In Silico Finding of Key Interaction Mediated α3β4 and α7 Nicotinic Acetylcholine Receptor Ligand Selectivity of Quinuclidine-Triazole Chemotype

 ,
, 
 , , and
, , and
Abstract
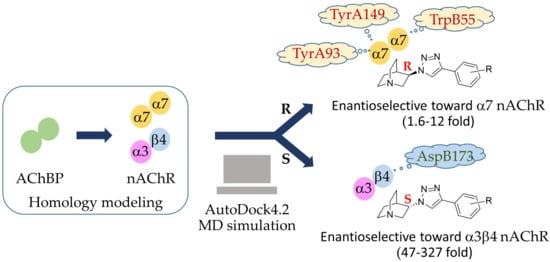
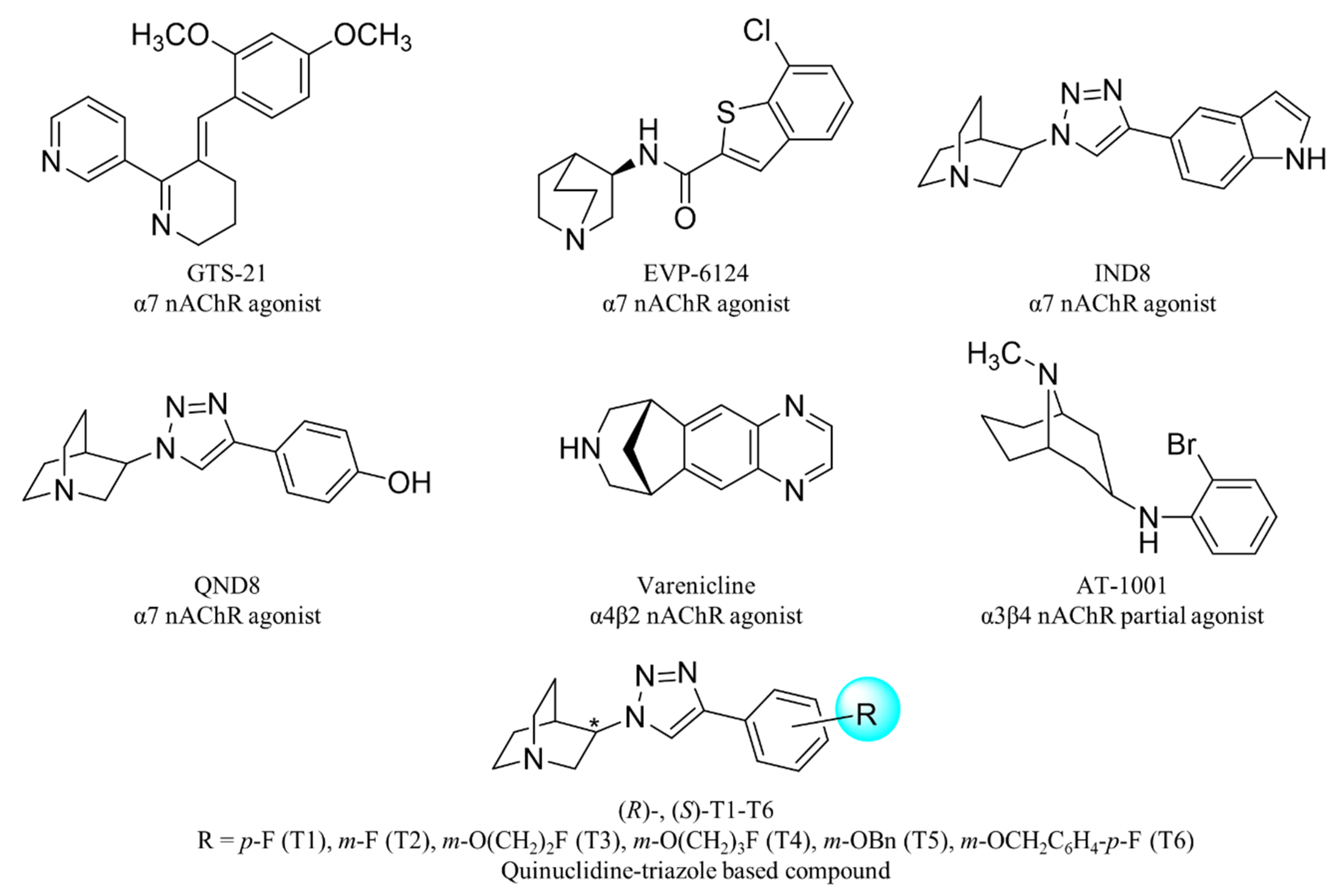
1. Introduction
2. Results and Discussion
2.1. nAChR Homology Model Templates (α7 and α3β4 Subtypes)
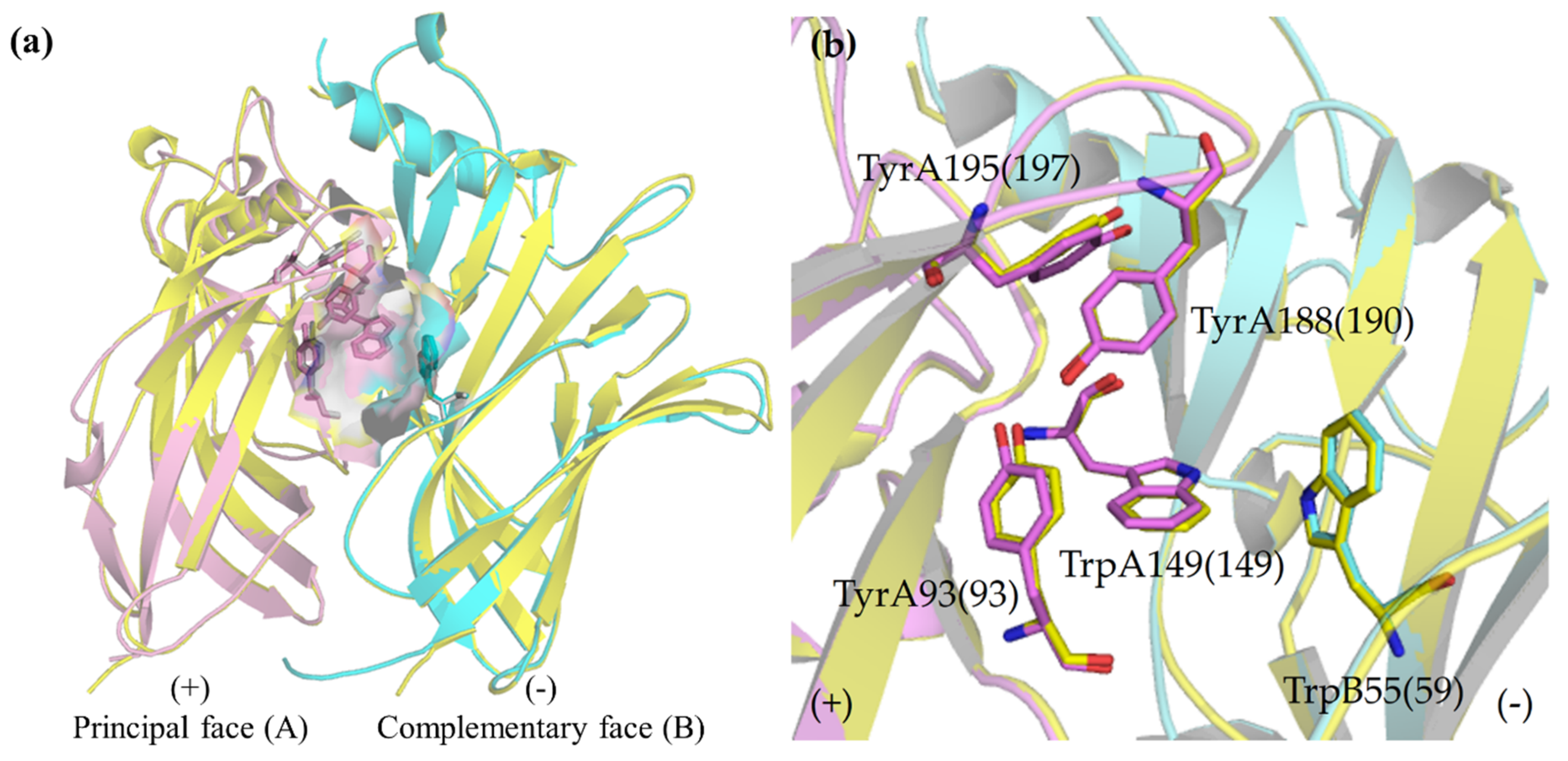
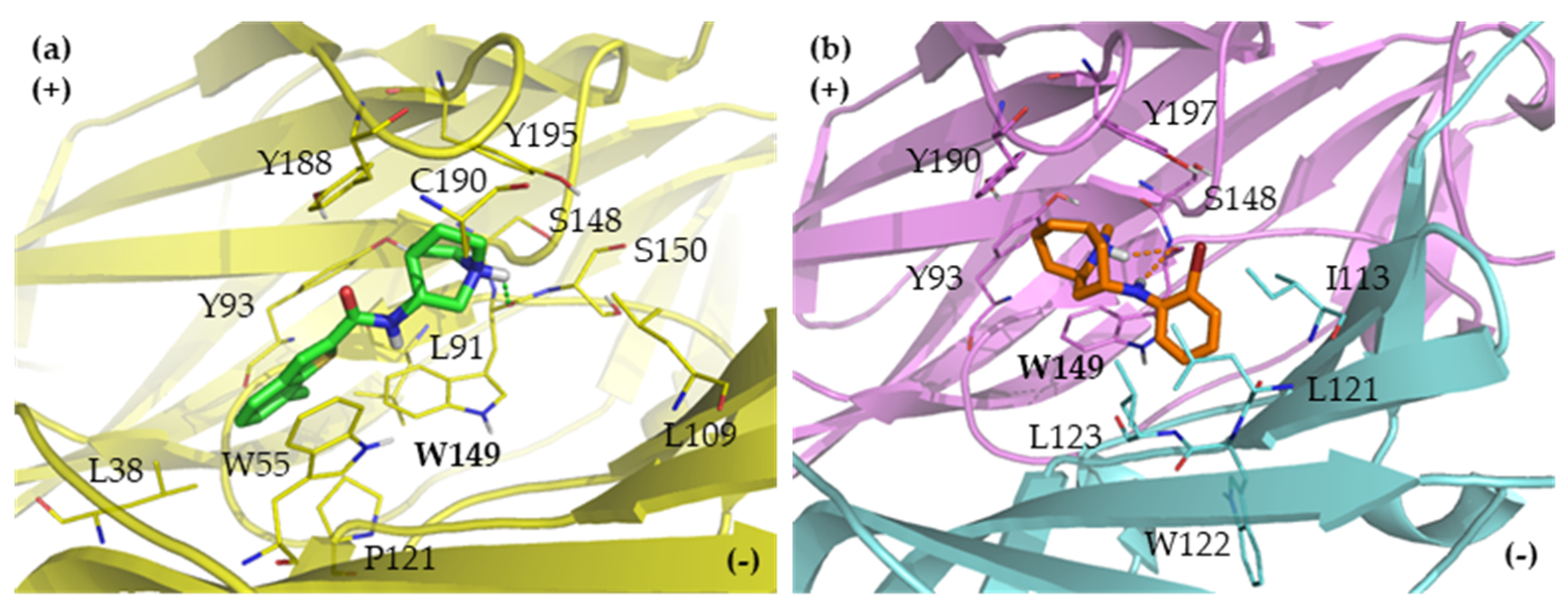
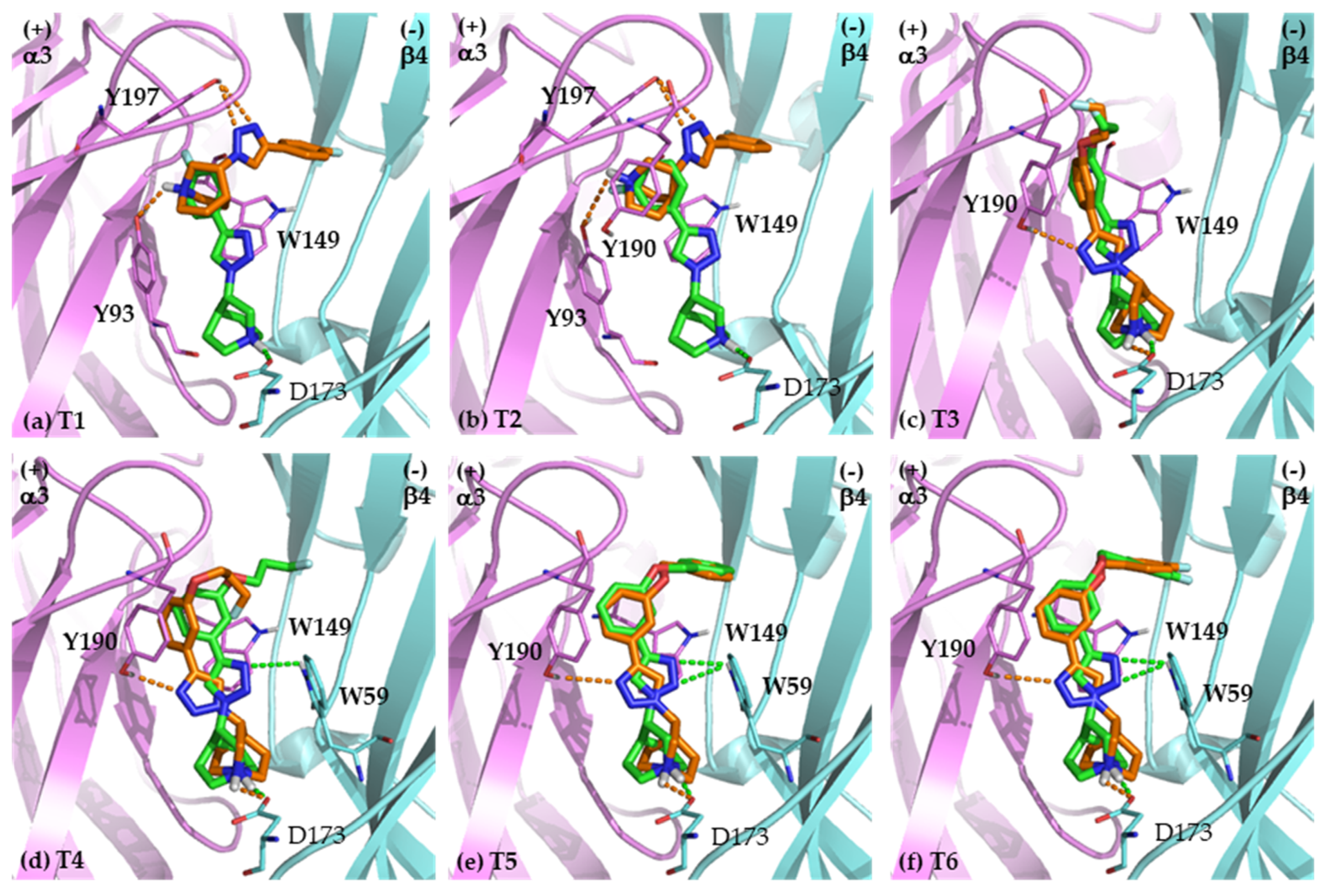
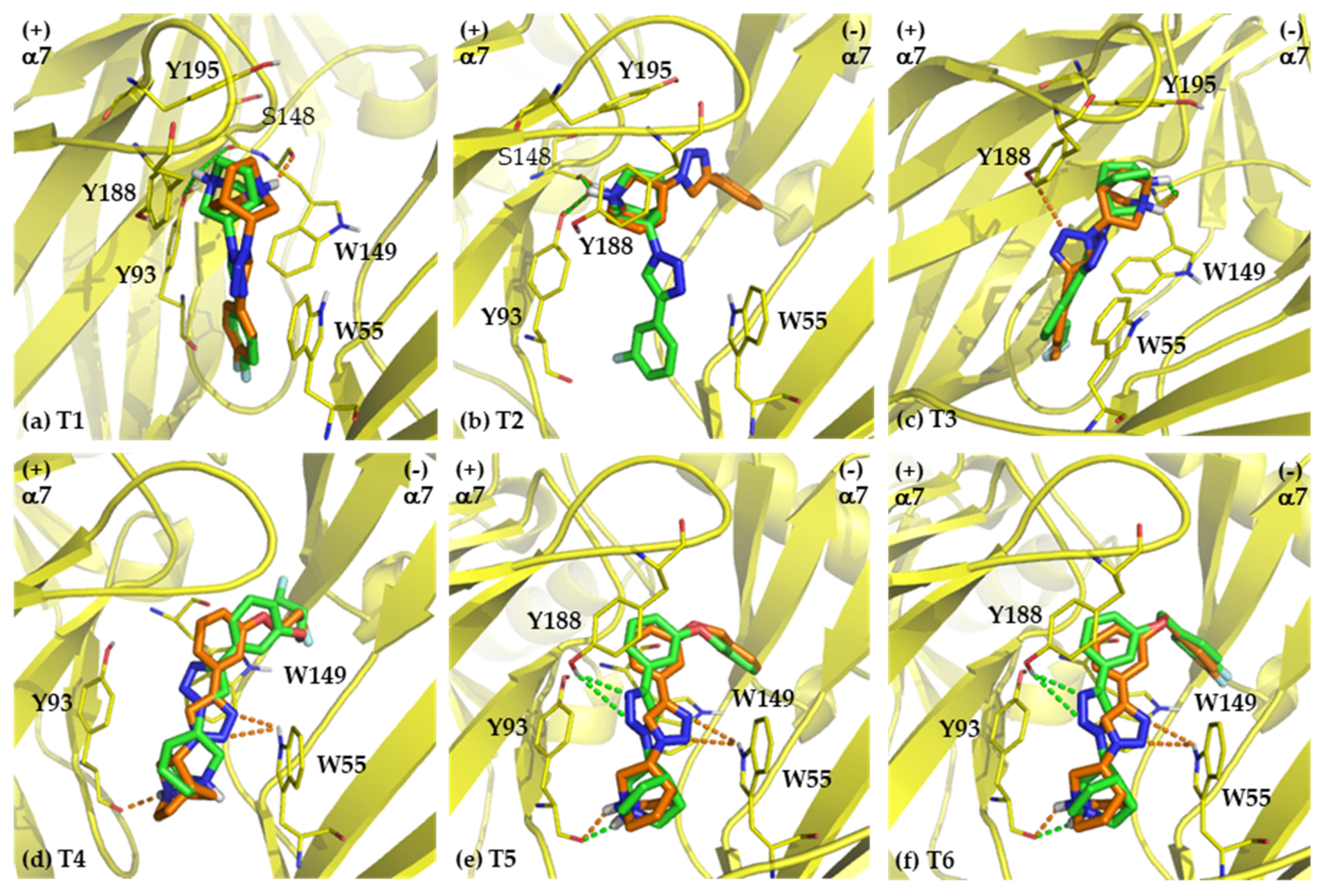
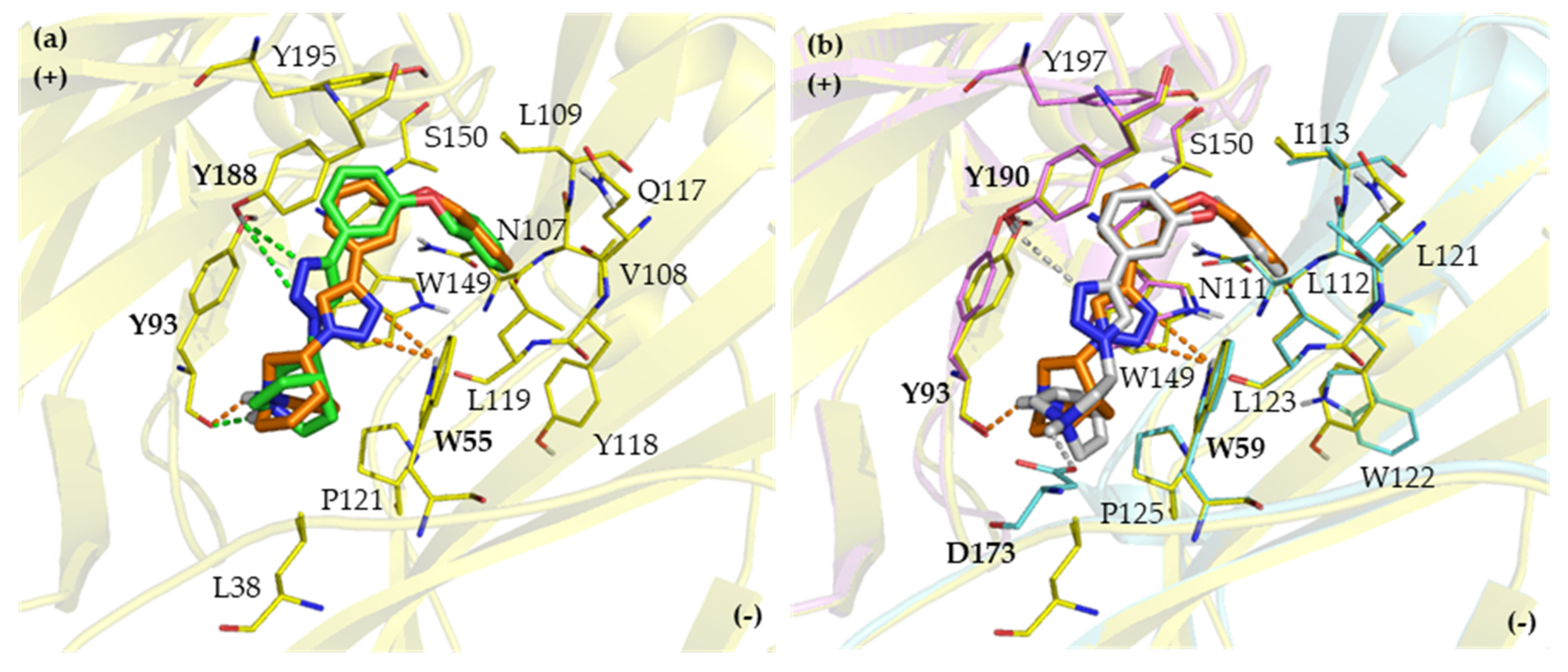
2.2. Binding Modes
2.2.1. Binding Modes of (S)-Enantiomers
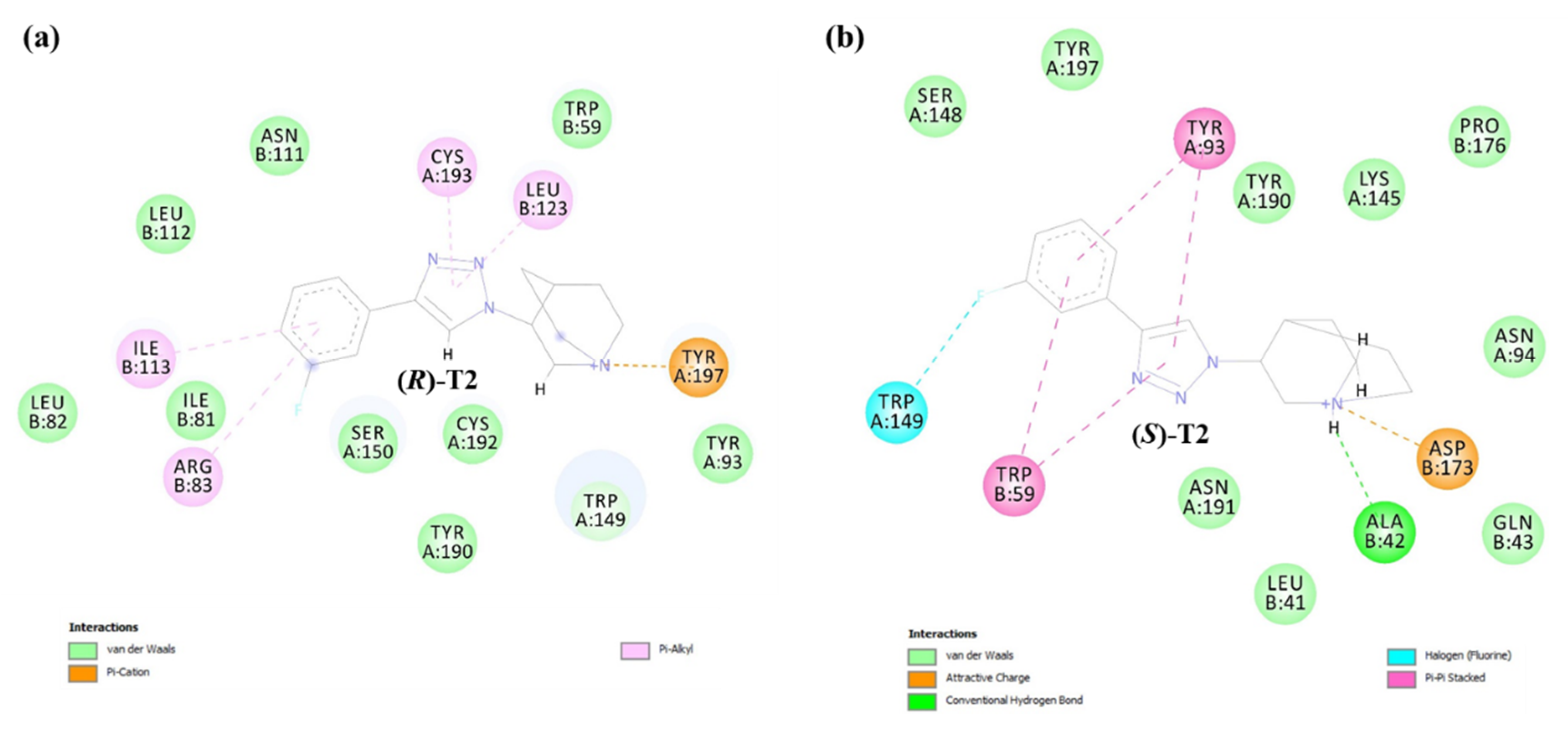
2.2.2. Binding Modes of (R)-Enantiomers
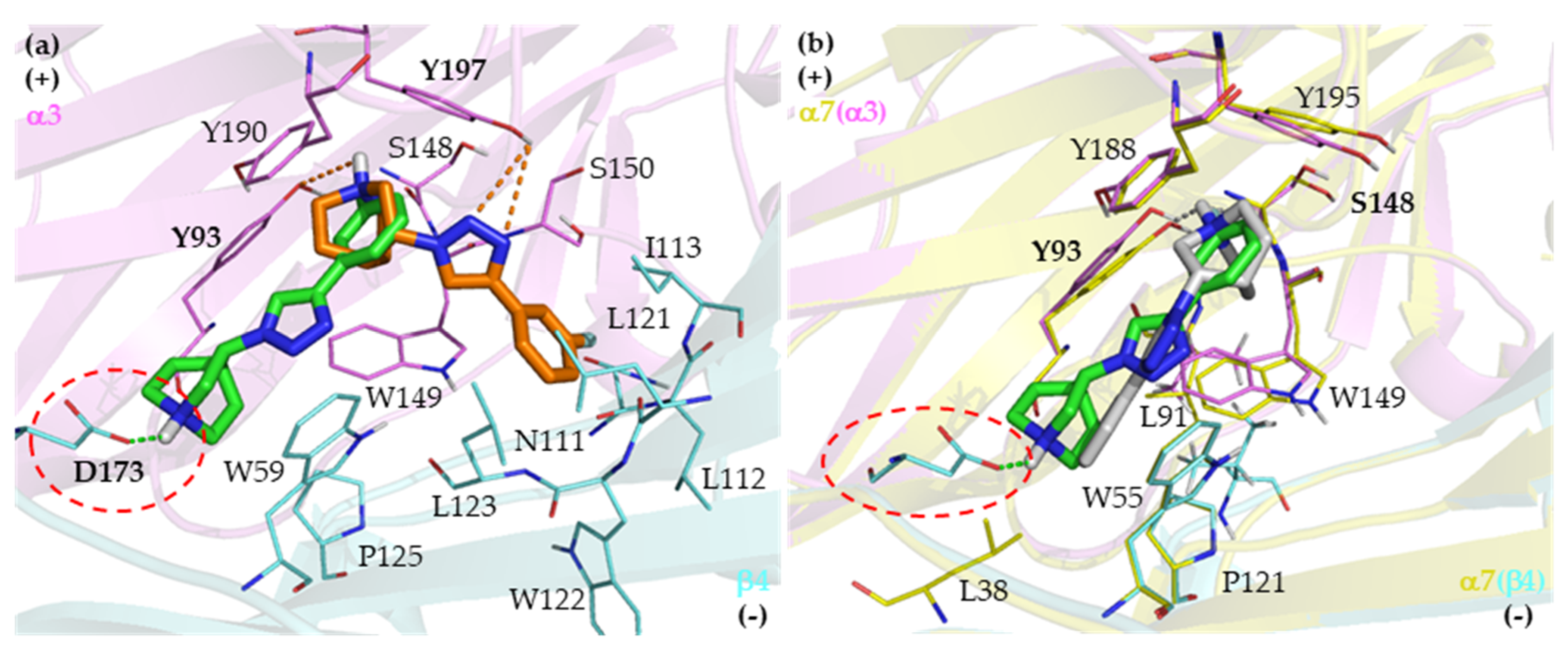
2.3. Identification of Amino Acid Determinants and Interactions for the Stereoselective Binding
2.3.1. Determinants of Selective α3β4 Binding
2.3.2. Determinants of Selective α7 Binding
3. Materials and Methods
3.1. Protein Template and Ligand Preparations
3.2. Molecular docking
3.3. Molecular Dynamics (MD) Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef]
- Jensen, A.A.; Frolund, B.; Liljefors, T.; Krogsgaard-Larsen, P. Neuronal nicotinic acetylcholine receptors: Structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 2005, 48, 4705–4745. [Google Scholar] [CrossRef]
- A Dani, J.; Bertrand, D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef]
- Schaaf, C.P. Nicotinic acetylcholine receptors in human genetic disease. Genet. Med. 2014, 16, 649–656. [Google Scholar] [CrossRef]
- Levin, E.D.; Bradley, A.; Addy, N.; Sigurani, N. Hippocampal alpha 7 and alpha 4 beta 2 nicotinic receptors and working memory. Neuroscience 2002, 109, 757–765. [Google Scholar] [CrossRef]
