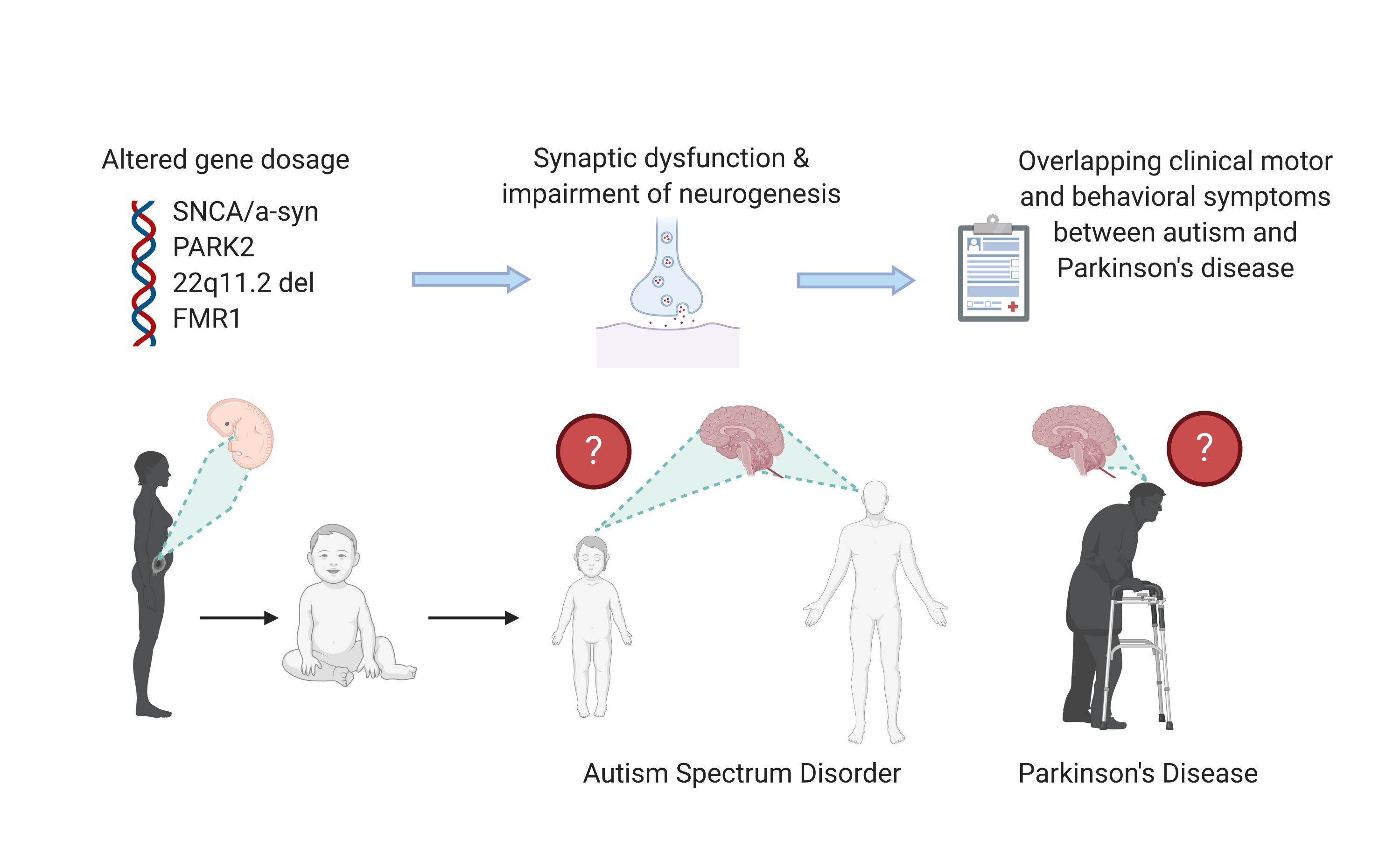
The Role of Alpha-Synuclein and Other Parkinson’s Genes in Neurodevelopmental and Neurodegenerative Disorders





Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction of Clinical Aspects of Parkinson’s Disease (PD) and Autism Spectrum Disorders (ASDs)
2. Overlapping Clinical Motor and Behavioral Phenomenology between ASD and PD
3. Genes Linked to Neurodegenerative Diseases Also Play a Role in Neurodevelopmental Disorders
3.1. PARK2 Copy Number Variants in PD and ASD and its Substrate GPR37 Are Linked to Autism
3.2. 22q11.2 Deletion/Di George Syndrome Linked to Parkinsonism
3.3. FMR1 Intermediate CGG Repeat Expansion Causes Autism and FXTAS/Parkinsonism
3.4. SNCA Genomic Region (4q22.1) Is a Multiplication/Deletion Syndrome for PD and ASD
3.5. Four PD-Associated Genes Are Linked with Neurodevelopmental Disorders
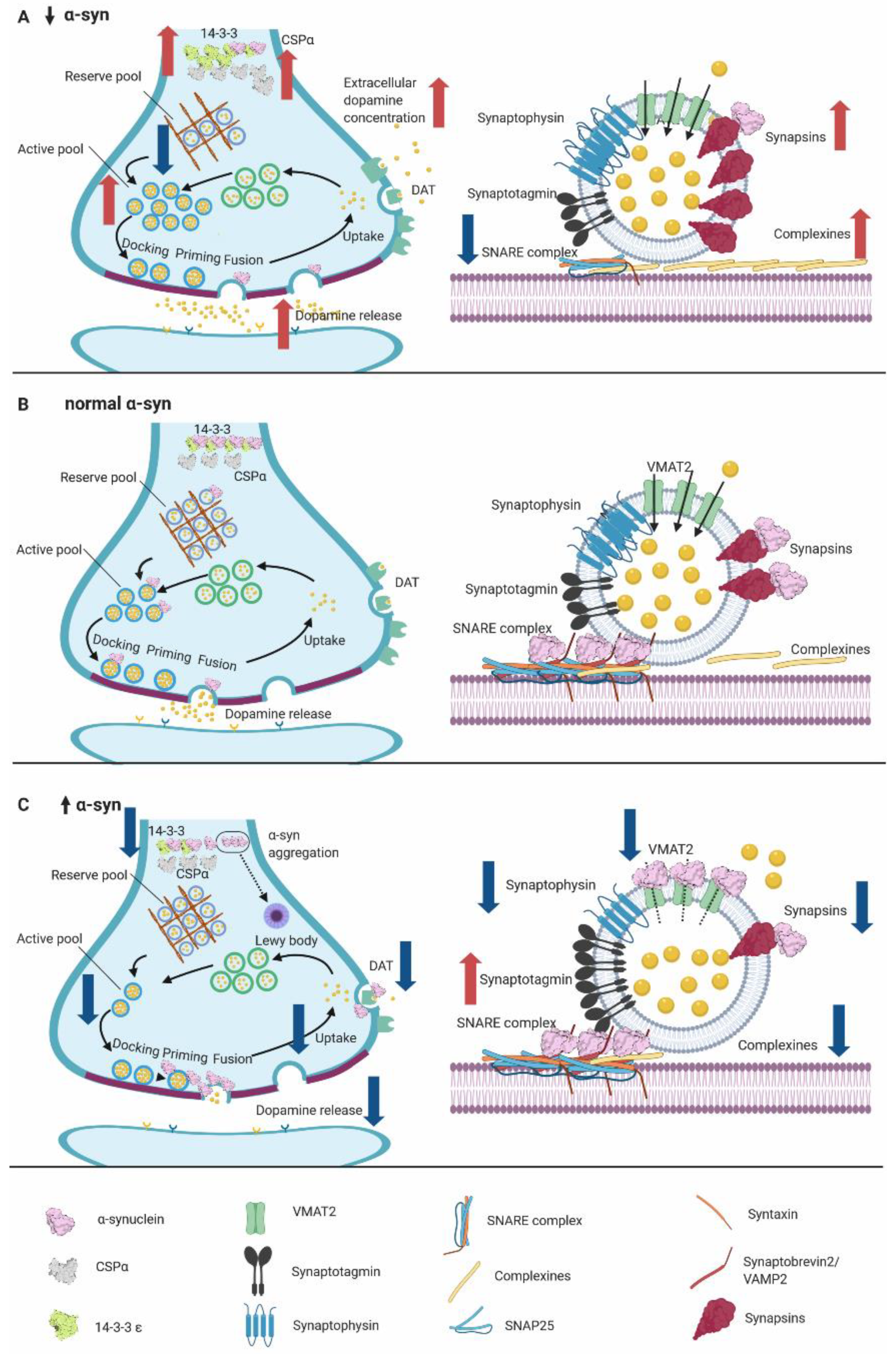
4. Physiological Function of α-syn
5. α-syn Knockout Mouse Models
5.1. α-Syn KO Model Accelerates the Recovery of Synaptic Vesicle Release
5.2. Double Synuclein KO Models Show Compensatory Increase of β- or γ-Syn
5.3. Triple SYN-KO Models Display Smaller Synapses
5.4. Syn-KOs Have Learning and Memory Deficits, Hyperdopaminergic-Like Behavior, and Resistance to MPTP
5.5. Overall Findings of α-Syn Murine Models
6. α-Syn Expression in the Developing Brain
6.1. α-Syn in Murine Development
6.2. α-Syn in Human Neurodevelopment
6.3. α-Syn Overexpression or Lack of α-Syn Impairs Adult Neurogenesis
7. Key Concepts and Future Directions: Synaptic Dysregulation and Impaired Neurogenesis Are Common Links between LBD and ASD
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine, serotonin |
| ADHD | Attention deficit hyperactivity disorder |
| ACMG | American College of Medical Genetics |
| AGP | Autism Genome Project |
| ArgK | Arginine kinase |
| ASD | Autism spectrum disorder |
| CGH | Comparative genomic hybridization |
| CNV | Copy number variant |
| COMT | Catechol-O-methyltransferase |
| CSPα | Cysteine-string protein alpha |
| DAT | Dopamine transporter, SLC6A3 |
| DLB | Dementia with Lewy bodies |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| DRG | Dorsal root ganglia |
| ENT | Entopeduncular nucleus |
| FC | Frontal cortex |
| FMR1 | Fragile X mental retardation 1, FMRP Translational Regulator 1 |
| FXPOI | Fragile X-associated primary ovarian insufficiency |
| FXS | Fragile X syndrome |
| FXTAS | Fragile X-associated tremor ataxia syndrome |
| GP | Globus pallidus |
| GPR37 | Parkin-associated endothelin-receptor like receptor, Pael-R |
| GWAS | Genome-wide association studies |
| HC | Hippocampus |
| HVA | Homovanillic acid, catecholamine metabolite |
| ICB | Impulse control behavior |
| IHC | Immunohistochemistry |
| KO | Knock-out |
| LBD | Lewy body disease |
| LCR | Low copy repeat |
| LDLR | Low-density lipoprotein receptor |
| L-Dopa | Levodopa, L-3,4-dihydroxyphenylalanine |
| LRP6 | Related protein 6 |
| MECP 2 | Methyl-CpG Binding Protein 2 |
| Mmrn1 | Multimerin 1 |
| MPTP | Methyl-phenyl-tetrahydropyridine |
| MSA | Multiple system atrophy |
| mTOR | Mammalian target of rapamycin |
| NAHR | Non-allelic homologous recombination |
| nDA | Dopaminergic nigral neurons |
| NLGN | Neuroligin |
| NPC | Neural precursors cells |
| NRXN | Neurexin |
| OCD | Obsessive-compulsive disorders |
| PARK2 | Parkin RBR E3 ubiquitin protein ligase, PRKN |
| PC | Posterior cortex |
| PD | Parkinson’s disease |
| PDD | Parkinson’s disease dementia |
| PKM 2 | Pyruvate kinase isozymes M2 |
| PSP | Progressive supranuclear palsy |
| RdhB | Reductive dehalogenase anchoring protein |
| SHANK | Multiple Ankyrin Repeat Domains |
| SN | Substantia nigra |
| SNARE | Soluble N-ethylmaleimide sensitive factor attachment protein receptor proteins |
| SNCA | Alpha-synuclein gene |
| SNP | Single nucleotide polymorphism |
| SNpc | Substantia nigra pars compacta |
| SSV | Small structural variants |
| SYT11 | Synaptotagmin XI |
| TH | Tyrosine hydroxylase |
| TSC1/2 | Tuberous Sclerosis 1/2 Protein |
| UCSC | University of California Santa Cruz |
| VAMP2 | Vesicle-associated membrane protein-2, synaptobrevin-2 |
| VEN | Von Economo neurons |
| VMAT2 | Vesicular monoamine transporter |
| VPS35,VPS4 | Vacuolar protein sorting-associated protein 35 and 4 |
| VTA | Ventral tegmental area |
| α-syn | Alpha-synuclein protein |
| α-Syn-KO | Alpha-synuclein knockout model |
| β-syn | Beta-synuclein |
| γ-syn | Gamma-synuclein |
References
- Balestrino, R.; Schapira, A.H.V. Parkinson Disease. Eur. J. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Ahn, Y.D.; Kim, W.S.; Shin, C.M.; Jeong, S.J.; Song, Y.S.; Bae, Y.J.; Kim, J.M. Psychiatric Manifestation in Patients with Parkinson’s Disease. J. Korean Med. Sci. 2018, 33, e300. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.Q.; Shao, S.M.; Zheng, Y.Y.; Fu, F.W.; Zheng, G.Q.; Liu, C.F. Sympathetic skin response and heart rate variability in predicting autonomic disorders in patients with Parkinson disease. Medicine (Baltimore) 2017, 96, e6523. [Google Scholar] [CrossRef] [PubMed]
- Moisan, F.; Kab, S.; Mohamed, F.; Canonico, M.; Le Guern, M.; Quintin, C.; Carcaillon, L.; Nicolau, J.; Duport, N.; Singh-Manoux, A.; et al. Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Schüle, B.; Rees, L.; Nichols, R.J.; Barlow, C. Multisystem Lewy body disease and the other parkinsonian disorders. Nat. Genet. 2015, 47, 1378–1384. [Google Scholar] [CrossRef]
- Ghebremedhin, E.; Del Tredici, K.; Langston, J.W.; Braak, H. Diminished tyrosine hydroxylase immunoreactivity in the cardiac conduction system and myocardium in Parkinson’s disease: An anatomical study. Acta. Neuropathol. 2009, 118, 777–784. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K. Alpha-Synuclein and Lewy pathology in Parkinson’s disease. Curr. Opin. Neurol. 2015, 28, 375–381. [Google Scholar] [CrossRef]
- Gomperts, S.N. Lewy Body Dementias: Dementia With Lewy Bodies and Parkinson Disease Dementia. Continuum (Minneap Minn) 2016, 22, 435–463. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 2009, 201, 1–119. [Google Scholar]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P. Environmental risk factors and Parkinson’s disease: An umbrella review of meta-analyses. Parkinsonism Relat. Disord. 2016, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Delamarre, A.; Meissner, W.G. Epidemiology, environmental risk factors and genetics of Parkinson’s disease. Presse. Med. 2017, 46, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Klein, C.; Schneider, S.A.; Lang, A.E. Hereditary parkinsonism: Parkinson disease look-alikes--an algorithm for clinicians to “PARK” genes and beyond. Mov. Disord. 2009, 24, 2042–2058. [Google Scholar] [CrossRef]
- Singleton, A.; Hardy, J. Progress in the genetic analysis of Parkinson’s disease. Hum. Mol. Genet. 2019, 28, R215–R218. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharm. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Tanner, C.M. Advances in environmental epidemiology. Mov. Disord. 2010, 25 (Suppl. 1), S58–S62. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci. Bull 2017, 33, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Emberti Gialloreti, L.; Curatolo, P. Autism Spectrum Disorder: Why Do We Know So Little? Front. Neurol. 2018, 9, 670. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Huguet, G.; Ey, E.; Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genom. Hum. Genet. 2013, 14, 191–213. [Google Scholar] [CrossRef]
- Lange, N.; Travers, B.G.; Bigler, E.D.; Prigge, M.B.; Froehlich, A.L.; Nielsen, J.A.; Cariello, A.N.; Zielinski, B.A.; Anderson, J.S.; Fletcher, P.T.; et al. Longitudinal volumetric brain changes in autism spectrum disorder ages 6-35 years. Autism Res. 2015, 8, 82–93. [Google Scholar] [CrossRef]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef]
- Allman, J.M.; Tetreault, N.A.; Hakeem, A.Y.; Manaye, K.F.; Semendeferi, K.; Erwin, J.M.; Park, S.; Goubert, V.; Hof, P.R. The von Economo neurons in the frontoinsular and anterior cingulate cortex. Ann. N. Y. Acad. Sci. 2011, 1225, 59–71. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta. Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Ecker, C.; Schmeisser, M.J.; Loth, E.; Murphy, D.G. Neuroanatomy and Neuropathology of Autism Spectrum Disorder in Humans. Adv. Anat. Embryol. Cell Biol. 2017, 224, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Shen, J.; Lincoln, S.; Miller, D.T. Advances in Genetic Discovery and Implications for Counseling of Patients and Families with Autism Spectrum Disorders. Curr. Genet. Med. Rep. 2014, 2, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Kleijer, K.T.E.; Huguet, G.; Tastet, J.; Bourgeron, T.; Burbach, J.P.H. Anatomy and Cell Biology of Autism Spectrum Disorder: Lessons from Human Genetics. Adv. Anat. Embryol. Cell Biol. 2017, 224, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Grabrucker, A.M. Environmental factors in autism. Front. Psychiatry 2012, 3, 118. [Google Scholar] [CrossRef]
- Subramanian, K.; Brandenburg, C.; Orsati, F.; Soghomonian, J.J.; Hussman, J.P.; Blatt, G.J. Basal ganglia and autism—a translational perspective. Autism Res. 2017, 10, 1751–1775. [Google Scholar] [CrossRef]
- Paval, D. A Dopamine Hypothesis of Autism Spectrum Disorder. Dev. Neurosci. 2017, 39, 355–360. [Google Scholar] [CrossRef]
- Fineberg, N.A.; Potenza, M.N.; Chamberlain, S.R.; Berlin, H.A.; Menzies, L.; Bechara, A.; Sahakian, B.J.; Robbins, T.W.; Bullmore, E.T.; Hollander, E. Probing compulsive and impulsive behaviors, from animal models to endophenotypes: A narrative review. Neuropsychopharmacology 2010, 35, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Hollander, E.; Wang, A.T.; Braun, A.; Marsh, L. Neurological considerations: Autism and Parkinson’s disease. Psychiatry Res. 2009, 170, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Starkstein, S.; Gellar, S.; Parlier, M.; Payne, L.; Piven, J. High rates of parkinsonism in adults with autism. J. Neurodev. Disord. 2015, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Strang, J.F.; Kenworthy, L.; Daniolos, P.; Case, L.; Wills, M.C.; Martin, A.; Wallace, G.L. Depression and Anxiety Symptoms in Children and Adolescents with Autism Spectrum Disorders without Intellectual Disability. Res. Autism Spectr. Disord. 2012, 6, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Postorino, V.; Kerns, C.M.; Vivanti, G.; Bradshaw, J.; Siracusano, M.; Mazzone, L. Anxiety Disorders and Obsessive-Compulsive Disorder in Individuals with Autism Spectrum Disorder. Curr. Psychiatry Rep. 2017, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Wijnhoven, L.A.; Niels-Kessels, H.; Creemers, D.H.; Vermulst, A.A.; Otten, R.; Engels, R.C. Prevalence of comorbid depressive symptoms and suicidal ideation in children with autism spectrum disorder and elevated anxiety symptoms. J. Child Adolesc. Ment. Health 2019, 31, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Pezzimenti, F.; Han, G.T.; Vasa, R.A.; Gotham, K. Depression in Youth with Autism Spectrum Disorder. Child Adolesc. Psychiatr. Clin. N. Am. 2019, 28, 397–409. [Google Scholar] [CrossRef]
- Dell’Osso, L.; Carpita, B.; Muti, D.; Morelli, V.; Salarpi, G.; Salerni, A.; Scotto, J.; Massimetti, G.; Gesi, C.; Ballerio, M.; et al. Mood symptoms and suicidality across the autism spectrum. Compr. Psychiatry 2019, 91, 34–38. [Google Scholar] [CrossRef]
- Rai, D.; Heuvelman, H.; Dalman, C.; Culpin, I.; Lundberg, M.; Carpenter, P.; Magnusson, C. Association Between Autism Spectrum Disorders With or Without Intellectual Disability and Depression in Young Adulthood. Jama. Netw. Open 2018, 1, e181465. [Google Scholar] [CrossRef]
- Hermanowicz, N.; Jones, S.A.; Hauser, R.A. Impact of non-motor symptoms in Parkinson’s disease: A PMDAlliance survey. Neuropsychiatr. Dis. Treat. 2019, 15, 2205–2212. [Google Scholar] [CrossRef]
- Chuquilin-Arista, F.; Alvarez-Avellon, T.; Menendez-Gonzalez, M. Prevalence of Depression and Anxiety in Parkinson Disease and Impact on Quality of Life: A Community-Based Study in Spain. J. Geriatr. Psychiatry Neurol. 2019, 891988719874130. [Google Scholar] [CrossRef] [PubMed]
- Poletti, M.; Lucetti, C.; Del Dotto, P.; Berti, C.; Logi, C.; Bonuccelli, U. Relationship between neuropsychiatric symptoms and cognitive performance in de novo Parkinson’s disease. J. Neuropsychiatry Clin. Neurosci. 2012, 24, E22–E23. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Luca, A.; Raciti, L.; Contrafatto, D.; Bruno, E.; Dibilio, V.; Sciacca, G.; Mostile, G.; Petralia, A.; Zappia, M. Obsessive compulsive personality disorder and Parkinson’s disease. PLoS ONE 2013, 8, e54822. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Barone, P.; Bonuccelli, U.; Annoni, K.; Asgharnejad, M.; Stanzione, P. ICARUS study: Prevalence and clinical features of impulse control disorders in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.M.; Aldinio, V. Impulse Control Disorders in Parkinson’s Disease. A Brief and Comprehensive Review. Front. Neurol. 2019, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Jeon, B.; Koh, S.B.; Yoon, W.T.; Lee, H.W.; Kwon, O.D.; Kim, J.W.; Kim, J.M.; Ma, H.I.; Kim, H.T.; et al. Behavioural and trait changes in parkinsonian patients with impulse control disorder after switching from dopamine agonist to levodopa therapy: Results of REIN-PD trial. J Neurol. Neurosurg. Psychiatry 2019, 90, 30–37. [Google Scholar] [CrossRef]
- Oudkerk, M.; Esselink, R.A.J.; Kan, C.C.; Tendolkar, I.; van Beek, M.H.C.T. Impact of Comorbid Autism Spectrum Disorder in an Individual with Idiopathic Young-Onset Parkinson’s Disease. Adv. Neurodev. Disord. 2019, 3, 91–94. [Google Scholar] [CrossRef]
- Sriwimol, W.; Limprasert, P. Significant Changes in Plasma Alpha-Synuclein and Beta-Synuclein Levels in Male Children with Autism Spectrum Disorder. Biomed. Res. Int. 2018, 2018, 4503871. [Google Scholar] [CrossRef]
- Piper, D.A.; Sastre, D.; Schüle, B. Advancing Stem Cell Models of Alpha-Synuclein Gene Regulation in Neurodegenerative Disease. Front. Neurosci. 2018, 12, 199. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Hedrich, K.; Eskelson, C.; Wilmot, B.; Marder, K.; Harris, J.; Garrels, J.; Meija-Santana, H.; Vieregge, P.; Jacobs, H.; Bressman, S.B.; et al. Distribution, type, and origin of Parkin mutations: Review and case studies. Mov. Disord. 2004, 19, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Denison, S.R.; Wang, F.; Becker, N.A.; Schüle, B.; Kock, N.; Phillips, L.A.; Klein, C.; Smith, D.I. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 2003, 22, 8370–8378. [Google Scholar] [CrossRef] [PubMed]
- Schüle, B.; Byrne, C.; Rees, L.; Langston, J.W. Is PARKIN parkinsonism a cancer predisposition syndrome? Neurol. Genet. 2015, 1, e31. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Denison, S.; Lai, J.P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; et al. Parkin gene alterations in hepatocellular carcinoma. Genes Chromosomes Cancer 2004, 40, 85–96. [Google Scholar] [CrossRef]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 2010, 42, 77–82. [Google Scholar] [CrossRef]
- Scheuerle, A.; Wilson, K. PARK2 copy number aberrations in two children presenting with autism spectrum disorder: Further support of an association and possible evidence for a new microdeletion/microduplication syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156B, 413–420. [Google Scholar] [CrossRef]
- Mariani, M.; Crosti, F.; Redaelli, S.; Fossati, C.; Piras, R.; Biondi, A.; Dalpra, L.; Selicorni, A. Partial duplication of the PARK2 gene in a child with developmental delay and her normal mother: A second report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162B, 485–486. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef]
- Yin, C.L.; Chen, H.I.; Li, L.H.; Chien, Y.L.; Liao, H.M.; Chou, M.C.; Chou, W.J.; Tsai, W.C.; Chiu, Y.N.; Wu, Y.Y.; et al. Genome-wide analysis of copy number variations identifies PARK2 as a candidate gene for autism spectrum disorder. Mol. Autism 2016, 7, 23. [Google Scholar] [CrossRef]
- Jarick, I.; Volckmar, A.L.; Putter, C.; Pechlivanis, S.; Nguyen, T.T.; Dauvermann, M.R.; Beck, S.; Albayrak, O.; Scherag, S.; Gilsbach, S.; et al. Genome-wide analysis of rare copy number variations reveals PARK2 as a candidate gene for attention-deficit/hyperactivity disorder. Mol. Psychiatry 2014, 19, 115–121. [Google Scholar] [CrossRef]
- Capkova, P.; Srovnal, J.; Capkova, Z.; Staffova, K.; Becvarova, V.; Trkova, M.; Adamova, K.; Santava, A.; Curtisova, V.; Hajduch, M.; et al. MLPA is a practical and complementary alternative to CMA for diagnostic testing in patients with autism spectrum disorders and identifying new candidate CNVs associated with autism. PeerJ 2019, 6, e6183. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.L.; Hovanes, K.; Dasouki, M.; Manzardo, A.M.; Butler, M.G. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 2014, 535, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Clements, C.C.; Wenger, T.L.; Zoltowski, A.R.; Bertollo, J.R.; Miller, J.S.; de Marchena, A.B.; Mitteer, L.M.; Carey, J.C.; Yerys, B.E.; Zackai, E.H.; et al. Critical region within 22q11.2 linked to higher rate of autism spectrum disorder. Mol. Autism 2017, 8, 58. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef]
- Liu, K.; Li, F.; Han, H.; Chen, Y.; Mao, Z.; Luo, J.; Zhao, Y.; Zheng, B.; Gu, W.; Zhao, W. Parkin Regulates the Activity of Pyruvate Kinase M2. J. Biol. Chem. 2016, 291, 10307–10317. [Google Scholar] [CrossRef]
- Martinez, A.; Lectez, B.; Ramirez, J.; Popp, O.; Sutherland, J.D.; Urbe, S.; Dittmar, G.; Clague, M.J.; Mayor, U. Quantitative proteomic analysis of Parkin substrates in Drosophila neurons. Mol. Neurodegener 2017, 12, 29. [Google Scholar] [CrossRef]
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 248. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nat. Med. 2001, 7, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Huynh, D.P.; Scoles, D.R.; Nguyen, D.; Pulst, S.M. The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum. Mol. Genet. 2003, 12, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kang, X.; Zhou, L.; Chai, Z.; Wu, Q.; Huang, R.; Xu, H.; Hu, M.; Sun, X.; Sun, S.; et al. Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson’s disease-like pathology. Nat. Commun. 2018, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, M.; Madara, J.; Pankow, S.; Liu, X.; Yates, J., 3rd; Sudhof, T.C.; Maximov, A. Synaptotagmin-11 mediates a vesicle trafficking pathway that is essential for development and synaptic plasticity. Genes Dev. 2019, 33, 365–376. [Google Scholar] [CrossRef]
- Leinartaite, L.; Svenningsson, P. Folding Underlies Bidirectional Role of GPR37/Pael-R in Parkinson Disease. Trends Pharm. Sci. 2017, 38, 749–760. [Google Scholar] [CrossRef]
- Marazziti, D.; Mandillo, S.; Di Pietro, C.; Golini, E.; Matteoni, R.; Tocchini-Valentini, G.P. GPR37 associates with the dopamine transporter to modulate dopamine uptake and behavioral responses to dopaminergic drugs. Proc. Natl. Acad. Sci. USA 2007, 104, 9846–9851. [Google Scholar] [CrossRef]
- Murakami, T.; Shoji, M.; Imai, Y.; Inoue, H.; Kawarabayashi, T.; Matsubara, E.; Harigaya, Y.; Sasaki, A.; Takahashi, R.; Abe, K. Pael-R is accumulated in Lewy bodies of Parkinson’s disease. Ann. Neurol. 2004, 55, 439–442. [Google Scholar] [CrossRef]
- Berger, B.S.; Acebron, S.P.; Herbst, J.; Koch, S.; Niehrs, C. Parkinson’s disease-associated receptor GPR37 is an ER chaperone for LRP6. Embo. Rep. 2017, 18, 712–725. [Google Scholar] [CrossRef]
- Fujita-Jimbo, E.; Yu, Z.L.; Li, H.; Yamagata, T.; Mori, M.; Momoi, T.; Momoi, M.Y. Mutation in Parkinson disease-associated, G-protein-coupled receptor 37 (GPR37/PaelR) is related to autism spectrum disorder. PLoS ONE 2012, 7, e51155. [Google Scholar] [CrossRef]
- Fung, W.; Peall, K.J. Does 22q11.2 deletion syndrome contribute to the genetic aetiology of Parkinson’s disease? J. Neurol. 2018, 265, 2463–2465. [Google Scholar] [CrossRef]
- Boot, E.; Bassett, A.S.; Marras, C. 22q11.2 Deletion Syndrome-Associated Parkinson’s Disease. Mov. Disord. Clin. Pr. 2019, 6, 11–16. [Google Scholar] [CrossRef]
- Kobrynski, L.J.; Sullivan, K.E. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet 2007, 370, 1443–1452. [Google Scholar] [CrossRef]
- Kirkpatrick, J.A., Jr.; DiGeorge, A.M. Congenital absence of the thymus. Am. J. Roentgenol. Radium Nucl. Med. 1968, 103, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Guna, A.; Butcher, N.J.; Bassett, A.S. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J Neurodev. Disord. 2015, 7, 18. [Google Scholar] [CrossRef]
- Schneider, M.; Debbane, M.; Bassett, A.S.; Chow, E.W.; Fung, W.L.; van den Bree, M.; Owen, M.; Murphy, K.C.; Niarchou, M.; Kates, W.R.; et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: Results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry 2014, 171, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H.; Kurahashi, H.; Saitta, S.C.; O’Hare, A.M.; Hu, P.; Roe, B.A.; Driscoll, D.A.; McDonald-McGinn, D.M.; Zackai, E.H.; Budarf, M.L.; et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000, 9, 489–501. [Google Scholar] [CrossRef]
- Demaerel, W.; Mostovoy, Y.; Yilmaz, F.; Vervoort, L.; Pastor, S.; Hestand, M.S.; Swillen, A.; Vergaelen, E.; Geiger, E.A.; Coughlin, C.R.; et al. The 22q11 low copy repeats are characterized by unprecedented size and structural variability. Genome Res. 2019, 29, 1389–1401. [Google Scholar] [CrossRef]
- Butcher, N.J.; Marras, C.; Pondal, M.; Rusjan, P.; Boot, E.; Christopher, L.; Repetto, G.M.; Fritsch, R.; Chow, E.W.C.; Masellis, M.; et al. Neuroimaging and clinical features in adults with a 22q11.2 deletion at risk of Parkinson’s disease. Brain 2017, 140, 1371–1383. [Google Scholar] [CrossRef]
- Boot, E.; Butcher, N.J.; Udow, S.; Marras, C.; Mok, K.Y.; Kaneko, S.; Barrett, M.J.; Prontera, P.; Berman, B.D.; Masellis, M.; et al. Typical features of Parkinson disease and diagnostic challenges with microdeletion 22q11.2. Neurology 2018, 90, e2059–e2067. [Google Scholar] [CrossRef]
- Boot, E.; Butcher, N.J.; van Amelsvoort, T.A.; Lang, A.E.; Marras, C.; Pondal, M.; Andrade, D.M.; Fung, W.L.; Bassett, A.S. Movement disorders and other motor abnormalities in adults with 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2015, 167A, 639–645. [Google Scholar] [CrossRef]
- Lindsay, E.A.; Botta, A.; Jurecic, V.; Carattini-Rivera, S.; Cheah, Y.C.; Rosenblatt, H.M.; Bradley, A.; Baldini, A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 1999, 401, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, A.; Horike, K.; Hirai, K.; Butcher, N.; Boot, E.; Sakurai, T.; Nucifora, F.C., Jr.; Bassett, A.S.; Sawa, A.; Tomoda, T. A mouse model of 22q11.2 deletions: Molecular and behavioral signatures of Parkinson’s disease and schizophrenia. Sci. Adv. 2018, 4, eaar6637. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, A.; Hartwig, C.; Freeman, A.A.H.; Bassell, J.L.; Zlatic, S.A.; Sapp Savas, C.; Vadlamudi, T.; Abudulai, F.; Pham, T.T.; Crocker, A.; et al. Systems Analysis of the 22q11.2 Microdeletion Syndrome Converges on a Mitochondrial Interactome Necessary for Synapse Function and Behavior. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 3561–3581. [Google Scholar] [CrossRef]
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol. Autism 2010, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Berry-Kravis, E.; Zhang, W.; Tassone, F.; Spector, E.; Zerbe, G.; Hagerman, P.J.; Ouyang, B.; Leehey, M.A. FMR1 gray-zone alleles: Association with Parkinson’s disease in women? Mov. Disord. 2011, 26, 1900–1906. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Howard, K.; Hagerman, R.; Leehey, M.A. Parkinsonism in FMR1 premutation carriers may be indistinguishable from Parkinson disease. Parkinsonism Relat. Disord. 2009, 15, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.Q.; Yang, J.C.; Hall, D.A.; Leehey, M.A.; Tassone, F.; Olichney, J.M.; Hagerman, R.J.; Zhang, L. Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): Revisited. Parkinsonism Relat. Disord. 2014, 20, 456–459. [Google Scholar] [CrossRef][Green Version]
- De Pablo-Fernandez, E.; Doherty, K.M.; Holton, J.L.; Revesz, T.; Djamshidian, A.; Limousin, P.; Bhatia, K.P.; Warner, T.T.; Lees, A.J.; Ling, H. Concomitant fragile X-associated tremor ataxia syndrome and Parkinson’s disease: A clinicopathological report of two cases. J. Neurol. Neurosurg. Psychiatry 2015, 86, 934–936. [Google Scholar] [CrossRef][Green Version]
- Krans, A.; Skariah, G.; Zhang, Y.; Bayly, B.; Todd, P.K. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta. Neuropathol. Commun. 2019, 7, 152. [Google Scholar] [CrossRef]
- Sacino, A.N.; Prokop, S.; Walsh, M.A.; Adamson, J.; Subramony, S.H.; Krans, A.; Todd, P.K.; Giasson, B.I.; Yachnis, A.T. Fragile X-associated tremor ataxia syndrome with co-occurrent progressive supranuclear palsy-like neuropathology. Acta. Neuropathol. Commun. 2019, 7, 158. [Google Scholar] [CrossRef]
- Paucar, M.; Nennesmo, I.; Svenningsson, P. Pathological Study of a FMR1 Premutation Carrier With Progressive Supranuclear Palsy. Front. Genet. 2018, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Greco, C.M.; Hunsaker, M.R.; Seritan, A.L.; Berman, R.F.; Gane, L.W.; Jacquemont, S.; Basuta, K.; Jin, L.W.; Hagerman, P.J.; et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 2012, 11, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernandez, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Arguero-Sanchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A Perspective on Cellular Structures and Organelles. Front. Neurosci. 2019, 13, 1399. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Nussbaum, R.L.; Polymeropoulos, M.H. Genetics of Parkinson’s disease. Hum. Mol. Genet. 1997, 6, 1687–1691. [Google Scholar] [CrossRef]
- Munoz, E.; Oliva, R.; Obach, V.; Marti, M.J.; Pastor, P.; Ballesta, F.; Tolosa, E. Identification of Spanish familial Parkinson’s disease and screening for the Ala53Thr mutation of the alpha-synuclein gene in early onset patients. Neurosci. Lett. 1997, 235, 57–60. [Google Scholar] [CrossRef]
- Mokretar, K.; Pease, D.; Taanman, J.W.; Soenmez, A.; Ejaz, A.; Lashley, T.; Ling, H.; Gentleman, S.; Houlden, H.; Holton, J.L.; et al. Somatic copy number gains of alpha-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 2018, 141, 2419–2431. [Google Scholar] [CrossRef]
- Fuchs, J.; Nilsson, C.; Kachergus, J.; Munz, M.; Larsson, E.M.; Schüle, B.; Langston, J.W.; Middleton, F.A.; Ross, O.A.; Hulihan, M.; et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 2007, 68, 916–922. [Google Scholar] [CrossRef]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef]
- Book, A.; Guella, I.; Candido, T.; Brice, A.; Hattori, N.; Jeon, B.; Farrer, M.J.; Consortium, S.M.I.O.t.G. A Meta-Analysis of alpha-Synuclein Multiplication in Familial Parkinsonism. Front. Neurol. 2018, 9, 1021. [Google Scholar] [CrossRef]
- Zafar, F.; Valappil, R.A.; Kim, S.; Johansen, K.K.; Chang, A.L.S.; Tetrud, J.W.; Eis, P.S.; Hatchwell, E.; Langston, J.W.; Dickson, D.W.; et al. Genetic fine-mapping of the Iowan SNCA gene triplication in a patient with Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.M.; Lawrence, C.E.; Raphael, B.J. Detecting non-allelic homologous recombination from high-throughput sequencing data. Genome Biol. 2015, 16, 72. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Lacaria, M.; Zhang, F.; Withers, M.; Hastings, P.J.; Lupski, J.R. Frequency of nonallelic homologous recombination is correlated with length of h Genetic fine-mapping of the Iowan SNCA gene triplication in a patient with Parkinson’s disease omology: Evidence that ectopic synapsis precedes ectopic crossing-over. Am. J. Hum. Genet. 2011, 89, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Vona, B.; Nanda, I.; Neuner, C.; Schroder, J.; Kalscheuer, V.M.; Shehata-Dieler, W.; Haaf, T. Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: Review of literature. BMC Med. Genet. 2014, 15, 72. [Google Scholar] [CrossRef]
- Lin, A.E.; Garver, K.L.; Diggans, G.; Clemens, M.; Wenger, S.L.; Steele, M.W.; Jones, M.C.; Israel, J. Interstitial and terminal deletions of the long arm of chromosome 4: Further delineation of phenotypes. Am. J. Med. Genet. 1988, 31, 533–548. [Google Scholar] [CrossRef]
- Sterling, L.; Walter, M.; Ting, D.; Schüle, B. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. F1000Res 2014, 3, 259. [Google Scholar] [CrossRef]
- Vulto-van Silfhout, A.T.; Hehir-Kwa, J.Y.; van Bon, B.W.; Schuurs-Hoeijmakers, J.H.; Meader, S.; Hellebrekers, C.J.; Thoonen, I.J.; de Brouwer, A.P.; Brunner, H.G.; Webber, C.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Hum. Mutat. 2013, 34, 1679–1687. [Google Scholar] [CrossRef]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; van Bon, B.W.; Vulto-van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Head, E.; Powell, D.; Gold, B.T.; Schmitt, F.A. Alzheimer’s Disease in Down Syndrome. Eur. J. Neurodegener. Dis. 2012, 1, 353–364. [Google Scholar]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. Febs. Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Lavedan, C. The synuclein family. Genome Res. 1998, 8, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- George, J.M. The synucleins. Genome Biol. 2002, 3, REVIEWS3002. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2012, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Ullman, O.; Fisher, C.K.; Stultz, C.M. Explaining the structural plasticity of alpha-synuclein. J. Am. Chem. Soc. 2011, 133, 19536–19546. [Google Scholar] [CrossRef]
- Bras, I.C.; Dominguez-Meijide, A.; Gerhardt, E.; Koss, D.; Lazaro, D.F.; Santos, P.I.; Vasili, E.; Xylaki, M.; Outeiro, T.F. Synucleinopathies: Where we are and where we need to go. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Masaracchia, C.; Hnida, M.; Gerhardt, E.; Lopes da Fonseca, T.; Villar-Pique, A.; Branco, T.; Stahlberg, M.A.; Dean, C.; Fernandez, C.O.; Milosevic, I.; et al. Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta. Neuropathol. Commun. 2018, 6, 79. [Google Scholar] [CrossRef]
- Villar-Pique, A.; Lopes da Fonseca, T.; Outeiro, T.F. Structure, function and toxicity of alpha-synuclein: The Bermuda triangle in synucleinopathies. J. Neurochem. 2016, 139 (Suppl. 1), 240–255. [Google Scholar] [CrossRef]
- Scott, D.; Roy, S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef]
- Vargas, K.J.; Schrod, N.; Davis, T.; Fernandez-Busnadiego, R.; Taguchi, Y.V.; Laugks, U.; Lucic, V.; Chandra, S.S. Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep. 2017, 18, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Wislet-Gendebien, S.; D’Souza, C.; Kawarai, T.; St George-Hyslop, P.; Westaway, D.; Fraser, P.; Tandon, A. Cytosolic proteins regulate alpha-synuclein dissociation from presynaptic membranes. J. Biol. Chem. 2006, 281, 32148–32155. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. alpha-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef]
- Soper, J.H.; Roy, S.; Stieber, A.; Lee, E.; Wilson, R.B.; Trojanowski, J.Q.; Burd, C.G.; Lee, V.M. Alpha-synuclein-induced aggregation of cytoplasmic vesicles in Saccharomyces cerevisiae. Mol. Biol. Cell 2008, 19, 1093–1103. [Google Scholar] [CrossRef]
- Diao, J.; Burre, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Sudhof, T.C.; Brunger, A.T. Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2013, 2, e00592. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Sulzer, D.; Edwards, R.H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef]
- Atias, M.; Tevet, Y.; Sun, J.; Stavsky, A.; Tal, S.; Kahn, J.; Roy, S.; Gitler, D. