Lessons from Recent Advances in Ischemic Stroke Management and Targeting Kv2.1 for Neuroprotection

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
2. Physiology of the Ischemic Penumbra and Penumbral Preservation
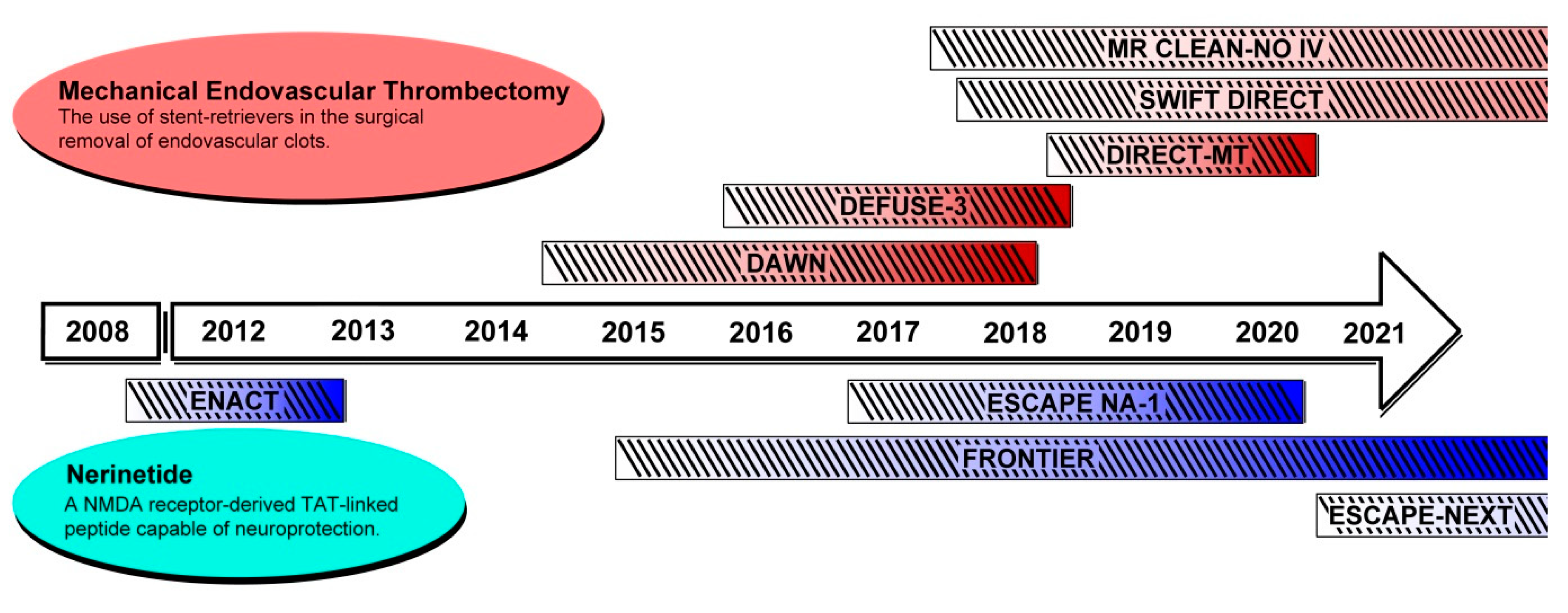
3. Recent Advances in Reperfusion Therapy
3.1. Canonical Management of Acute Ischemic Stroke
3.2. DAWN Phase II/III Trial
3.3. DEFUSE-3 Phase III Trial
4. Recent Advances in Stroke Neuroprotective Therapy
4.1. Neuroprotective Agents in Stroke
4.2. PSD-95 Inhibition, Excitotoxicity, and Nerinetide (NoNO Inc.)
4.3. ENACT Phase II Trial
4.4. ESCAPE-NA1 Phase III Trial
5. The Emerging Landscape of Ischemic Stroke Therapy
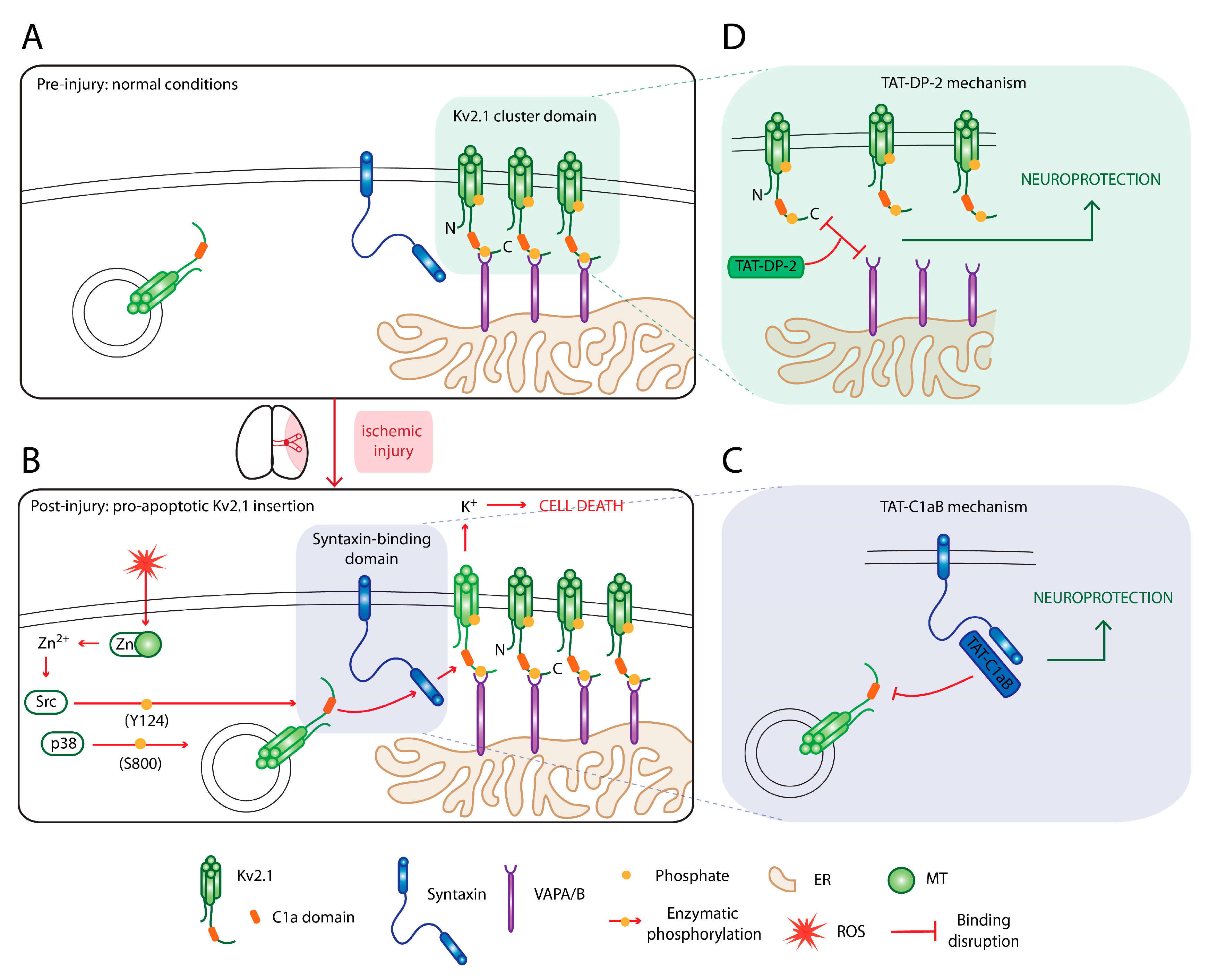
6. Targeting Kv2.1 for Neuroprotection
6.1. An Omnipresent Cell Death Mechanism in Neurodegeneration
6.2. Disrupting the Kv2.1–Syntaxin Interaction
6.3. Disruption of Kv2.1-ER Cluster Junctions
7. Other Recent and Ongoing Clinical Trials for Ischemic Stroke Therapy
7.1. Mechanical Endovascular Thrombectomy
7.2. NMDA-Related Neuroprotective Therapies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DALYs | Disability-adjusted life years |
| MR | Magnetic resonance |
| DW | Diffusion-weighted |
| PW | Perfusion-weighted |
| CT | Computed tomography |
| ROS | Reactive oxygen species |
| rt-PA | Recombinate tissue plasminogen activator |
| NIHSS | NIH stroke scale |
| ICA | Internal carotid artery |
| MCA | Middle cerebral artery |
| LVO | Large vessel occlusion |
| ATP | Adenosine triphosphate |
| NMDA | N-methyl-D-aspartate |
| PSD | Postsynaptic density |
| MAGUK | Membrane-associated guanylate-kinase |
| nNOS | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| O− | Superoxide |
| ONOO− | Peroxynitrite |
| TAT | Transactivator of transcription |
| MCAO | Middle cerebral artery occlusion |
| FLAIR | Fluid-attenuated inversion recovery MR imaging |
| MT | Metallothionine |
| SNARE | SNAP receptor |
| ER-PM | Endoplasmic reticulum-plasma membrane |
| VAP | VAMP-associated protein |
References
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; Floyd, J.; Fornage, M.; Gillespie, C.; Isasi, C. Heart disease and stroke statistics-2017 update: A report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Savitz, S.I.; Baron, J.-C.; Yenari, M.A.; Sanossian, N.; Fisher, M. Reconsidering Neuroprotection in the Reperfusion Era. Stroke 2017, 48, 3413–3419. [Google Scholar] [CrossRef] [PubMed]
- Savitz, S.I.; Fisher, M. Future of neuroprotection for acute stroke: In the aftermath of the SAINT trials. Ann. Neurol. 2007, 61, 396–402. [Google Scholar] [CrossRef]