- Shimohama, S.; Kawamata, J.; Akaike, A.; Misu, Y. Roles of Nicotinic Acetylcholine Receptors in the Pathology and Treatment of Alzheimer’s and Parkinson’s Diseases. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Springer: Singapore, 2018; pp. 137–158. [Google Scholar]
- Terry, A.V., Jr.; Callahan, P.M. Alpha7 nicotinic acetylcholine receptors as therapeutic targets in schizophrenia: Update on animal and clinical studies and strategies for the future. Neuropharmacology 2020, 170. [Google Scholar] [CrossRef]
- Buckingham, S.D.; Jones, A.K.; Brown, L.A.; Sattelle, D.B. Nicotinic acetylcholine receptor signalling: Roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol. Rev. 2009, 61, 39–61. [Google Scholar] [CrossRef]
- Hoskin, J.L.; Al-Hasan, Y.; Sabbagh, M.N. Nicotinic Acetylcholine Receptor Agonists for the Treatment of Alzheimer’s Dementia: An Update. Nicotine Tob. Res. 2018, 21, 370–376. [Google Scholar] [CrossRef]
- Gotti, C.; Clementi, F.; Fornari, A.; Gaimarri, A.; Guiducci, S.; Manfredi, I.; Moretti, M.; Pedrazzi, P.; Pucci, L.; Zoli, M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [Google Scholar] [CrossRef]
- Chatterjee, S.; Steensland, P.; A Simms, J.; Holgate, J.; Coe, J.W.; Hurst, R.S.; Shaffer, C.L.; Lowe, J.; Rollema, H.; Bartlett, S.E. Partial Agonists of the α3β4* Neuronal Nicotinic Acetylcholine Receptor Reduce Ethanol Consumption and Seeking in Rats. Neuropsychopharmacology 2010, 36, 603–615. [Google Scholar] [CrossRef]
- Glick, S.D.; Maisonneuve, I.M.; A Kitchen, B.; Fleck, M.W. Antagonism of alpha 3 beta 4 nicotinic receptors as a strategy to reduce opioid and stimulant self-administration. Eur. J. Pharmacol. 2002, 438, 99–105. [Google Scholar] [CrossRef]
- Rahman, S.; Engleman, E.A.; Bell, R.L. Nicotinic receptor modulation to treat alcohol and drug dependence. Front. Mol. Neurosci. 2015, 8, 426. [Google Scholar] [CrossRef]
