Bile Acid Signaling in Neurodegenerative and Neurological Disorders



Abstract
1. Introduction
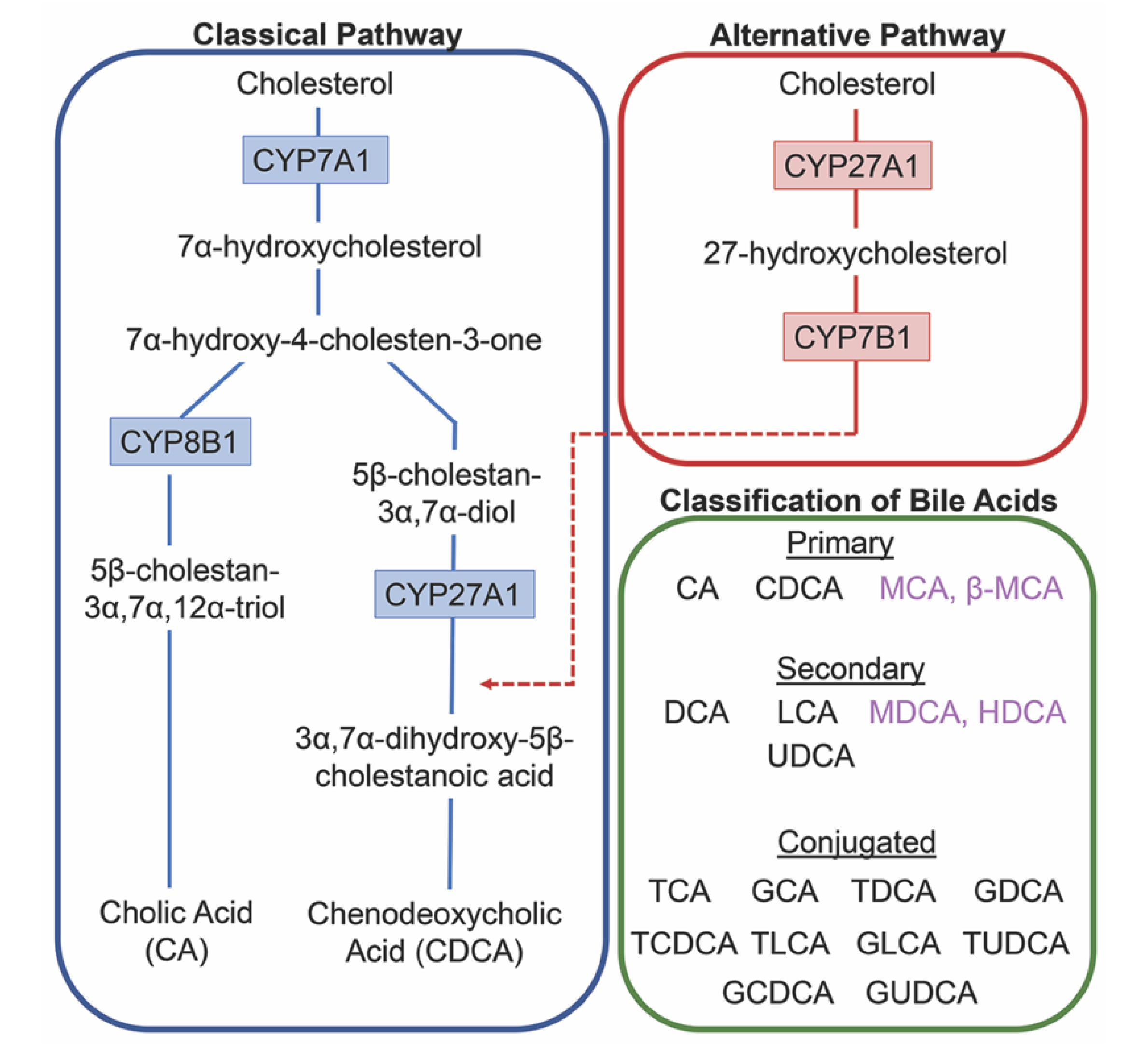
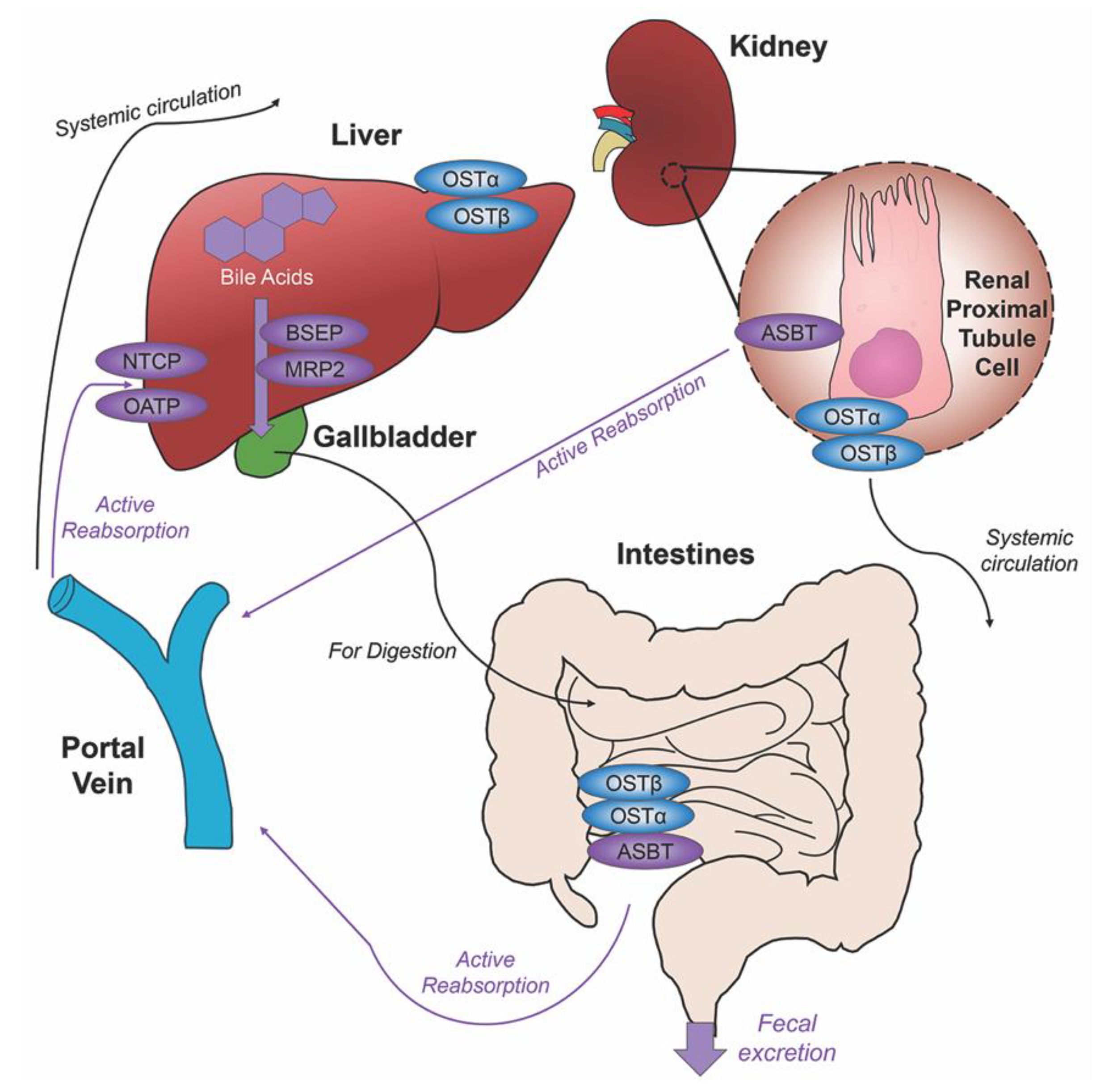
2. Bile Acids Synthesis, Metabolism and Enterohepatic Circulation
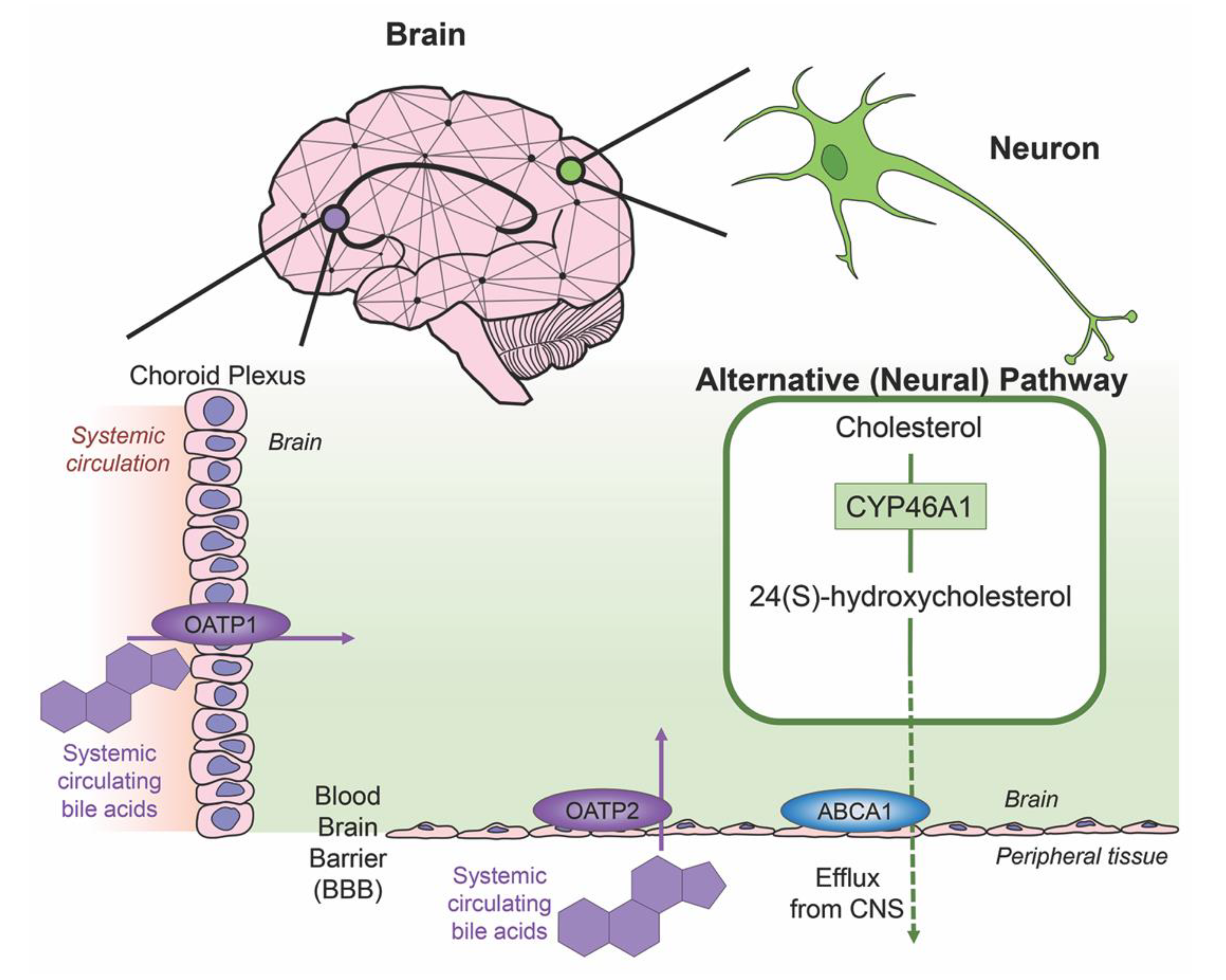
3. Bile Acids in the Brain
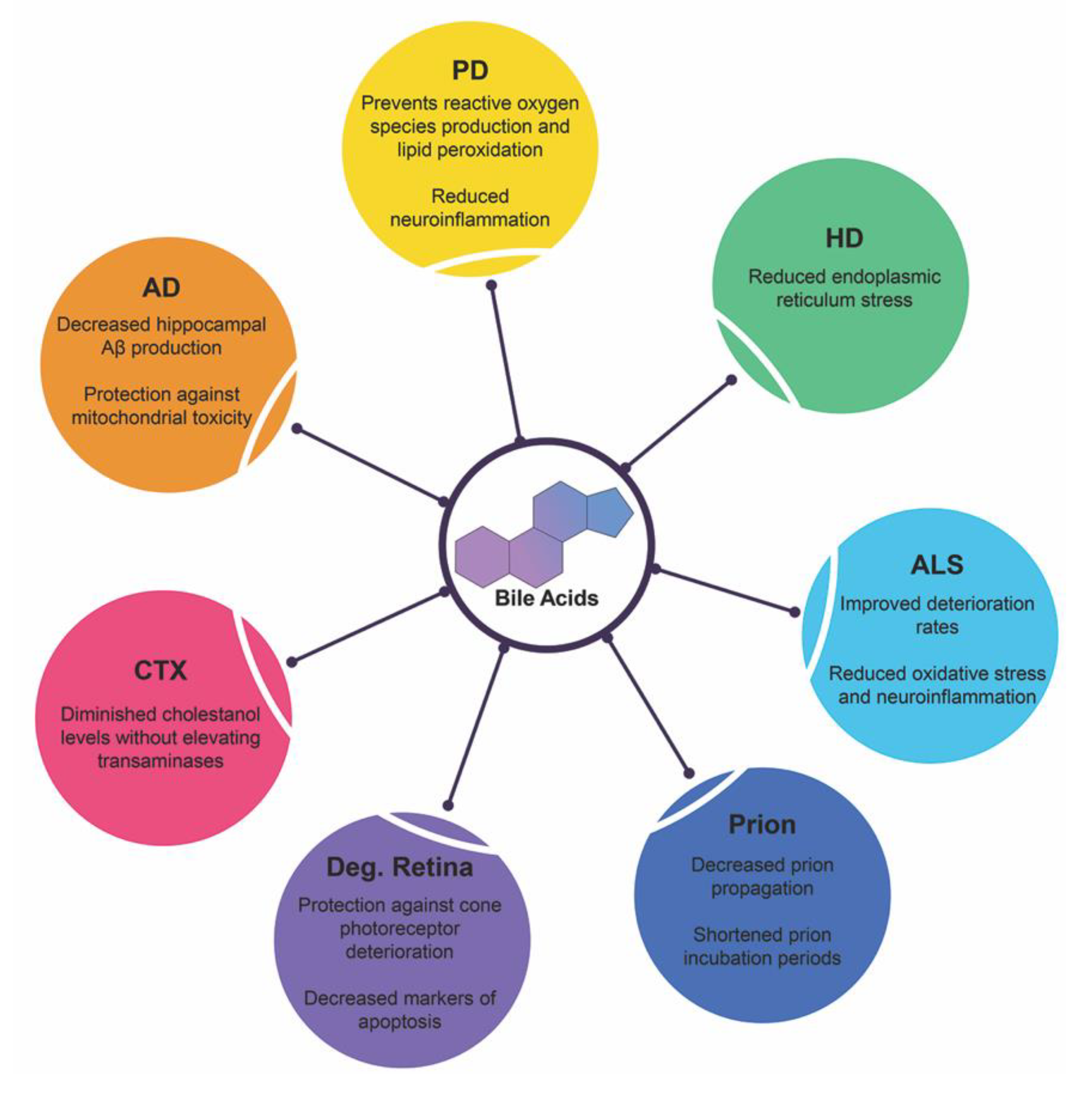
4. Bile Acids in Neurodegenerative Diseases
4.1. Alzheimer’s Disease
4.2. Parkinson’s Disease
4.3. Huntington’s Disease
4.4. Amyotrophic Lateral Sclerosis
4.5. Prion Diseases
4.6. Degenerative Retina Diseases
4.7. Cerebrotendinous Xanthomatosis
5. Neurological Disorders and Bile Acids
5.1. Multiple Sclerosis
5.2. Hepatic Encephalopathy
5.3. Miscellaneous Neurological Disorders
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6ECDCA | 6α-ethyl-chenodeoxycholic acid |
| AAV | Associated virus |
| Aβ | Amyloid β peptides |
| ABCA1 | ATP-binding cassette transporter 1 |
| AD | Alzheimer’s Disease |
| Akt | Protein kinase B |
| ALS | Amyotrophic Lateral Sclerosis |
| AMD | Age-related macular degeneration |
| ApoE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| ASBT | Apical sodium-dependent bile acid transporter |
| Bax | BCL2 associated X apoptosis regulator |
| BBB | Blood brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| BiP | Binding immunoglobulin protein |
| BSEP | Bile salt export pump |
| C9orf72 | Chromosome 9 open reading frame 72 |
| CA | Cholic acid |
| CAG | Cytosine-adenine-guanine |
| CDCA | Chenodeoxycholic acid |
| CJD | Creutzfeldt-Jakob disease |
| CCL2 | Chemokine ligand 2 |
| CPA | Cyclopiazonic acid |
| CREB | cAMP-response element-binding protein |
| CTX | Cerebrotendinous Xanthomatosis |
| CYP | Cytochrome P450 |
| DCA | Deoxycholic acid |
| Drp1 | Dynamin-related protein 1 |
| eIF2α | Eukaryotic translation initiation factor |
| FUS | Fused in sarcoma |
| FXR | Farnesoid X receptor |
| GCA | Glycocholic acid |
| GCDCA | Glycochenodeoxycholic acid |
| GR | Glucocorticoid receptor |
| GRP78 | 78-kDa glucose-regulated protein |
| GUDCA | Glycoursodeoxycholic acid |
| HD | Huntington’s disease |
| HDCA | Hyodeoxycholic acid |
| HE | Hepatic Encephalopathy |
| HPA | Hypothalamic pituitary adrenal |
| IBA1 | Ionized calcium binding adaptor molecule 1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| LCA | Lithocholic acid |
| MCA | α-Muricholic acid |
| β-MCA | β-Muricholic acid |
| MDCA | Murideoxycholic acid |
| MMP-9 | Metalloproteinase-9 |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MRP1 | Multidrug resistance-associated protein 1 |
| MRP2 | Multidrug resistance-associated protein 2 |
| MRP3 | Multidrug resistance-associated protein 3 |
| MS | Multiple Sclerosis |
| NeuN | Neuronal nuclei |
| NF-κB | Nuclear factor-κB |
| NLRP3 | NOD-like receptor family pyrin domain containing receptor 3 |
| NPHP4 | Nephrocystin-4 |
| Nrf2 | Nuclear factor erythroid 2 related factor 2 |
| NTCP | Sodium taurocholate cotransporting polypeptide |
| OATP | Organic anion transport polypeptide |
| OST | Organic solute transporter |
| PD | Parkinson’s Disease |
| PDI | Protein disulphide isomerase |
| PMS | Primary progressive Multiple Sclerosis |
| PSD95 | Postsynaptic density protein 95 |
| PXR | Pregnane X receptor |
| RD | Retinal degeneration |
| RRMS | Relapsing-remitting Multiple Sclerosis |
| ROS | Reactive oxygen species |
| RP | Retinitis pigmentosa |
| RPE | Retinal pigment epithelium |
| RPGR | Retinitis pigmentosa GTPase regulator |
| S1PR2 | Sphingosine-1-phospphate receptor |
| SOD | Superoxide dismutase |
| SPMS | Secondary progressive Multiple Sclerosis |
| TBI | Traumatic brain injury |
| TCA | Taurocholic acid |
| TCDCA | Taurochenodeoxycholic acid |
| TDP-43 | Transactive response DNA-binding protein 43 |
| TGR5 | Takeda G-protein-coupled bile acid receptor 5 |
| TLCA | Taurolithocholic acid |
| TNF-α | Tumor necrosis factor-α |
| TUDCA | Tauroursodeoxycholic acid |
| UDCA | Ursodeoxycholic acid |
| UDP | Uridine 5′-disphosphate |
| UGT | Uridine 5′-disphoasphate-glucuronosyltransferase |
| UPR | Unfolded protein response |
| VDR | Vitamin D receptor |
| XALD | X-linked adrenoleukodystrophy |
References