- Straka, M.; Albers, G.W.; Bammer, R. Real-time diffusion-perfusion mismatch analysis in acute stroke. J. Magn. Reson. Imaging 2010, 32, 1024–1037. [Google Scholar] [CrossRef]
- Hakim, A.M. The cerebral ischemic penumbra. Can. J. Neurol. Sci. 1987, 14, 557–559. [Google Scholar]
- van der Worp, H.B.; van Gijn, J. Acute Ischemic Stroke. N. Engl. J. Med. 2007, 357, 572–579. [Google Scholar] [CrossRef]
- Bråtane, B.T.; Cui, H.; Cook, D.J.; Bouley, J.; Tymianski, M.; Fisher, M. Neuroprotection by freezing ischemic penumbra evolution without cerebral blood flow augmentation with a postsynaptic density-95 protein inhibitor. Stroke 2011, 42, 3265–3270. [Google Scholar] [CrossRef]
- Baron, J.-C. Protecting the ischaemic penumbra as an adjunct to thrombectomy for acute stroke. Nat. Rev. Neurol. 2018, 14, 325–337. [Google Scholar] [CrossRef]
- Wufuer, A.; Wubuli, A.; Mijiti, P.; Zhou, J.; Tuerxun, S.; Cai, J.; Ma, J.; Zhang, X. Impact of collateral circulation status on favorable outcomes in thrombolysis treatment: A systematic review and meta-analysis. Exp. Ther. Med. 2018, 15, 707–718. [Google Scholar] [CrossRef]
- Iwasawa, E.; Ichijo, M.; Ishibashi, S.; Yokota, T. Acute development of collateral circulation and therapeutic prospects in ischemic stroke. Neural Regen. Res. 2016, 11, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, N.M.; Saver, J.L.; Diener, H.C.; Gorelick, P.B.; Shuaib, A.; Solberg, Y.; Thackeray, L.; Savic, M.; Janelidze, T.; Zarqua, N.; et al. An injectable implant to stimulate the sphenopalatine ganglion for treatment of acute ischaemic stroke up to 24 h from onset (ImpACT-24B): An international, randomised, double-blind, sham-controlled, pivotal trial. Lancet Lond. Engl. 2019, 394, 219–229. [Google Scholar] [CrossRef]
- Bang, O.Y.; Chung, J.W.; Kim, S.K.; Kim, S.J.; Lee, M.J.; Hwang, J.; Seo, W.K.; Ha, Y.S.; Sung, S.M.; Kim, E.G.; et al. Therapeutic-induced hypertension in patients with noncardioembolic acute stroke. Neurology 2019, 93, e1955–e1963. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol. Neurobiol. 2001, 24, 107–129. [Google Scholar] [CrossRef]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef]
- Nour, M.; Scalzo, F.; Liebeskind, D.S. Ischemia-reperfusion injury in stroke. Interv. Neurol. 2012, 1, 185–199. [Google Scholar] [CrossRef]
- Disorders, N.I.o.N.; Group, S.r.-P.S.S. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995, 333, 1581–1588. [Google Scholar]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar]
- Emberson, J.; Lees, K.R.; Lyden, P.; Blackwell, L.; Albers, G.; Bluhmki, E.; Brott, T.; Cohen, G.; Davis, S.; Donnan, G. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: A meta-analysis of individual patient data from randomised trials. Lancet 2014, 384, 1929–1935. [Google Scholar] [CrossRef]