- Mazurov, A.; Hauser, T.; Miller, C. Selective α7 Nicotinic Acetylcholine Receptor Ligands. Curr. Med. Chem. 2006, 13, 1567–1584. [Google Scholar] [CrossRef]
- A Briggs, C.; Anderson, D.J.; Brioni, J.D.; Buccafusco, J.J.; Buckley, M.J.; E Campbell, J.; Decker, M.W.; Donnelly–Roberts, D.; Elliott, R.L.; Gopalakrishnan, M.; et al. Functional characterization of the novel neuronal nicotinic acetylcholine receptor ligand GTS-21 in vitro and in vivo. Pharmacol. Biochem. Behav. 1997, 57, 231–241. [Google Scholar] [CrossRef]
- Preskorn, S.H.; Gawryl, M.; Dgetluck, N.; Palfreyman, M.; Bauer, L.O.; Hilt, D.C. Normalizing Effects of EVP-6124, an Alpha-7 Nicotinic Partial Agonist, on Event-Related Potentials and Cognition. J. Psychiatr. Pract. 2014, 20, 12–24. [Google Scholar] [CrossRef]
- Prickaerts, J.; Van Goethem, N.P.; Chesworth, R.; Shapiro, G.; Boess, F.G.; Methfessel, C.; Reneerkens, O.A.H.; Flood, D.; Hilt, D.; Gawryl, M.; et al. EVP-6124, a novel and selective α7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of α7 nicotinic acetylcholine receptors. Neuropharmacology 2012, 62, 1099–1110. [Google Scholar] [CrossRef]
- Kaur, K.; Kaushal, S.; Chopra, S.C. Varenicline for smoking cessation: A review of the literature. Curr. Ther. Res. 2009, 70, 35–54. [Google Scholar] [CrossRef][Green Version]
- Mihalak, K.B.; Carroll, F.I.; Luetje, C.W. Varenicline Is a Partial Agonist at α4β2 and a Full Agonist at α7 Neuronal Nicotinic Receptors. Mol. Pharmacol. 2006, 70, 801–805. [Google Scholar] [CrossRef]
- Cippitelli, A.; Wu, J.; Gaiolini, K.A.; Mercatelli, D.; Schoch, J.; Gorman, M.; Ramirez, A.; Ciccocioppo, R.; Khroyan, T.V.; Yasuda, D.; et al. AT-1001: A high-affinity alpha3beta4 nAChR ligand with novel nicotine-suppressive pharmacology. Brit. J. Pharmacol. 2015, 172, 1834–1845. [Google Scholar] [CrossRef]
