Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
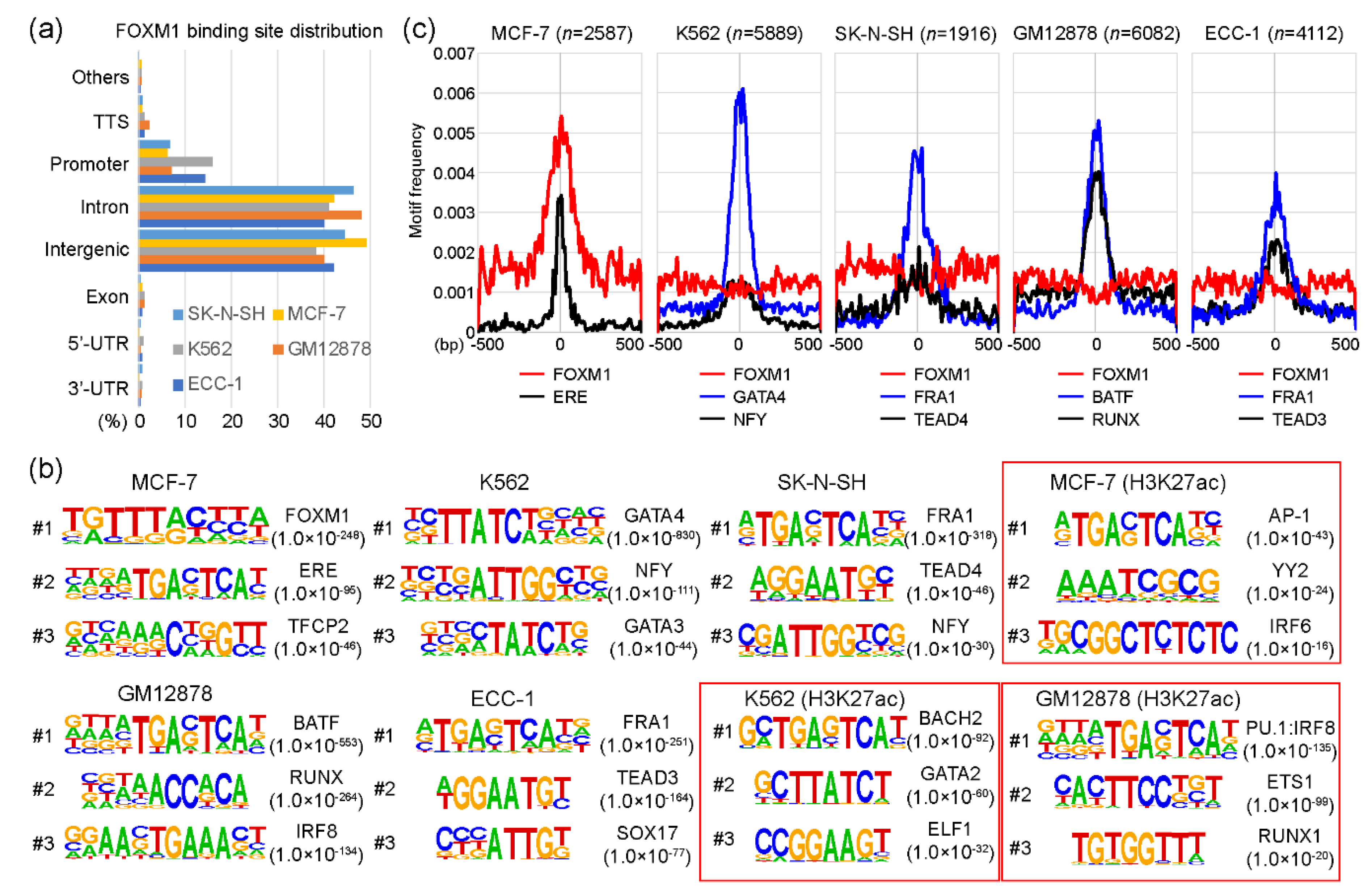
2.1. Canonical and non-Canonical FOXM1 Binding Motifs
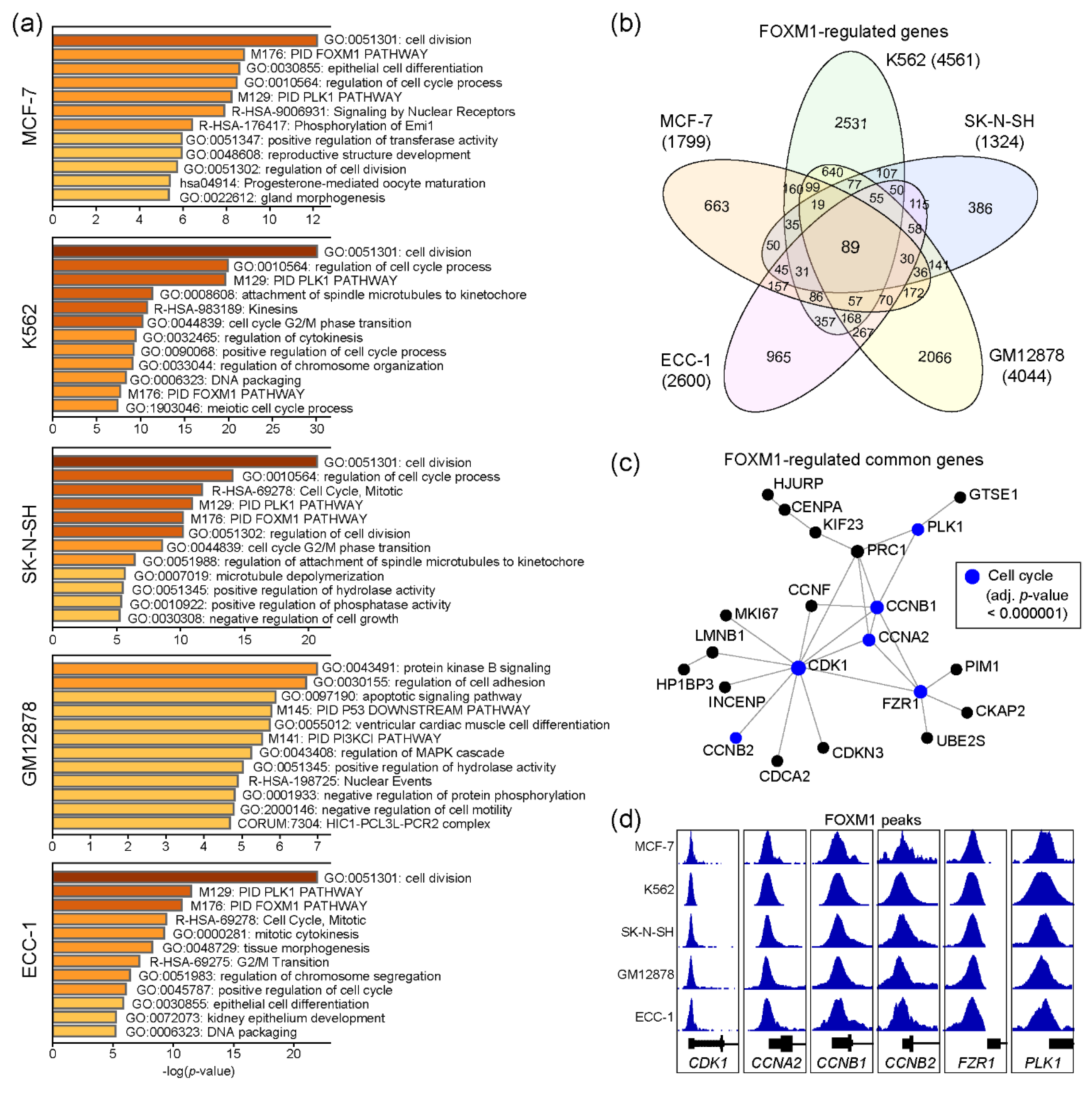
2.2. Functional Prediction of FOXM1 Target Genes
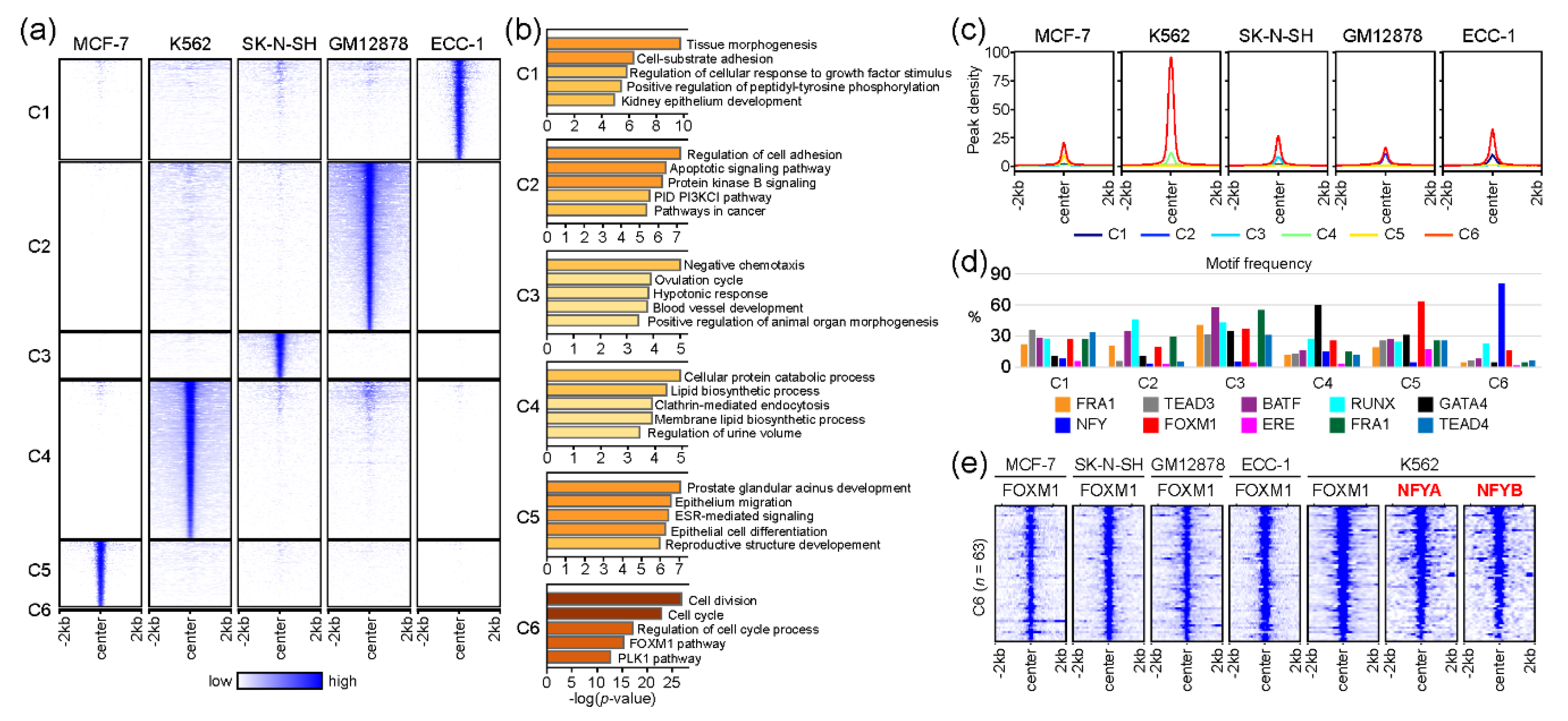
2.3. The association of FOXM1 and NFY in Regulating Cell-Cycle-Related Genes
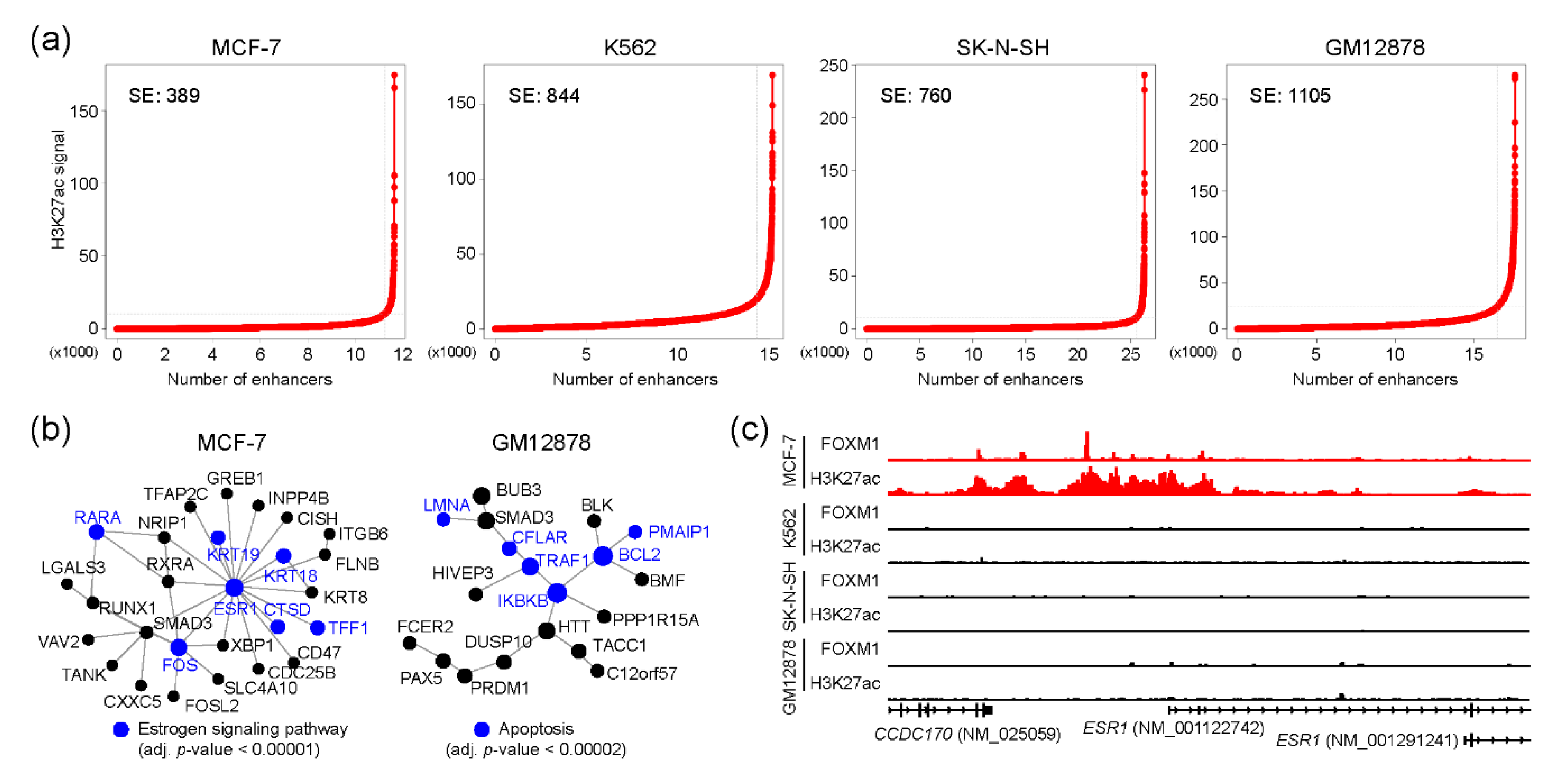
2.4. FOXM1 and Super-Enhancers
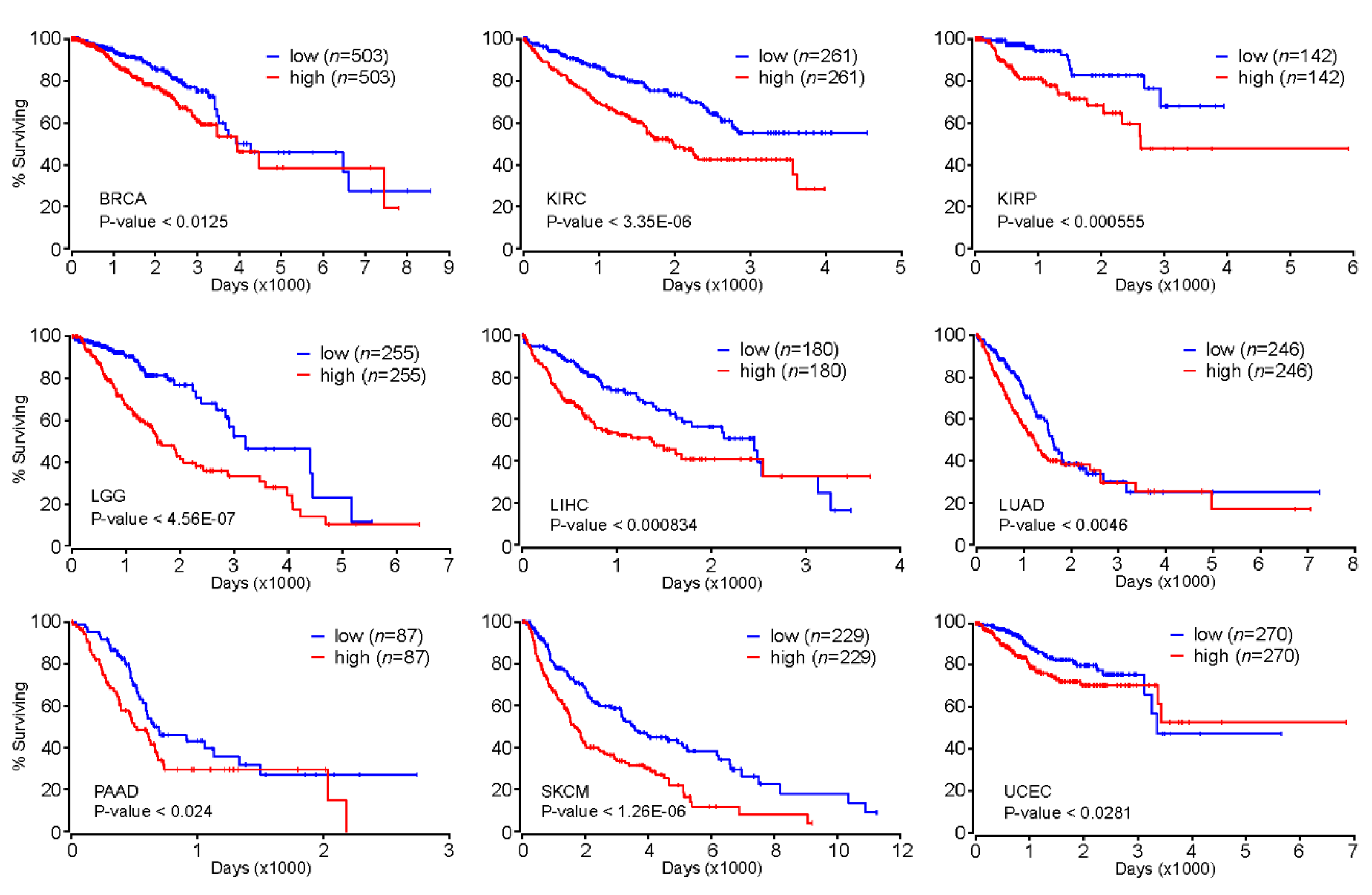
2.5. FOXM1 as a Prognostic Marker for Various Types of Cancer
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. ChIP-seq Data Analysis
5.2. Identification of Super-Enhancers
5.3. Motif Analysis
5.4. Gene Ontology, Protein–Protein Interaction-Based Network Analysis, and Visualization
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FKH | Forkhead |
| DBD | DNA binding domain |
| CHR | Cell cycle genes homology region |
| ChIP-seq | Chromatin immunoprecipitation followed by sequencing |
| ENCODE | The encyclopedia of DNA elements |
| GO | Gene ontology |
| PPI | Protein–protein interaction |
| SE | Super-enhancer |
| BRCA | Breast invasive carcinoma |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LGG | Brain lower grade glioma |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung adenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| SKCM | Skin cutaneous melanoma |
| UCEC | Uterine corpus endometrial carcinoma |
References
- Wang, X.; Kiyokawa, H.; Dennewitz, M.B.; Costa, R.H. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 16881–16886. [Google Scholar] [CrossRef]
