynabeads in the presence or absence of 100 μg/mL platelet-derived mitochondria (n = 3) or PBMC-derived mitochondria (n = 3), respectively. Untreated CD4+ T cells served as negative control. Cells were collected for flow cytometry analysis after the treatment at 37 °C and 5% CO2 for 48 h.
4.5. Confocal Detection for Interaction between Mitochondria and CD4+ T Cells
To explore the interaction of mitochondria and CD4+ T cells, the platelets derived mitochondria were stained with MitoTracker Deep Red FM (100 nM) (Thermo Fisher Scientific, Waltham, MA, USA) and cocultured with MitoTracker Green FM (100 nM) (Thermo Fisher Scientific, Waltham, MA, USA) labeled CD4+ T cells for 2 h, at room temperature. Hoechst 33,342 (Sigma, Saint Louis, MO, USA) was used to stain the nuclei. Briefly, the purified CD4+ T cells were initially labeled with MitoTracker Green FM (100 nM) (Thermo Fisher Scientific, Waltham, MA, USA) and Hoechst 33,342 (Sigma, Saint Louis, MO, USA), and then planted in the 96-well tissue culture-treated plates at 2 × 105 cells/well with serum-free culture medium X-VIVO 15 in the presence or absence of MitoTracker Deep Red-labeled platelet-derived mitochondria. The interaction between mitochondria and CD4+ T cells was directly observed and photographed under a confocal microscope with Nikon A1R confocal microscope on Nikon Eclipse Ti2 inverted base at room temperature.
4.6. Quantitative Real Time PCR
Total RNAs from each sample were extracted using a Qiagen kit (Valencia, CA, USA). First-strand cDNAs were synthesized from total RNA using an iScript gDNA Clear cDNA Synthesis Kit according to the manufacturer’s protocol (Bio-Rad, Hercules, CA, USA). The quantification of human mitochondrial DNA by real-time PCR was performed by using the StepOnePlus Real-Time PCR System (Applied Biosystem, CA, USA) [
16]. Human mitochondrial DNA (mtDNA) monitoring primer sets (catalogue No. 7246), including ND1 and ND5 (Takara Bio, Mountain view, CA, USA), were used to detect the mitochondrial DNA. SLCO2B1 Primer Mix and SERPINA1 Primer Mix (Takara Bio, Mountain view, CA, USA) were used for detection of nuclear DNA (nDNA). The data analysis of the mtDNA Copy Number was performed by using the mtDNA Copy Number Calculation Tool (
https://www.takarabio.com/resourcedocument/x102669), as recommended by the manufacturer.
The validated primers of Carboxypeptidase M (CPM) (Catalogue No. PPH17190E-200) and β-actin (Catalogue No. PPH00073G-200) were purchased from Qiagen (Valencia, CA, USA) and running under following conditions: 95 °C for 10 min, then 40 cycles of 95 °C for 15 s, and 60 °C for 60 s. The expression level of Carboxypeptidase M was determined relative to β-actin as an internal control.
4.7. Blocking Experiments with CXCR4 Receptor Antagonist AMD 3100 and SDF-1 Antibody
To determine whether the SDF-1/CXCR4 signal was contributing to the interaction between mitochondria and CD4+ T cells, we performed the blocking experiments with CXCR4 receptor antagonist AMD3100 and SDF-1 Ab. The isolated CD4+ T cells were treated with MitoTracker Deep Red-labeled purified mitochondria in the presence or absence of AMD 3100 (30 μM) for 24 h. The equal concentration of DMSO served as control. The isolated CD4+ T cells were treated with MitoTracker Deep Red-labeled purified mitochondria in the presence or absence of SDF-1 Ab (20 μg/mL) for 2 h. To determine the blocking effects of SDF-1 Ab on the immune modulation of platelet-derived mitochondria, the purified CD4+ T cells (1 × 105 cells/well) from PBMC of healthy donors (n = 3) were activated with T-cell activator anti-CD3/CD28 Dynabeads in the presence or absence of 100 μg/mL platelet-derived mitochondria or 20 μg/mL SDF-1 Ab. Untreated CD4+ T cells served as negative control. Cells were collected for flow cytometry analysis after the treatment at 37 °C and 5% CO2 for 48 h. CD4+ T cells were washed twice with PBS and prepared for flow cytometry detection.
4.8. Evaluation of Cytokine Levels in Serum
To detect the cytokine production by CD4+ T cells, 1 × 105 CD4+ T cells were stimulated with anti-CD3/anti-CD28 beads (Thermo Fisher Scientific, Waltham, MA, USA) in the presence or absence of mitochondria at 100 μg/mL in a 96-well plate with a total of 200 μL X-VIVO 15 serum-free culture medium (Lonza, Walkersville, MD, USA) per well. After the treatment for 48 h, the supernatants were collected to examine the level of inflammatory cytokines (IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, IL-17F, IL-21, IL-22, IFN-γ, and TNF-α) using a LEGENDplex™ Human Th Panel (Biolegend, San Diego, CA, USA) by Gallios Flow Cytometer according to the manufacturer’s protocols.
4.9. Western Blot
Purified mitochondria or cells were treated with RIPA buffer, as previously described [
16,
30]. Proteins (20 μg/sample) were separated by 4–15% Criterion Tris-HCl gel (Bio-Rad, Hercules, CA, USA) and transferred to the PVDF membrane, blotted overnight with mouse anti-human cytochrome C (Biolegend, Dedham, MA, USA) monoclonal antibody (mAb) and mouse anti-human Calnexin mAb (Santa Cruz, Dallas, TX, USA), and followed by anti-mouse HRP-conjugated secondary mAb (Cell Signaling, Danvers, MA, USA). Membranes were incubated with chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA) and chemiluminescent signal was detected by using ChemiDoc Imaging System (Bio-Rad, Hercules, CA, USA). β-actin served as an internal loading control [
31].
4.10. RNA-seq
RNA sequencing (RNA-seq) analysis was performed between the mitochondrion-treated and untreated CD4
+ T cells in the presence of anti-CD3/anti-CD28 beads stimulation in three preparations, as previously described [
21]. Total RNAs from each sample were extracted by a Qiagen kit (Valencia, CA, USA) and shipped to Genewiz (South Plainfield, NJ, USA) in dry ice for standard RNA sequencing and profiling gene expression by using Illumina NovaSeq™ 6000 Sequencing System (Genewiz, South Plainfield, NJ, USA), with 2 × 150 bp configuration, single index, per lane.
4.11. Statistical Analysis
Statistical analyses of data were performed with GraphPad Prism 8 (version 8.0.1) software. The normality test of samples was evaluated using the Shapiro–Wilk test. Statistical analyses of data were performed using the two-tailed paired Student’s t-test to determine statistical significance between untreated and treated groups. The Mann–Whitney U test was utilized for non-parametric data. Values were given as mean ± SD (standard deviation). Statistical significance was defined as p < 0.05. For RNA-seq data analysis, the Wald test with DESeq2 software was utilized to generate p-values and log2 fold changes. Genes with a p-value < 0.05 and absolute log2 fold change >1 were determined as differentially expressed genes for each comparison.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