- Toll, L.; Zaveri, N.T.; Polgar, W.E.; Jiang, F.; Khroyan, T.V.; Zhou, W.; Xie, X.S.; Stauber, G.B.; Costello, M.R.; Leslie, F.M.; et al. AT-1001: A high affinity and selective alpha3beta4 nicotinic acetylcholine receptor antagonist blocks nicotine self-administration in rats. Neuropsychopharmacology 2012, 37, 1367–1376. [Google Scholar] [CrossRef]
- Sarasamkan, J.; Scheunemann, M.; Apaijai, N.; Palee, S.; Parichatikanond, W.; Arunrungvichian, K.; Fischer, S.; Chattipakorn, S.; Deuther-Conrad, W.; Schüürmann, G.; et al. Varying Chirality Across Nicotinic Acetylcholine Receptor Subtypes: Selective Binding of Quinuclidine Triazole Compounds. ACS Med. Chem. Lett. 2016, 7, 890–895. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Adv. Struct. Saf. Stud. 2014, 1137, 1–15. [Google Scholar] [CrossRef]
- Spurny, R.; Debaveye, S.; Farinha, A.; Veys, K.; Vos, A.M.; Gossas, T.; Atack, J.R.; Bertrand, S.; Bertrand, D.; Danielson, U.H.; et al. Molecular blueprint of allosteric binding sites in a homologue of the agonist-binding domain of the α7 nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2015, 112, E2543–E2552. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of Neuronal α7-nAChR by α-Conotoxin ImI Explained by Computational Scanning and Energy Calculations. PLoS Comput. Boil. 2011, 7, e1002011. [Google Scholar] [CrossRef]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human alpha4beta2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega. Curr. Protoc. Bioinform. 2014, 48, 3.13.1–3.13.16. [Google Scholar] [CrossRef]
- Melo, F.; Sali, A. Fold assessment for comparative protein structure modeling. Protein Sci. 2007, 16, 2412–2426. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Celie, P.H.; E Van Rossum-Fikkert, S.; Van Dijk, W.J.; Brejc, K.; Smit, A.B.; Sixma, T.K. Nicotine and Carbamylcholine Binding to Nicotinic Acetylcholine Receptors as Studied in AChBP Crystal Structures. Neuron 2004, 41, 907–914. [Google Scholar] [CrossRef]
- Hansen, S.B.; Sulzenbacher, G.; Huxford, T.; Marchot, P.; Taylor, P.; Bourne, Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005, 24, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Rucktooa, P.; Smit, A.B.; Sixma, T.K. Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem. Pharmacol. 2009, 78, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Van Arnam, E.B.; Dougherty, D.A. Functional Probes of Drug–Receptor Interactions Implicated by Structural Studies: Cys-Loop Receptors Provide a Fertile Testing Ground. J. Med. Chem. 2014, 57, 6289–6300. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef]
- Harpsøe, K.; Hald, H.; Timmermann, D.B.; Jensen, M.L.; Dyhring, T.; Nielsen, E.Ø.; Peters, D.; Balle, T.; Gajhede, M.; Kastrup, J.S.; et al. Molecular Determinants of Subtype-selective Efficacies of Cytisine and the Novel Compound NS3861 at Heteromeric Nicotinic Acetylcholine Receptors*. J. Boil. Chem. 2012, 288, 2559–2570. [Google Scholar] [CrossRef]
- Parker, M.J.; Harvey, S.C.; Luetje, C.W. Determinants of agonist binding affinity on neuronal nicotinic receptor beta subunits. J. Pharmacol. Exp. Ther. 2001, 299, 385–391. [Google Scholar]
- Cuny, H.; Yu, R.; Tae, H.-S.; Kompella, S.N.; Adams, D.J. α-Conotoxins active at α3-containing nicotinic acetylcholine receptors and their molecular determinants for selective inhibition. Brit. J. Pharmacol. 2017, 175, 1855–1868. [Google Scholar] [CrossRef]
- Cuny, H.; Kompella, S.N.; Tae, H.S.; Yu, R.; Adams, D.J. Key structural determinants in the agonist binding loops of human beta2 and beta4 nicotinic acetylcholine receptor subunits contribute to alpha3beta4 subtype selectivity of alpha-conotoxins. J. Biol. Chem. 2016, 291, 23779–23792. [Google Scholar] [CrossRef]
- Abraham, N.; Healy, M.; Ragnarsson, L.; Brust, A.; Alewood, P.F.; Lewis, R.J. Structural mechanisms for alpha-conotoxin activity at the human alpha3beta4 nicotinic acetylcholine receptor. Sci. Rep. 2017, 7, 45466. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Hooft, R.W.; Sander, C.; Vriend, G. Objectively judging the quality of a protein structure from a Ramachandran plot. Comput. Appl. Biosci. CABIOS 1997, 13, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Jaikhan, P.; Boonyarat, C.; Arunrungvichian, K.; Taylor, P.; Vajragupta, O. Design and Synthesis of Nicotinic Acetylcholine Receptor Antagonists and their Effect on Cognitive Impairment. Chem. Boil. Drug Des. 2015, 87, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.E.M.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef]
- Tom, D.; Darrin, Y.; Lee, P. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 R | Ki in nM (Mean ± SD) 1 | Eudismic Ratio 2 | Subtype Selectivity Ratio 3 | ||||
|---|---|---|---|---|---|---|---|
| Cpd | α3β4 | α7 | α3β4 (S/R) | α7 (R/S) | α3β4 | α7 | |
| (R)-T1 | p-F | 1010 ± 162 | 73.0 ± 15 | 2.4 | 14 | ||
| (S)-T1 | 3.09 ± 0.10 | 174.5 ± 66 | 327 | 56 | |||
| (R)-T2 | m-F | 362 ± 27 | 117 ± 4 | 5.7 | 3.1 | ||
| (S)-T2 | 2.25 ± 0.42 | 660.5 ± 139 | 161 | 294 | |||
| (R)-T3 | m-O(CH2)2F | 558 ± 34 | 38.8 ± 8 | 1.9 | 14 | ||
| (S)-T3 | 11.8 ± 0.3 | 74.9 ± 20 | 47 | 6.3 | |||
| (R)-T4 | m-O(CH2)3F | 1628 ± 11 | 62.3 ± 10 | 1.6 | 26 | ||
| (S)-T4 | 19.5 ± 0.4 | 96.9 ± 17 | 83 | 5 | |||
| (R)-T5 | m-OBn | 631 ± 206 | 22.5 ± 9 | 12 | 28 | ||
| (S)-T5 | 6.67 ± 0.7 | 279 ± 31 | 95 | 42 | |||
| (R)-T6 | m-OCH2C6H4-p-F | 1090 ± 163 | 33.2 ± 7 | 4.5 | 33 | ||
| (S)-T6 | 7.17 ± 1.2 | 149 ± 42 | 152 | 21 | |||
| Selected nAChR Model | Modeller Scoring Function | Ramachandran Plot | ||||
|---|---|---|---|---|---|---|
| DOPE Score | GA341 Score | Most Favored Region | Additional Allowed Region | Generously Allowed Region | Disallowed Region | |
| α7 | −46,953.29688 | 1 | 92.3 | 6.6 | 0.5 | 0.5 |
| α3β4 | −46,290.92188 | 0.99998 | 91.1 | 7.6 | 1.0 | 0.3 |
 | α3β4 nAChR Interaction | α7 nAChR Interaction | ||||||
|---|---|---|---|---|---|---|---|---|
| Hydrogen Bond (Distance in Å) | Salt Bridge Opportunity | Cation-π | π-π | Hydrogen Bond (Distance in Å) | Cation-π | π-π | ||
| Cpd | R | |||||||
| (R)-T1 | p-F | TyrA93 (2.45) TyrA197 (3.02, 3.16) | - | TrpA149 TyrA197 | - | TrpA149 (1.80) | TrpA149 TyrA195 | TrpB55 |
| (S)-T1 | AspB173 (1.78) | AspB173 | - | TrpA149 | TyrA93 (2.30) SerA148 (3.36) | TyrA188 TyrA195 | - | |
| (R)-T2 | m-F | TyrA93 (3.36) TyrA197 (3.23, 3.36) | - | TyrA190 TyrA197 | - | TyrA93 (2.24) SerA148 (2.82) | TyrA195 | - |
| (S)-T2 | AspB173 (1.77) | AspB173 | - | TrpA149 TyrA190 | TyrA93 (2.11) SerA148 (3.22) | TyrA188 TyrA195 | TrpB55 | |
| (R)-T3 | m-O(CH2)2F | AspB173 (2.05) TyrA190 (2.77) | AspB173 | - | TyrA190 | TrpA149 (1.90) TyrA188 (3.01) | TrpA149 TyrA195 | - |
| (S)-T3 | AspB173 (1.78) | AspB173 | - | TrpA149TyrA190 | TrpA149 (1.99) | TrpA149 TyrA195 | TrpA149 TrpB55 | |
| (R)-T4 | m-O(CH2)3F | AspB173 (2.08) TyrA190 (2.66) | AspB173 | - | TrpA149TyrA190 | TyrA93 (2.18) TrpB55 (2.68, 3.12) | - | TrpA149 |
| (S)-T4 | AspB173 (1.70) TrpB59 (3.39) | AspB173 | - | TrpA149 | - | TrpB55 | TrpA149 | |
| (R)-T5 | m-OBn | AspB173 (1.97) TyrA190 (3.19) | AspB173 | - | TrpA149 | TyrA93 (1.94) TrpB55 (3.01, 3.09) | - | TrpA149 |