- Wonsey, D.R.; Follettie, M.T. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005, 65, 5181–5189. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, P.; Park, H.J. FoxM1: A master regulator of tumor metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ahmad, A.; Li, Y.; Banerjee, S.; Kong, D.; Sarkar, F.H. Forkhead box M1 transcription factor: A novel target for cancer therapy. Cancer Treat. Rev. 2010, 36, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Bektas, N.; Haaf, A.; Veeck, J.; Wild, P.J.; Luscher-Firzlaff, J.; Hartmann, A.; Knuchel, R.; Dahl, E. Tight correlation between expression of the Forkhead transcription factor FOXM1 and HER2 in human breast cancer. BMC Cancer 2008, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Hui, W.W.; Cai, P.C.; Liu, M.X.; Yung, M.M.; Mak, C.S.; Leung, T.H.; Chan, K.K.; Ngan, H.Y. Targeting GRB7/ERK/FOXM1 signaling pathway impairs aggressiveness of ovarian cancer cells. PLoS ONE 2012, 7, e52578. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.Y.; Zhu, Z.M.; Chen, L.B.; Wang, J.H.; Su, Q.S.; Yang, J.R.; Lin, Y.; Xue, L.J.; Liu, X.B.; Mo, X.B. FOXM1 expression correlates with tumor invasion and a poor prognosis of colorectal cancer. Acta Histochem. 2012, 114, 755–762. [Google Scholar] [CrossRef]
- Xu, N.; Jia, D.; Chen, W.; Wang, H.; Liu, F.; Ge, H.; Zhu, X.; Song, Y.; Zhang, X.; Zhang, D.; et al. FoxM1 is associated with poor prognosis of non-small cell lung cancer patients through promoting tumor metastasis. PLoS ONE 2013, 8, e59412. [Google Scholar] [CrossRef]