- Del Zoppo, G.J.; Poeck, K.; Pessin, M.S.; Wolpert, S.M.; Furlan, A.J.; Ferbert, A.; Alberts, M.J.; Zivin, J.A.; Wechsler, L.; Busse, O. Recombinant tissue plasminogen activator in acute thrombotic and embolic stroke. Ann. Neurol. 1992, 32, 78–86. [Google Scholar] [CrossRef]
- Mori, E.; Yoneda, Y.; Tabuchi, M.; Yoshida, T.; Ohkawa, S.; Ohsumi, Y.; Kitano, K.; Tsutsumi, A.; Yamadori, A. Intravenous recombinant tissue plasminogen activator in acute carotid artery territory stroke. Neurology 1992, 42, 976. [Google Scholar] [CrossRef] [PubMed]
- Menon, B.K.; Al-Ajlan, F.S.; Najm, M.; Puig, J.; Castellanos, M.; Dowlatshahi, D.; Calleja, A.; Sohn, S.-I.; Ahn, S.H.; Poppe, A. Association of clinical, imaging, and thrombus characteristics with recanalization of visible intracranial occlusion in patients with acute ischemic stroke. JAMA 2018, 320, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Adeoye, O.; Hornung, R.; Khatri, P.; Kleindorfer, D. Recombinant tissue-type plasminogen activator use for ischemic stroke in the United States: A doubling of treatment rates over the course of 5 years. Stroke 2011, 42, 1952–1955. [Google Scholar] [CrossRef]
- Munich, S.A.; Vakharia, K.; Levy, E.I. Overview of mechanical thrombectomy techniques. Neurosurgery 2019, 85, S60–S67. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L.; Goyal, M.; Bonafe, A.; Diener, H.-C.; Levy, E.I.; Pereira, V.M.; Albers, G.W.; Cognard, C.; Cohen, D.J.; Hacke, W. Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. N. Engl. J. Med. 2015, 372, 2285–2295. [Google Scholar] [CrossRef]
- Campbell, B.C.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N. Engl. J. Med. 2015, 372, 1009–1018. [Google Scholar] [CrossRef]
- Goyal, M.; Demchuk, A.M.; Menon, B.K.; Eesa, M.; Rempel, J.L.; Thornton, J.; Roy, D.; Jovin, T.G.; Willinsky, R.A.; Sapkota, B.L. Randomized assessment of rapid endovascular treatment of ischemic stroke. N. Engl. J. Med. 2015, 372, 1019–1030. [Google Scholar] [CrossRef]
- Jovin, T.G.; Chamorro, A.; Cobo, E.; de Miquel, M.A.; Molina, C.A.; Rovira, A.; San Román, L.; Serena, J.; Abilleira, S.; Ribó, M. Thrombectomy within 8 hours after symptom onset in ischemic stroke. N. Engl. J. Med. 2015, 372, 2296–2306. [Google Scholar] [CrossRef]
- Berkhemer, O.A.; Fransen, P.S.; Beumer, D.; Van Den Berg, L.A.; Lingsma, H.F.; Yoo, A.J.; Schonewille, W.J.; Vos, J.A.; Nederkoorn, P.J.; Wermer, M.J. A randomized trial of intraarterial treatment for acute ischemic stroke. N. Engl. J. Med. 2015, 372, 11–20. [Google Scholar] [CrossRef]
- Goyal, M.; Menon, B.K.; van Zwam, W.H.; Dippel, D.W.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Majoie, C.B.; van der Lugt, A.; De Miquel, M.A. Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Chia, N.H.; Leyden, J.M.; Newbury, J.; Jannes, J.; Kleinig, T.J. Determining the number of ischemic strokes potentially eligible for endovascular thrombectomy: A population-based study. Stroke 2016, 47, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- McMeekin, P.; White, P.; James, M.A.; Price, C.I.; Flynn, D.; Ford, G.A. Estimating the number of UK stroke patients eligible for endovascular thrombectomy. Eur. Stroke J. 2017, 2, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Albers, G.W.; Marks, M.P.; Kemp, S.; Christensen, S.; Tsai, J.P.; Ortega-Gutierrez, S.; McTaggart, R.A.; Torbey, M.T.; Kim-Tenser, M.; Leslie-Mazwi, T. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N. Engl. J. Med. 2018, 378, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef]
- Jauch, E.C.; Saver, J.L.; Adams Jr, H.P.; Bruno, A.; Connors, J.; Demaerschalk, B.M.; Khatri, P.; McMullan Jr, P.W.; Qureshi, A.I.; Rosenfield, K. Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 870–947. [Google Scholar] [CrossRef]
- Committee, E.S.O.E.; Committee, E.W. Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc. Dis. 2008, 25, 457–507. [Google Scholar]
- Hill, K. Australian Clinical Guidelines for Acute Stroke Management 2007: Acute Stroke Guidelines Writing Subgroup on behalf of the National Stroke Foundation Clinical Guidelines for Acute Stroke Management Expert Working Group. Int. J. Stroke 2008, 3, 120–129. [Google Scholar] [CrossRef]
- Casaubon, L.K.; Boulanger, J.-M.; Blacquiere, D.; Boucher, S.; Brown, K.; Goddard, T.; Gordon, J.; Horton, M.; Lalonde, J.; LaRivière, C. Canadian stroke best practice recommendations: Hyperacute stroke care guidelines, update 2015. Int. J. Stroke 2015, 10, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Albers, G.W.; Goyal, M.; Jahan, R.; Bonafe, A.; Diener, H.C.; Levy, E.I.; Pereira, V.M.; Cognard, C.; Cohen, D.J.; Hacke, W. Ischemic core and hypoperfusion volumes predict infarct size in SWIFT PRIME. Ann. Neurol. 2016, 79, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, H.M.; Mlynash, M.; Inoue, M.; Tipirneni, A.; Liggins, J.; Zaharchuk, G.; Straka, M.; Kemp, S.; Bammer, R.; Lansberg, M.G. Early diffusion-weighted imaging and perfusion-weighted imaging lesion volumes forecast final infarct size in DEFUSE 2. Stroke 2013, 44, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Lansberg, M.G.; Christensen, S.; Kemp, S.; Mlynash, M.; Mishra, N.; Federau, C.; Tsai, J.P.; Kim, S.; Nogueria, R.G.; Jovin, T. Computed tomographic perfusion to predict response to recanalization in ischemic stroke. Ann. Neurol. 