| (S)-T5 | AspB173 (1.65) TrpB59 (3.08, 3.22) | AspB173 | - | TrpA149 | TyrA93 (1.85) TyrA188 (2.73, 3.37) | - | - | |
| (R)-T6 | m-OCH2C6H4-p-F | AspB173 (1.95) TyrA190 (3.15) | AspB173 | - | TrpA149 TyrA190 | TyrA93 (1.90) TrpB55 (2.83, 3.02) | - | TrpA149 |
| (S)-T6 | AspB173 (1.66) TrpB59 (3.00, 3.18) | AspB173 | - | TrpA149 | TyrA93 (1.96) TyrA188 (2.66, 3.32) | - | TyrA188 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arunrungvichian, K.; Chongruchiroj, S.; Sarasamkan, J.; Schüürmann, G.; Brust, P.; Vajragupta, O. In Silico Finding of Key Interaction Mediated α3β4 and α7 Nicotinic Acetylcholine Receptor Ligand Selectivity of Quinuclidine-Triazole Chemotype. Int. J. Mol. Sci. 2020, 21, 6189. https://doi.org/10.3390/ijms21176189
Arunrungvichian K, Chongruchiroj S, Sarasamkan J, Schüürmann G, Brust P, Vajragupta O. In Silico Finding of Key Interaction Mediated α3β4 and α7 Nicotinic Acetylcholine Receptor Ligand Selectivity of Quinuclidine-Triazole Chemotype. International Journal of Molecular Sciences. 2020; 21(17):6189. https://doi.org/10.3390/ijms21176189
Chicago/Turabian StyleArunrungvichian, Kuntarat, Sumet Chongruchiroj, Jiradanai Sarasamkan, Gerrit Schüürmann, Peter Brust, and Opa Vajragupta. 2020. "In Silico Finding of Key Interaction Mediated α3β4 and α7 Nicotinic Acetylcholine Receptor Ligand Selectivity of Quinuclidine-Triazole Chemotype" International Journal of Molecular Sciences 21, no. 17: 6189. https://doi.org/10.3390/ijms21176189
APA StyleArunrungvichian, K., Chongruchiroj, S., Sarasamkan, J., Schüürmann, G., Brust, P., & Vajragupta, O. (2020). In Silico Finding of Key Interaction Mediated α3β4 and α7 Nicotinic Acetylcholine Receptor Ligand Selectivity of Quinuclidine-Triazole Chemotype. International Journal of Molecular Sciences, 21(17), 6189. https://doi.org/10.3390/ijms21176189