- Sanders, D.A.; Ross-Innes, C.S.; Beraldi, D.; Carroll, J.S.; Balasubramanian, S. Genome-wide mapping of FOXM1 binding reveals co-binding with estrogen receptor alpha in breast cancer cells. Genome Biol. 2013, 14, R6. [Google Scholar] [CrossRef]
- Khan, S.A.; Rogers, M.A.; Khurana, K.K.; Meguid, M.M.; Numann, P.J. Estrogen receptor expression in benign breast epithelium and breast cancer risk. J. Natl. Cancer Inst. 1998, 90, 37–42. [Google Scholar] [CrossRef]
- Karadedou, C.T. Regulation of the FOXM1 transcription factor by the estrogen receptor alpha at the protein level, in breast cancer. Hippokratia 2006, 10, 128–132. [Google Scholar] [PubMed]
- Miao, L.; Xiong, X.; Lin, Y.; Cheng, Y.; Lu, J.; Zhang, J.; Cheng, N. Down-regulation of FoxM1 leads to the inhibition of the epithelial-mesenchymal transition in gastric cancer cells. Cancer Genet. 2014, 207, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Bhat, U.G.; Halasi, M.; Gartel, A.L. FoxM1 is a general target for proteasome inhibitors. PLoS ONE 2009, 4, e6593. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhu, X.; Zhang, K.; Zhu, L.; Zhou, F. FoxM1 inhibition enhances chemosensitivity of docetaxel-resistant A549 cells to docetaxel via activation of JNK/mitochondrial pathway. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 804–809. [Google Scholar] [CrossRef]
- Laoukili, J.; Stahl, M.; Medema, R.H. FoxM1: At the crossroads of ageing and cancer. Biochim. Biophys. Acta 2007, 1775, 92–102. [Google Scholar] [CrossRef]
- Chen, X.; Muller, G.A.; Quaas, M.; Fischer, M.; Han, N.; Stutchbury, B.; Sharrocks, A.D.; Engeland, K. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell. Biol. 2013, 33, 227–236. [Google Scholar] [CrossRef]
- Sanders, D.A.; Gormally, M.V.; Marsico, G.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. FOXM1 binds directly to non-consensus sequences in the human genome. Genome Biol. 2015, 16, 130. [Google Scholar] [CrossRef]
- Muller, G.A.; Engeland, K. The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription. FEBS J. 2010, 277, 877–893. [Google Scholar] [CrossRef]
- Linhart, C.; Elkon, R.; Shiloh, Y.; Shamir, R. Deciphering transcriptional regulatory elements that encode specific cell cycle phasing by comparative genomics analysis. Cell Cycle 2005, 4, 1788–1797. [Google Scholar] [CrossRef]
- Wang, Y.; Ung, M.H.; Xia, T.; Cheng, W.; Cheng, C. Cancer cell line specific co-factors modulate the FOXM1 cistrome. Oncotarget 2017, 8, 76498–76515. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Oldfield, A.J.; Yang, P.; Conway, A.E.; Cinghu, S.; Freudenberg, J.M.; Yellaboina, S.; Jothi, R. Histone-fold domain protein NF-Y promotes chromatin accessibility for cell type-specific master transcription factors. Mol. Cell 2014, 55, 708–722. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Song, R.X.; Santen, R.J. Membrane initiated estrogen signaling in breast cancer. Biol. Reprod. 2006, 75, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.L.; Yoshida, H.; Yamaguchi, M. Nuclear transcription factor Y and its roles in cellular processes related to human disease. Am. J. Cancer Res. 2013, 3, 339–346. [Google Scholar] [PubMed]
- Costessi, A.; Mahrour, N.; Tijchon, E.; Stunnenberg, R.; Stoel, M.A.; Jansen, P.W.; Sela, D.; Martin-Brown, S.; Washburn, M.P.; Florens, L.; et al. The tumour antigen PRAME is a subunit of a Cul2 ubiquitin ligase and associates with active NFY promoters. EMBO J. 2011, 30, 3786–3798. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Niederriter, A.R.; Varshney, A.; Parker, S.C.; Martin, D.M. Super Enhancers in Cancers, Complex Disease, and Developmental Disorders. Genes 2015, 6, 1183–1200. [Google Scholar] [CrossRef] [PubMed]
- Peeters, J.G.; Vervoort, S.J.; Tan, S.C.; Mijnheer, G.; de Roock, S.; Vastert, S.J.; Nieuwenhuis, E.E.; van Wijk, F.; Prakken, B.J.; Creyghton, M.P.; et al. Inhibition of Super-Enhancer Activity in Autoinflammatory Site-Derived T Cells Reduces Disease-Associated Gene Expression. Cell Rep. 2015, 12, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Gormally, M.V.; Dexheimer, T.S.; Marsico, G.; Sanders, D.A.; Lowe, C.; Matak-Vinkovic, D.; Michael, S.; Jadhav, A.; Rai, G.; Maloney, D.J.; et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5, 5165. [Google Scholar] [CrossRef]
- Costa, R.H. FoxM1 dances with mitosis. Nat. Cell Biol. 2005, 7, 108–110. [Google Scholar] [CrossRef]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Xue, L.; Chiang, L.; He, B.; Zhao, Y.Y.; Winoto, A. FoxM1, a forkhead transcription factor is a master cell cycle regulator for mouse mature T cells but not double positive thymocytes. PLoS ONE 2010, 5, e9229. [Google Scholar] [CrossRef]
- Kang, K.; Robinson, G.W.; Hennighausen, L. Comprehensive meta-analysis of Signal Transducers and Activators of Transcription (STAT) genomic binding patterns discerns cell-specific cis-regulatory modules. BMC Genom. 2013, 14, 4. [Google Scholar] [CrossRef]
- Han, T.; Oh, S.; Kang, K. ETS family protein GABP is a novel co-factor strongly associated with genomic YY1 binding sites in various cell lines. Genes Genom. 2016, 38, 119–125. [Google Scholar] [CrossRef]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Littler, D.R.; Alvarez-Fernandez, M.; Stein, A.; Hibbert, R.G.; Heidebrecht, T.; Aloy, P.; Medema, R.H.; Perrakis, A. Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res. 2010, 38, 4527–4538. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.T.; Leung, M.H.; Qin, J.; Qin, Y.; Wang, J.; Lee, Y.L.; Yao, K.M. The Forkhead box transcription factor FOXM1 is required for the maintenance of cell proliferation and protection against oxidative stress in human embryonic stem cells. Stem. Cell Res. 2016, 16, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Caretti, G.; Salsi, V.; Vecchi, C.; Imbriano, C.; Mantovani, R. Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J. Biol. Chem. 2003, 278, 30435–30440. [Google Scholar] [CrossRef]
- Wang, I.C.; Chen, Y.J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol. Cell. Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef]
- Sadasivam, S.; Duan, S.; DeCaprio, J.A. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012, 26, 474–489. [Google Scholar] [CrossRef]
- Madureira, P.A.; Varshochi, R.; Constantinidou, D.; Francis, R.E.; Coombes, R.C.; Yao, K.M.; Lam, E.W. The Forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J. Biol. Chem. 2006, 281, 25167–25176. [Google Scholar] [CrossRef]
- Lu, X.F.; Zeng, L.W.Q.; Chen, C.F.; Sun, S.M.; Lin, H.Y. FoxM1 is a promising candidate target in the treatment of breast cancer. Oncotarget 2018, 9, 842–852. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Madak-Erdogan, Z.; Kim, Y.J.; Choi, Y.L.; Lu, H.; Katzenellenbogen, B.S. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014, 16, 436. [Google Scholar] [CrossRef]
- Khongkow, P.; Karunarathna, U.; Khongkow, M.; Gong, C.; Gomes, A.R.; Yague, E.; Monteiro, L.J.; Kongsema, M.; Zona, S.; Man, E.P.; et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 2014, 33, 4144–4155. [Google Scholar] [CrossRef]
- Varghese, V.; Magnani, L.; Harada-Shoji, N.; Mauri, F.; Szydlo, R.M.; Yao, S.; Lam, E.W.; Kenny, L.M. FOXM1 modulates 5-FU resistance in colorectal cancer through regulating TYMS expression. Sci. Rep. 2019, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, X.; Jiang, L.; Zhang, L.; Xiang, M.; Ren, H. FoxM1 Induced Paclitaxel Resistance via Activation of the FoxM1/PHB1/RAF-MEK-ERK Pathway and Enhancement of the ABCA2 Transporter. Mol. Ther. Oncolytics 2019, 14, 196–212. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, L.; Zhao, S.H.; Ai, Z.H.; Guo, J.Z.; Liu, W.C. FoxM1 expression is significantly associated with cisplatin-based chemotherapy resistance and poor prognosis in advanced non-small cell lung cancer patients. Lung Cancer 2013, 79, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ren, X.; Wang, I.C.; Pradhan, A.; Zhang, Y.; Flood, H.M.; Han, B.; Whitsett, J.A.; Kalin, T.V.; Kalinichenko, V.V. The FOXM1 inhibitor RCM-1 suppresses goblet cell metaplasia and prevents IL-13 and STAT6 signaling in allergen-exposed mice. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Stromberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, K.; Choi, Y.; Kim, H.H.; Yoo, K.H.; Yu, S. Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines. Int. J. Mol. Sci. 2020, 21, 6141. https://doi.org/10.3390/ijms21176141
Kang K, Choi Y, Kim HH, Yoo KH, Yu S. Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines. International Journal of Molecular Sciences. 2020; 21(17):6141. https://doi.org/10.3390/ijms21176141
Chicago/Turabian StyleKang, Keunsoo, Yoonjung Choi, Hoo Hyun Kim, Kyung Hyun Yoo, and Sungryul Yu. 2020. "Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines" International Journal of Molecular Sciences 21, no. 17: 6141. https://doi.org/10.3390/ijms21176141
APA StyleKang, K., Choi, Y., Kim, H. H., Yoo, K. H., & Yu, S. (2020). Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines. International Journal of Molecular Sciences, 21(17), 6141. https://doi.org/10.3390/ijms21176141