2017, 81, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Lansberg, M.G.; Straka, M.; Kemp, S.; Mlynash, M.; Wechsler, L.R.; Jovin, T.G.; Wilder, M.J.; Lutsep, H.L.; Czartoski, T.J.; Bernstein, R.A. MRI profile and response to endovascular reperfusion after stroke (DEFUSE 2): A prospective cohort study. Lancet Neurol. 2012, 11, 860–867. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K. The fate of medications evaluated for ischemic stroke pharmacotherapy over the period 1995–2015. Acta Pharm. Sin. B 2016, 6, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Lyden, P.; Hemmen, T.; Grotta, J.; Rapp, K.; Ernstrom, K.; Rzesiewicz, T.; Parker, S.; Concha, M.; Hussain, S.; Agarwal, S. Results of the ICTuS 2 trial (intravascular cooling in the treatment of stroke 2). Stroke 2016, 47, 2888–2895. [Google Scholar] [CrossRef]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; Milot, G.; Clark, W.M.; MacDonald, R.L.; Kelly, M.E.; Boulton, M. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef]
- Hankey, G.J. Nerinetide before reperfusion in acute ischaemic stroke: Déjà vu or new insights? Lancet Lond. Engl. 2020, 395, 843–844. [Google Scholar] [CrossRef]
- Tanaka, E.; Yamamoto, S.; Kudo, Y.; Mihara, S.; Higashi, H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J. Neurophysiol. 1997, 78, 891–902. [Google Scholar] [CrossRef]
- Siemkowicz, E.; Hansen, A. Brain extracellular ion composition and EEG activity following 10 minutes ischemia in normo-and hyperglycemic rats. Stroke 1981, 12, 236–240. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef]
- Choi, D.W.; Rothman, S.M. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Ramdial, K.; Franco, M.C.; Estevez, A.G. Cellular mechanisms of peroxynitrite-induced neuronal death. Brain Res. Bull. 2017, 133, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Ballarin, B.; Tymianski, M. Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharmacol. Sin. 2018, 39, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.-T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 2012, 483, 213–217. [Google Scholar] [CrossRef]
- Cook, D.J.; Teves, L.; Tymianski, M. A translational paradigm for the preclinical evaluation of the stroke neuroprotectant Tat-NR2B9c in gyrencephalic nonhuman primates. Sci. Transl. Med. 2012, 4, ra133–ra154. [Google Scholar] [CrossRef]
- Docagne, F.; Parcq, J.; Lijnen, R.; Ali, C.; Vivien, D. Understanding the functions of endogenous and exogenous tissue-type plasminogen activator during stroke. Stroke 2015, 46, 314–320. [Google Scholar] [CrossRef]
- Shah, N.H.; Aizenman, E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke Res. 2014, 5, 38–58. [Google Scholar] [CrossRef]
- Wu, K.; Yang, P.; Li, S.; Liu, C.; Sun, F. VEGF attenuated increase of outward delayed-rectifier potassium currents in hippocampal neurons induced by focal ischemia via PI3-K pathway. Neuroscience 2015, 298, 94–101. [Google Scholar] [CrossRef]
- Yu, W.; Parakramaweera, R.; Teng, S.; Gowda, M.; Sharad, Y.; Thakker-Varia, S.; Alder, J.; Sesti, F. Oxidation of KCNB1 potassium channels causes neurotoxicity and cognitive impairment in a mouse model of traumatic brain injury. J. Neurosci. 2016, 36, 11084–11096. [Google Scholar] [CrossRef] [PubMed]
- Redman, P.T.; Jefferson, B.S.; Ziegler, C.B.; Mortensen, O.V.; Torres, G.E.; Levitan, E.S.; Aizenman, E. A vital role for voltage-dependent potassium channels in dopamine transporter-mediated 6-hydroxydopamine neurotoxicity. Neuroscience 2006, 143, 1–6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frazzini, V.; Guarnieri, S.; Bomba, M.; Navarra, R.; Morabito, C.; Mariggiò, M.; Sensi, S. Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis. 2016, 7, e2100. [Google Scholar] [CrossRef] [PubMed]
- McCord, M.C.; Aizenman, E. The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 77. [Google Scholar] [CrossRef]
- Aizenman, E.; Stout, A.K.; Hartnett, K.A.; Dineley, K.E.; McLaughlin, B.; Reynolds, I.J. Induction of neuronal apoptosis by thiol oxidation: Putative role of intracellular zinc release. J. Neurochem. 2000, 75, 1878–1888. [Google Scholar] [CrossRef]
- Redman, P.T.; Hartnett, K.A.; Aras, M.A.; Levitan, E.S.; Aizenman, E. Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J. Physiol. 2009, 587, 4393–4404. [Google Scholar] [CrossRef]
- Redman, P.T.; He, K.; Hartnett, K.A.; Jefferson, B.S.; Hu, L.; Rosenberg, P.A.; Levitan, E.S.; Aizenman, E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc. Natl. Acad. Sci. USA 2007, 104, 3568–3573. [Google Scholar] [CrossRef]
- He, K.; McCord, M.C.; Hartnett, K.A.; Aizenman, E. Regulation of pro-apoptotic phosphorylation of Kv2.1 K+ channels. PLoS ONE 2015, 10, e0129498. [Google Scholar] [CrossRef]
- Pal, S.; Takimoto, K.; Aizenman, E.; Levitan, E.S. Apoptotic surface delivery of K+ channels. Cell Death Differ. 2006, 13, 661–667. [Google Scholar] [CrossRef]
- Graham, A.J. Toxic effects of tetraethylammonium bromide. Br. Med. J. 1950, 2, 321. [Google Scholar] [CrossRef]
- Yeh, C.-Y.; Ye, Z.; Moutal, A.; Gaur, S.; Henton, A.M.; Kouvaros, S.; Saloman, J.L.; Hartnett-Scott, K.A.; Tzounopoulos, T.; Khanna, R. Defining the Kv2.1–syntaxin molecular interaction identifies a first-in-class small molecule neuroprotectant. Proc. Natl. Acad. Sci. USA 2019, 116, 15696–15705. [Google Scholar] [CrossRef]