- Zwicker, B.L.; Agellon, L.B. Transport and biological activities of bile acids. Int. J. Biochem. Cell Biol. 2013, 45, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Parry, G.J.; Rodrigues, C.M.; Aranha, M.M.; Hilbert, S.J.; Davey, C.; Kelkar, P.; Low, W.C.; Steer, C.J. Safety, tolerability, and cerebrospinal fluid penetration of ursodeoxycholic Acid in patients with amyotrophic lateral sclerosis. Clin. Neuropharmacol. 2010, 33, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.; Sola, S.; Nan, Z.; Castro, R.E.; Ribeiro, P.S.; Low, W.C.; Steer, C.J. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc. Natl. Acad. Sci. USA 2003, 100, 6087–6092. [Google Scholar] [CrossRef] [PubMed]
- Min, J.H.; Hong, Y.H.; Sung, J.J.; Kim, S.M.; Lee, J.B.; Lee, K.W. Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: A randomized cross-over trial. J. Korean Med. Sci. 2012, 27, 200–206. [Google Scholar] [CrossRef]
- Mondelli, M.; Sicurelli, F.; Scarpini, C.; Dotti, M.T.; Federico, A. Cerebrotendinous xanthomatosis: 11-Year treatment with chenodeoxycholic acid in five patients. An electrophysiological study. J. Neurol. Sci. 2001, 190, 29–33. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- Xiang, X.; Backman, J.T.; Neuvonen, P.J.; Niemi, M. Gender, but not CYP7A1 or SLCO1B1 polymorphism, affects the fasting plasma concentrations of bile acids in human beings. Basic Clin. Pharmacol. Toxicol. 2012, 110, 245–252. [Google Scholar] [CrossRef]
- Trottier, J.; Caron, P.; Straka, R.J.; Barbier, O. Profile of serum bile acids in noncholestatic volunteers: Gender-related differences in response to fenofibrate. Clin. Pharmacol. Ther. 2011, 90, 279–286. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid metabolism in liver pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Phelps, T.; Snyder, E.; Rodriguez, E.; Child, H.; Harvey, P. The influence of biological sex and sex hormones on bile acid synthesis and cholesterol homeostasis. Biol. Sex Differ. 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2018, 16, 4–14. [Google Scholar] [CrossRef]
- Dawson, P.A. Role of the Intestinal Bile acid Transporters in Bile Acid and Drug Disposition. In Drug Tranporters; Handbook of Experimental Pharmacology, Vol. 201; Springer: Berlin/Heidelberg, Germany, 2011; pp. 169–203. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef]
- Chen, I.; Cassaro, S. Physiology, Bile Acids; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef]
- McMillin, M.; Frampton, G.; Quinn, M.; Ashfaq, S.; de los Santos 3rd, M.; Grant, S.; DeMorrow, S. Bile acid signaling is involved in the neurological decline in a murine model of acute liver failure. Am. J. Pathol. 2016, 186, 312–323. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Jiang, R.; Zhao, A.; Yan, J.; Zheng, X.; Huang, F.; Liu, X.; Panee, J.; Rajani, C.; et al. Dysregulated bile acid signaling contributes to the neurological impairment in murine models of acute and chronic liver failure. EBioMedicine 2018, 37, 294–306. [Google Scholar] [CrossRef]
- Barbier, O.; Trottier, J.; Kaeding, J.; Caron, P.; Verreault, M. Lipid-activated transcription factors control bile acid glucuronidation. Mol. Cell. Biochem. 2009, 326, 3–8. [Google Scholar] [CrossRef]
- Pauli-Magnus, C.; Meier, P.J. Hepatocellular transporters and cholestasis. J. Clin. Gastroenterol. 2005, 39, S103–S110. [Google Scholar] [CrossRef]
- Takikawa, H.; Otsuka, H.; Beppu, T.; Seyama, Y.; Yamakawa, T. Serum concentrations of bile acid glucuronides in hepatobiliary diseases. Digestion 1983, 27, 189–195. [Google Scholar] [CrossRef]
- Perreault, M.; Bialek, A.; Trottier, J.; Verreault, M.; Caron, P.; Milkiewicz, P.; Barbier, O. Role of glucuronidation for hepatic detoxification and urinary elimination of toxic bile acids during biliary obstruction. PLoS ONE 2013, 8, e80994. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, P.I.; Bock, K.W.; Burchell, B.; Guillemette, C.; Ikushiro, S.; Iyanagi, T.; Miners, J.O.; Owens, I.S.; Nebert, D.W. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet. Genom. 2005, 15, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Trottier, J.; Milkiewicz, P.; Kaeding, J.; Verreault, M.; Barbier, O. Coordinate regulation of hepatic bile acid oxidation and conjugation by nuclear receptors. Mol. Pharm. 2006, 3, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Nakajima, M.; Yamanaka, H.; Fujiwara, R.; Yokoi, T. Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab. Dispos. 2008, 36, 1461–1464. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Katoh, M.; Imai, K.; Kondo, Y.; Asai, Y.; Ikushiro, S.; Nadai, M. Expression of UGT1A subfamily in rat brain. Biopharm. Drug Dispos. 2016, 37, 314–319. [Google Scholar] [CrossRef]
- King, C.D.; Rios, G.R.; Assouline, J.A.; Tephly, T.R. Expression of UDP-glucuronosyltransferases (UGTs) 2B7 and 1A6 in the human brain and identification of 5-hydroxytryptamine as a substrate. Arch. Biochem. Biophys. 1999, 365, 156–162. [Google Scholar] [CrossRef]
- Suleman, F.G.; Abid, A.; Gradinaru, D.; Daval, J.L.; Magdalou, J.; Minn, A. Identification of the uridine diphosphate glucuronosyltransferase isoform UGT1A6 in rat brain and in primary cultures of neurons and astrocytes. Arch. Biochem. Biophys. 1998, 358, 63–67. [Google Scholar] [CrossRef]
- Gradinaru, D.; Minn, A.L.; Artur, Y.; Minn, A.; Heydel, J.M. Effect of oxidative stress on UDP-glucuronosyltransferases in rat astrocytes. Toxicol. Lett. 2012, 213, 316–324. [Google Scholar] [CrossRef]
- Togna, A.R.; Antonilli, L.; Dovizio, M.; Salemme, A.; De Carolis, L.; Togna, G.I.; Patrignani, P.; Nencini, P. In vitro morphine metabolism by rat microglia. Neuropharmacology 2013, 75, 391–398. [Google Scholar] [CrossRef]
- Bjorkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W. Cholesterol homeostasis and function in neurons of the central nervous system. Cell. Mol. Life Sci. 2003, 60, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Nieweg, K.; Schaller, H.; Pfrieger, F.W. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J. Neurochem. 2009, 109, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef]
- Gao, B.; Hagenbuch, B.; Kullak-Ublick, G.A.; Benke, D.; Aguzzi, A.; Meier, P.J. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 2000, 294, 73–79. [Google Scholar]
- McNeilly, A.D.; Macfarlane, D.P.; O’Flaherty, E.; Livingstone, D.E.; Mitic, T.; McConnell, K.M.; McKenzie, S.M.; Davies, E.; Reynolds, R.M.; Thiesson, H.C.; et al. Bile acids modulate glucocorticoid metabolism and the hypothalamic-pituitary-adrenal axis in obstructive jaundice. J. Hepatol. 2010, 52, 705–711. [Google Scholar] [CrossRef]
- McMillin, M.; Frampton, G.; Quinn, M.; Divan, A.; Grant, S.; Patel, N.; Newell-Rogers, K.; DeMorrow, S. Suppression of the HPA axis during cholestasis can be attributed to hypothalamic bile acid signaling. Mol. Endocrinol. 2015, 29, 1720–1730. [Google Scholar] [CrossRef]
- Copple, B.L.; Li, T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol. Res. 2016, 104, 9–21. [Google Scholar] [CrossRef]
- Ananthanarayanan, M.; Balasubramanian, N.; Makishima, M.; Mangelsdorf, D.J.; Suchy, F.J. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 2001, 276, 28857–28865. [Google Scholar] [CrossRef]
- Plass, J.R.; Mol, O.; Heegsma, J.; Geuken, M.; Faber, K.N.; Jansen, P.L.; Muller, M. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 2002, 35, 589–596. [Google Scholar] [CrossRef]
- Barbier, O.; Torra, I.P.; Sirvent, A.; Claudel, T.; Blanquart, C.; Duran-Sandoval, D.; Kuipers, F.; Kosykh, V.; Fruchart, J.C.; Staels, B. FXR induces the UGT2B4 enzyme in hepatocytes: A potential mechanism of negative feedback control of FXR activity. Gastroenterology 2003, 124, 1926–1940. [Google Scholar] [CrossRef]
- Yu, J.; Lo, J.L.; Huang, L.; Zhao, A.; Metzger, E.; Adams, A.; Meinke, P.T.; Wright, S.D.; Cui, J. Lithocholic acid decreases expression of bile salt export pump through farnesoid X receptor antagonist activity. J. Biol. Chem. 2002, 277, 31441–31447. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Goldberg, A.A.; Beach, A.; Davies, G.F.; Harkness, T.A.; Leblanc, A.; Titorenko, V.I. Lithocholic bile acid selectively kills neuroblastoma cells, while sparing normal neuronal cells. Oncotarget 2011, 2, 761–782. [Google Scholar] [CrossRef] [PubMed]
- Luu, T.H.; Bard, J.M.; Carbonnelle, D.; Chaillou, C.; Huvelin, J.M.; Bobin-Dubigeon, C.; Nazih, H. Lithocholic bile acid inhibits lipogenesis and induces apoptosis in breast cancer cells. Cell. Oncol. 2018, 41, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- Gohlke, H.; Schmitz, B.; Sommerfeld, A.; Reinehr, R.; Haussinger, D. α5β1-integrins are sensors for tauroursodeoxycholic acid in hepatocytes. Hepatology 2013, 57, 1117–1129. [Google Scholar] [CrossRef]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acids as metabolic regulators and nutrient sensors. Annu. Rev. Nutr. 2019, 39, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Nuclear receptors in bile acid metabolism. Drug Metab. Rev. 2013, 45, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, J.; Hu, W.; Wang, C.; Lu, X.; Tong, L.; Wu, F.; Zhang, W. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 2016, 590, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wang, T.; Lan, Y.; Yang, L.; Pan, W.; Zhu, Y.; Lv, B.; Wei, Y.; Shi, H.; Wu, H.; et al. Deletion of mouse FXR gene disturbs multiple neurotransmitter systems and alters neurobehavior. Front. Behav. Neurosci. 2015, 9, 70. [Google Scholar] [CrossRef]
- McMillin, M.; Grant, S.; Frampton, G.; Petrescu, A.D.; Kain, J.; Williams, E.; Haines, R.; Canady, L.; DeMorrow, S. FXR-Mediated cortical cholesterol accumulation contributes to the pathogenesis of type A hepatic encephalopathy. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 47–63. [Google Scholar] [CrossRef]
- Keitel, V.; Gorg, B.; Bidmon, H.J.; Zemtsova, I.; Spomer, L.; Zilles, K.; Haussinger, D. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 2010, 58, 1794–1805. [Google Scholar] [CrossRef]
- McMillin, M.; Frampton, G.; Tobin, R.; Dusio, G.; Smith, J.; Shin, H.; Newell-Rogers, K.; Grant, S.; DeMorrow, S. TGR5 signaling reduces neuroinflammation during hepatic encephalopathy. J. Neurochem. 2015, 135, 565–576. [Google Scholar] [CrossRef]
- Kempf, A.; Tews, B.; Arzt, M.E.; Weinmann, O.; Obermair, F.J.; Pernet, V.; Zagrebelsky, M.; Delekate, A.; Iobbi, C.; Zemmar, A.; et al. The sphingolipid receptor S1PR2 is a receptor for Nogo-a repressing synaptic plasticity. PLoS Biol. 2014, 12, e1001763. [Google Scholar] [CrossRef]
- McMillin, M.; Frampton, G.; Grant, S.; Khan, S.; Diocares, J.; Petrescu, A.; Wyatt, A.; Kain, J.; Jefferson, B.; DeMorrow, S. Bile Acid-Mediated Sphingosine-1-Phosphate Receptor 2 Signaling Promotes Neuroinflammation during Hepatic Encephalopathy in Mice. Front. Cell. Neurosci. 2017, 11, 191. [Google Scholar] [CrossRef]
- Lemmen, J.; Tozakidis, I.E.; Galla, H.J. Pregnane X receptor upregulates ABC-transporter Abcg2 and Abcb1 at the blood-brain barrier. Brain Res. 2013, 1491, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. RXRα, PXR and CAR xenobiotic receptors mediate the apoptotic and neurotoxic actions of nonylphenol in mouse hippocampal cells. J. Steroid Biochem. Mol. Biol. 2016, 156, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.W.; Liu, P.Y.; Josh, P.; Cui, X. Intracellular distribution of the vitamin D receptor in the brain: Comparison with classic target tissues and redistribution with development. Neuroscience 2014, 268, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the vitamin D receptor and 1 α-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Bonus, M.; Sommerfeld, A.; Qvartskhava, N.; Gorg, B.; Ludwig, B.S.; Kessler, H.; Gohlke, H.; Haussinger, D. Evidence for functional selectivity in TUDC- and norUDCA-induced signal transduction via α5β1 integrin towards choleresis. Sci. Rep. 2020, 10, 5795. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Escuin, S.; van der Flier, A.; De Arcangelis, A.; Hynes, R.O.; Georges-Labouesse, E. Integrin α5β1 is necessary for regulation of radial migration of cortical neurons during mouse brain development. Eur. J. Neurosci. 2010, 31, 399–409. [Google Scholar] [CrossRef]
- Roberts, J.; de Hoog, L.; Bix, G.J. Mice deficient in endothelial α5 integrin are profoundly resistant to experimental ischemic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 85–96. [Google Scholar] [CrossRef]
- Miura, T.; Ouchida, R.; Yoshikawa, N.; Okamoto, K.; Makino, Y.; Nakamura, T.; Morimoto, C.; Makino, I.; Tanaka, H. Functional modulation of the glucocorticoid receptor and suppression of NF-κB-dependent transcription by ursodeoxycholic acid. J. Biol. Chem. 2001, 276, 47371–47378. [Google Scholar] [CrossRef]
- Sun, X.C.; Ren, X.F.; Chen, L.; Gao, X.Q.; Xie, J.X.; Chen, W.F. Glucocorticoid receptor is involved in the neuroprotective effect of ginsenoside Rg1 against inflammation-induced dopaminergic neuronal degeneration in substantia nigra. J. Steroid. Biochem. Mol. Biol. 2016, 155, 94–103. [Google Scholar] [CrossRef]
- Sola, S.; Amaral, J.D.; Borralho, P.M.; Ramalho, R.M.; Castro, R.E.; Aranha, M.M.; Steer, C.J.; Rodrigues, C.M. Functional modulation of nuclear steroid receptors by tauroursodeoxycholic acid reduces amyloid β-peptide-induced apoptosis. Mol. Endocrinol. 2006, 20, 2292–2303. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Alzheimer’s, Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef] [PubMed]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Guell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Marksteiner, J.; Blasko, I.; Kemmler, G.; Koal, T.; Humpel, C. Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in Alzheimer’s disease. Metabolomics 2018, 14, 1. [Google Scholar] [CrossRef]
- Wu, X.; Lv, Y.G.; Du, Y.F.; Chen, F.; Reed, M.N.; Hu, M.; Suppiramaniam, V.; Tang, S.S.; Hong, H. Neuroprotective effects of INT-777 against Aβ1–42-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice. Brain Behav. Immun. 2018, 73, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ma, H.; Guo, X.; Liu, J.; Gui, T.; Gai, Z. Farnesoid X Receptor (FXR) Aggravates Amyloid-β-Triggered Apoptosis by Modulating the cAMP-Response Element-Binding Protein (CREB)/Brain-Derived Neurotrophic Factor (BDNF) Pathway In Vitro. Med. Sci. Monit. 2019, 25, 9335–9345. [Google Scholar] [CrossRef] [PubMed]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Chenodeoxycholic acid ameliorates AlCl3-induced Alzheimer’s disease neurotoxicity and cognitive deterioration via enhanced insulin signaling in rats. Molecules 2019, 24, 1992. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid β-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; Clemmens, H.; Al-Rafiah, A.R.; Al-Ofi, E.A.; Leech, V.; Bandmann, O.; Shaw, P.J.; Blackburn, D.J.; Ferraiuolo, L.; et al. Ursodeoxycholic acid improves mitochondrial function and redistributes drp1 in fibroblasts from patients with either sporadic or familial Alzheimer’s disease. J. Mol. Biol. 2018, 430, 3942–3953. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal models for Parkinson’s disease research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, P.; Seppi, K.; Poewe, W. The concept of prodromal Parkinson’s disease. J. Parkinsons Dis. 2015, 5, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Rey, N.L.; Yilmaz, A.; Kumar, P.; Madaj, Z.; Maddens, M.; Bahado-Singh, R.O.; Becker, K.; Schulz, E.; Meyerdirk, L.K.; et al. Biochemical profiling of the brain and blood metabolome in a mouse model of prodromal Parkinson’s disease reveals distinct metabolic profiles. J. Proteome Res. 2018, 17, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, S.M.; Crowley, E.K.; Brown, J.R.; O’Sullivan, O.; O’Leary, O.F.; Timmons, S.; Nolan, Y.M.; Clarke, D.J.; Hyland, N.P.; Joyce, S.A.; et al. Nigral overexpression of α-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol. Motil. 2020, 32, e13726. [Google Scholar] [CrossRef]
- Graham, S.F.; Rey, N.L.; Ugur, Z.; Yilmaz, A.; Sherman, E.; Maddens, M.; Bahado-Singh, R.O.; Becker, K.; Schulz, E.; Meyerdirk, L.K.; et al. Metabolomic profiling of bile acids in an experimental model of prodromal Parkinson’s disease. Metabolites 2018, 8, 71. [Google Scholar] [CrossRef]
- Rosa, A.I.; Duarte-Silva, S.; Silva-Fernandes, A.; Nunes, M.J.; Carvalho, A.N.; Rodrigues, E.; Gama, M.J.; Rodrigues, C.M.P.; Maciel, P.; Castro-Caldas, M. Tauroursodeoxycholic acid improves motor symptoms in a mouse model of Parkinson’s disease. Mol. Neurobiol. 2018, 55, 9139–9155. [Google Scholar] [CrossRef]
- Moreira, S.; Fonseca, I.; Nunes, M.J.; Rosa, A.; Lemos, L.; Rodrigues, E.; Carvalho, A.N.; Outeiro, T.F.; Rodrigues, C.M.P.; Gama, M.J.; et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 2017, 295, 77–87. [Google Scholar] [CrossRef]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic acid ameliorates apoptotic cascade in the Rotenone model of Parkinson’s disease: Modulation of mitochondrial perturbations. Mol. Neurobiol. 2016, 53, 810–817. [Google Scholar] [CrossRef]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Boussicault, L.; Alves, S.; Lamaziere, A.; Planques, A.; Heck, N.; Moumne, L.; Despres, G.; Bolte, S.; Hu, A.; Pages, C.; et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016, 139, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Di Pardo, A.; Amico, E.; Basit, A.; Armirotti, A.; Joshi, P.; Neely, M.D.; Vuono, R.; Castaldo, S.; Digilio, A.F.; Scalabri, F.; et al. Defective Sphingosine-1-phosphate metabolism is a druggable target in Huntington’s disease. Sci. Rep. 2017, 7, 5280. [Google Scholar] [CrossRef] [PubMed]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, G.; Kaplan, A.; Gaschler, M.M.; Zhang, X.; Hou, Z.; Jiang, M.; Zott, R.; Cremers, S.; Stockwell, B.R.; et al. Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Upagupta, C.; Carlisle, R.E.; Dickhout, J.G. Analysis of the potency of various low molecular weight chemical chaperones to prevent protein aggregation. Biochem. Biophys. Res. Commun. 2017, 486, 163–170. [Google Scholar] [CrossRef]
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar]
- Philips, T.; Rothstein, J.D. Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef]
- Elia, A.E.; Lalli, S.; Monsurro, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 45–52. [Google Scholar] [CrossRef]
- Vaz, A.R.; Cunha, C.; Gomes, C.; Schmucki, N.; Barbosa, M.; Brites, D. Glycoursodeoxycholic acid reduces matrix metalloproteinase-9 and caspase-9 activation in a cellular model of superoxide dismutase-1 neurodegeneration. Mol. Neurobiol. 2015, 51, 864–877. [Google Scholar] [CrossRef]
- Thams, S.; Lowry, E.R.; Larraufie, M.H.; Spiller, K.J.; Li, H.; Williams, D.J.; Hoang, P.; Jiang, E.; Williams, L.A.; Sandoe, J.; et al. A stem cell-based screening platform identifies compounds that desensitize motor neurons to endoplasmic reticulum stress. Mol. Ther. 2019, 27, 87–101. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Bartz, J.C.; Glatzel, M. Cellular and molecular mechanisms of prion disease. Annu. Rev. Pathol. 2019, 14, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Diack, A.B.; Alibhai, J.D.; Manson, J.C. Gene targeted transgenic mouse models in prion research. Prog. Mol. Biol. Transl. Sci. 2017, 150, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Ladner-Keay, C.L.; Ross, L.; Perez-Pineiro, R.; Zhang, L.; Bjorndahl, T.C.; Cashman, N.; Wishart, D.S. A simple in vitro assay for assessing the efficacy, mechanisms and kinetics of anti-prion fibril compounds. Prion 2018, 12, 280–300. [Google Scholar] [CrossRef]
- Cortez, L.M.; Campeau, J.; Norman, G.; Kalayil, M.; Van der Merwe, J.; McKenzie, D.; Sim, V.L. Bile acids reduce prion conversion, reduce neuronal loss, and prolong male survival in models of prion disease. J. Virol. 2015, 89, 7660–7672. [Google Scholar] [CrossRef]
- Norman, G.; Campeau, J.; Sim, V.L. High dose and delayed treatment with bile acids ineffective in RML prion-infected mice. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Cuenca, N.; Fernandez-Sanchez, L.; Campello, L.; Maneu, V.; De la Villa, P.; Lax, P.; Pinilla, I. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog. Retin. Eye Res. 2014, 43, 17–75. [Google Scholar] [CrossRef]
- Veleri, S.; Lazar, C.H.; Chang, B.; Sieving, P.A.; Banin, E.; Swaroop, A. Biology and therapy of inherited retinal degenerative disease: Insights from mouse models. Dis. Model. Mech. 2015, 8, 109–129. [Google Scholar] [CrossRef]
- Lobysheva, E.; Taylor, C.M.; Marshall, G.R.; Kisselev, O.G. Tauroursodeoxycholic acid binds to the G-protein site on light activated rhodopsin. Exp. Eye Res. 2018, 170, 51–57. [Google Scholar] [CrossRef]
- Lawson, E.C.; Bhatia, S.K.; Han, M.K.; Aung, M.H.; Ciavatta, V.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic Acid Protects Retinal Function and Structure in rd1 Mice. Adv Exp Med Biol. 2016, 854, 431–436. [Google Scholar] [CrossRef]
- Tao, Y.; Dong, X.; Lu, X.; Qu, Y.; Wang, C.; Peng, G.; Zhang, J. Subcutaneous delivery of tauroursodeoxycholic acid rescues the cone photoreceptors in degenerative retina: A promising therapeutic molecule for retinopathy. Biomed. Pharmacother. 2019, 117, 109021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shahani, U.; Reilly, J.; Shu, X. Disease mechanisms and neuroprotection by tauroursodeoxycholic acid in Rpgr knockout mice. J. Cell. Physiol. 2019, 234, 18801–18812. [Google Scholar] [CrossRef] [PubMed]
- Warden, C.; Barnett, J.M.; Brantley, M.A., Jr. Taurocholic acid inhibits features of age-related macular degeneration in vitro. Exp. Eye Res. 2020, 193, 107974. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Chen, G.; Cao, X.; Zhang, Y. Cerebrotendinous xanthomatosis: A comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet. J. Rare Dis. 2014, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Bavner, A.; Shafaati, M.; Hansson, M.; Olin, M.; Shpitzen, S.; Meiner, V.; Leitersdorf, E.; Bjorkhem, I. On the mechanism of accumulation of cholestanol in the brain of mice with a disruption of sterol 27-hydroxylase. J. Lipid Res. 2010, 51, 2722–2730. [Google Scholar] [CrossRef]
- Mandia, D.; Chaussenot, A.; Besson, G.; Lamari, F.; Castelnovo, G.; Curot, J.; Duval, F.; Giral, P.; Lecerf, J.M.; Roland, D.; et al. Cholic acid as a treatment for cerebrotendinous xanthomatosis in adults. J. Neurol. 2019, 266, 2043–2050. [Google Scholar] [CrossRef]
- Mignarri, A.; Magni, A.; Del Puppo, M.; Gallus, G.N.; Bjorkhem, I.; Federico, A.; Dotti, M.T. Evaluation of cholesterol metabolism in cerebrotendinous xanthomatosis. J. Inherit. Metab. Dis. 2016, 39, 75–83. [Google Scholar] [CrossRef]
- Diaz, C.; Zarco, L.A.; Rivera, D.M. Highly active multiple sclerosis: An update. Mult. Scler. Relat. Disord. 2019, 30, 215–224. [Google Scholar] [CrossRef]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of Multiple Sclerosis. Eur. J. Pharmacol. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Crick, P.J.; Griffiths, W.J.; Zhang, J.; Beibel, M.; Abdel-Khalik, J.; Kuhle, J.; Sailer, A.W.; Wang, Y. Reduced plasma levels of 25-hydroxycholesterol and increased cerebrospinal fluid levels of bile acid precursors in multiple sclerosis patients. Mol. Neurobiol. 2017, 54, 8009–8020. [Google Scholar] [CrossRef]
- Bhargava, P.; Smith, M.D.; Mische, L.; Harrington, E.; Fitzgerald, K.C.; Martin, K.; Kim, S.; Reyes, A.A.; Gonzalez-Cardona, J.; Volsko, C.; et al. Bile acid metabolism is altered in multiple sclerosis and supplementation ameliorates neuroinflammation. J. Clin. Investig. 2020, 130, 3467–3482. [Google Scholar] [CrossRef] [PubMed]
- Hucke, S.; Herold, M.; Liebmann, M.; Freise, N.; Lindner, M.; Fleck, A.K.; Zenker, S.; Thiebes, S.; Fernandez-Orth, J.; Buck, D.; et al. The farnesoid-X-receptor in myeloid cells controls CNS autoimmunity in an IL-10-dependent fashion. Acta Neuropathol. 2016, 132, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.P.; Steinman, L. Obeticholic acid, a synthetic bile acid agonist of the farnesoid X receptor, attenuates experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2016, 113, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Kandiah, P.A.; Kumar, G. Hepatic encephalopathy—The old and the new. Crit. Care Clin. 2016, 32, 311–329. [Google Scholar] [CrossRef]
- Liere, V.; Sandhu, G.; DeMorrow, S. Recent advances in hepatic encephalopathy. F1000Research 2017, 6, 1637. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Norenberg, M.D.; Felipo, V.; Ferenci, P.; Albrecht, J.; Blei, A.T.; Members of the ISHEN Commission on Experimental Models of HE. Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int. 2009, 29, 783–788. [Google Scholar] [CrossRef]
- Weiss, N.; Barbier Saint Hilaire, P.; Colsch, B.; Isnard, F.; Attala, S.; Schaefer, A.; Amador, M.D.; Rudler, M.; Lamari, F.; Sedel, F.; et al. Cerebrospinal fluid metabolomics highlights dysregulation of energy metabolism in overt hepatic encephalopathy. J. Hepatol. 2016, 65, 1120–1130. [Google Scholar] [CrossRef]
- Quinn, M.; McMillin, M.; Galindo, C.; Frampton, G.; Pae, H.Y.; DeMorrow, S. Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig. Liver Dis. 2014, 46, 527–534. [Google Scholar] [CrossRef]
- Karababa, A.; Groos-Sahr, K.; Albrecht, U.; Keitel, V.; Shafigullina, A.; Gorg, B.; Haussinger, D. Ammonia attenuates LPS-induced upregulation of pro-inflammatory cytokine mRNA in co-cultured astrocytes and microglia. Neurochem. Res. 2017, 42, 737–749. [Google Scholar] [CrossRef]
- McMillin, M.; Frampton, G.; Thompson, M.; Galindo, C.; Standeford, H.; Whittington, E.; Alpini, G.; DeMorrow, S. Neuronal CCL2 is upregulated during hepatic encephalopathy and contributes to microglia activation and neurological decline. J. Neuroinflamm. 2014, 11, 121. [Google Scholar] [CrossRef]
- Wang, K.K.; Yang, Z.; Zhu, T.; Shi, Y.; Rubenstein, R.; Tyndall, J.A.; Manley, G.T. An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert Rev. Mol. Diagn. 2018, 18, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gu, G.; Wang, J.; Chai, Y.; Fan, Y.; Yang, M.; Xu, X.; Gao, W.; Li, F.; Yin, D.; et al. Administration of tauroursodeoxycholic acid attenuates early brain injury via akt pathway activation. Front. Cell. Neurosci. 2017, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Nizamutdinov, D.; DeMorrow, S.; McMillin, M.; Kain, J.; Mukherjee, S.; Zeitouni, S.; Frampton, G.; Bricker, P.C.; Hurst, J.; Shapiro, L.A. Hepatic alterations are accompanied by changes to bile acid transporter-expressing neurons in the hypothalamus after traumatic brain injury. Sci. Rep. 2017, 7, 40112. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Forss-Petter, S.; Eichler, F.S. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie 2014, 98, 135–142. [Google Scholar] [CrossRef]
- Platek, T.; Orso, E.; Zapala, B.; Polus, A.; Kiec-Wilk, B.; Piwowar, M.; Chojnacka, M.; Cialowicz, U.; Malczewska-Malec, M.; Schmitz, G.; et al. Case report of dysregulation of primary bile acid synthesis in a family with X-linked adrenoleukodystrophy. Medicine. 2018, 97, e13353. [Google Scholar] [CrossRef]
- Launay, N.; Ruiz, M.; Grau, L.; Ortega, F.J.; Ilieva, E.V.; Martinez, J.J.; Galea, E.; Ferrer, I.; Knecht, E.; Pujol, A.; et al. Tauroursodeoxycholic bile acid arrests axonal degeneration by inhibiting the unfolded protein response in X-linked adrenoleukodystrophy. Acta Neuropathol. 2017, 133, 283–301. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Bile Acid Ligands | Cellular Localization | Expression/Functionality | References |
|---|---|---|---|---|
| FXR | CDCA, CA, DCA, LCA | Cortical neurons | Nuclear and cytoplasmic expression in cortical neurons; transcriptional activity via SHP activation. FXR deletion elevates cerebellar neurotransmitter concentrations. FXR modulates cholesterol metabolism in a rodent model of type A hepatic encephalopathy. | [56,57,58] |
| TGR5 | LCA, DCA, CDCA, CA | Neurons, astrocytes, microglia | Response to neurosteroids resulting in increased intracellular cAMP. TGR5 signaling is neuroprotective and diminishes inflammation against CCL2 in a rodent model of type A hepatic encephalopathy | [59,60] |
| S1P2R | TCA, GCA, TDCA, GDCA, TUDCA | Cortical neurons, microglia, hippocampal pyramidal cells, retinal ganglion cells | Mediates synaptic neuroplasticity, repair and neurite outgrowth. TCA activation promotes inflammation in a type A rodent model of hepatic encephalopathy | [61,62] |
| PXR | LCA | Brain endothelial cells, hippocampal neurons | BBB regulation via ABC-transporters, nonyphenol toxicity activates PXR-mediated apoptosis and neurotoxicity | [63,64] |
| VDR | LCA | Neurons, glia | Location of VDR indicates involvement with neurosteroids, confirmation of nuclear location | [65,66] |
| α5β1 integrin | TUDCA, norUDCA (UDCA homolog) | Cortical neurons, brain endothelial cells | Regulates neural morphology and migration during development, α5 influence BBB permeability | [67,68,69] |
| GR | UDCA, TCA, GCDCA, TUDCA | Neurons, microglia, cortical neurons | UDCA-bound GR modulates NF-κB-dependent transcription, GR-signaling in ginseng has protective implications in neurodegenerative models, GR-mediated HPA axis suppression is induced via injection of bile acids, GR attenuates amyloid-beta-induced apoptosis in cortical neurons through TUDCA | [39,70,71,72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grant, S.M.; DeMorrow, S. Bile Acid Signaling in Neurodegenerative and Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 5982. https://doi.org/10.3390/ijms21175982
Grant SM, DeMorrow S. Bile Acid Signaling in Neurodegenerative and Neurological Disorders. International Journal of Molecular Sciences. 2020; 21(17):5982. https://doi.org/10.3390/ijms21175982
Chicago/Turabian StyleGrant, Stephanie M., and Sharon DeMorrow. 2020. "Bile Acid Signaling in Neurodegenerative and Neurological Disorders" International Journal of Molecular Sciences 21, no. 17: 5982. https://doi.org/10.3390/ijms21175982
APA StyleGrant, S. M., & DeMorrow, S. (2020). Bile Acid Signaling in Neurodegenerative and Neurological Disorders. International Journal of Molecular Sciences, 21(17), 5982. https://doi.org/10.3390/ijms21175982