- McCord, M.C.; Kullmann, P.H.; He, K.; Hartnett, K.A.; Horn, J.P.; Lotan, I.; Aizenman, E. Syntaxin-binding domain of Kv2.1 is essential for the expression of apoptotic K+ currents. J. Physiol. 2014, 592, 3511–3521. [Google Scholar] [CrossRef] [PubMed]
- Schulien, A.J.; Yeh, C.-Y.; Orange, B.N.; Pav, O.J.; Hopkins, M.P.; Moutal, A.; Khanna, R.; Sun, D.; Justice, J.A.; Aizenman, E. Targeted disruption of Kv2.1-VAPA association provides neuroprotection against ischemic stroke in mice by declustering Kv2.1 channels. Sci. Adv. 2020, 6, eaaz8110. [Google Scholar] [CrossRef]
- Yeh, C.-Y.; Bulas, A.M.; Moutal, A.; Saloman, J.L.; Hartnett, K.A.; Anderson, C.T.; Tzounopoulos, T.; Sun, D.; Khanna, R.; Aizenman, E. Targeting a Potassium Channel/Syntaxin Interaction Ameliorates Cell Death in Ischemic Stroke. J. Neurosci. 2017, 37, 5648–5658. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.M.; Loftus, R.; Tamkun, M.M. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc. Natl. Acad. Sci. USA 2010, 107, 12351–12356. [Google Scholar] [CrossRef]
- O’Connell, K.M.; Rolig, A.S.; Whitesell, J.D.; Tamkun, M.M. Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J. Neurosci. 2006, 26, 9609–9618. [Google Scholar] [CrossRef]
- Tamkun, M.M.; O’Connell, K.M.; Rolig, A.S. A cytoskeletal-based perimeter fence selectively corrals a sub-population of cell surface Kv2.1 channels. J. Cell Sci. 2007, 120, 2413–2423. [Google Scholar] [CrossRef]
- Deutsch, E.; Weigel, A.V.; Akin, E.J.; Fox, P.; Hansen, G.; Haberkorn, C.J.; Loftus, R.; Krapf, D.; Tamkun, M.M. Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol. Biol. Cell 2012, 23, 2917–2929. [Google Scholar] [CrossRef]
- Fox, P.D.; Haberkorn, C.J.; Akin, E.J.; Seel, P.J.; Krapf, D.; Tamkun, M.M. Induction of stable ER–plasma-membrane junctions by Kv2.1 potassium channels. J. Cell Sci. 2015, 128, 2096–2105. [Google Scholar] [CrossRef]
- Johnson, B.; Leek, A.N.; Solé, L.; Maverick, E.E.; Levine, T.P.; Tamkun, M.M. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc. Natl. Acad. Sci. USA 2018, 115, E7331–E7340. [Google Scholar] [CrossRef]
- Kirmiz, M.; Vierra, N.C.; Palacio, S.; Trimmer, J.S. Identification of VAPA and VAPB as Kv2 channel-interacting proteins defining endoplasmic reticulum–plasma membrane junctions in mammalian brain neurons. J. Neurosci. 2018, 38, 7562–7584. [Google Scholar] [CrossRef] [PubMed]
- Baver, S.B.; O’Connell, K.M. The C-terminus of neuronal Kv2.1 channels is required for channel localization and targeting but not for NMDA-receptor-mediated regulation of channel function. Neuroscience 2012, 217, 56–66. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Justice, J.A.; Schulien, A.J.; He, K.; Hartnett, K.A.; Aizenman, E.; Shah, N.H. Disruption of KV2. 1 somato-dendritic clusters prevents the apoptogenic increase of potassium currents. Neuroscience 2017, 354, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, Y.; Zhang, L.; Zhang, Y.; Treurniet, K.M.; Chen, W.; Peng, Y.; Han, H.; Wang, J.; Wang, S. Endovascular Thrombectomy with or without Intravenous Alteplase in Acute Stroke. N. Engl. J. Med. 2020, 382, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N. Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef]
- Hong, J.M.; Choi, M.H.; Sohn, S.-I.; Hwang, Y.-H.; Ahn, S.H.; Lee, Y.-B.; Shin, D.-I.; Chamorro, Á.; Choi, D.W. Safety and Optimal Neuroprotection of neu2000 in acute Ischemic stroke with reCanalization: Study protocol for a randomized, double-blinded, placebo-controlled, phase-II trial. Trials 2018, 19, 375. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, K.B.; Park, J.H.; Sung, S.M.; Oh, K.; Kim, E.G.; Chang, D.i.; Hwang, Y.H.; Lee, E.J.; Kim, W.K. Safety and Efficacy of Otaplimastat in Patients with Acute Ischemic Stroke Requiring tPA (SAFE-TPA): A Multicenter, Randomized, Double-Blind, Placebo-Controlled Phase 2 Study. Ann. Neurol. 2019, 87, 233–245. [Google Scholar] [CrossRef]
- Darsalia, V.; Klein, T.; Nyström, T.; Patrone, C. Glucagon-like receptor 1 agonists and DPP-4 inhibitors: Anti-diabetic drugs with anti-stroke potential. Neuropharmacology 2018, 136, 280–286. [Google Scholar] [CrossRef]
- Darsalia, V.; Johansen, O.E.; Lietzau, G.; Nyström, T.; Klein, T.; Patrone, C. Dipeptidyl Peptidase-4 Inhibitors for the Potential Treatment of Brain Disorders; A Mini-Review With Special Focus on Linagliptin and Stroke. Front. Neurol. 2019, 10, 493. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, C.-Y.; Schulien, A.J.; Molyneaux, B.J.; Aizenman, E. Lessons from Recent Advances in Ischemic Stroke Management and Targeting Kv2.1 for Neuroprotection. Int. J. Mol. Sci. 2020, 21, 6107. https://doi.org/10.3390/ijms21176107
Yeh C-Y, Schulien AJ, Molyneaux BJ, Aizenman E. Lessons from Recent Advances in Ischemic Stroke Management and Targeting Kv2.1 for Neuroprotection. International Journal of Molecular Sciences. 2020; 21(17):6107. https://doi.org/10.3390/ijms21176107
Chicago/Turabian StyleYeh, Chung-Yang, Anthony J. Schulien, Bradley J. Molyneaux, and Elias Aizenman. 2020. "Lessons from Recent Advances in Ischemic Stroke Management and Targeting Kv2.1 for Neuroprotection" International Journal of Molecular Sciences 21, no. 17: 6107. https://doi.org/10.3390/ijms21176107
APA StyleYeh, C.-Y., Schulien, A. J., Molyneaux, B. J., & Aizenman, E. (2020). Lessons from Recent Advances in Ischemic Stroke Management and Targeting Kv2.1 for Neuroprotection. International Journal of Molecular Sciences, 21(17), 6107. https://doi.org/10.3390/ijms21176107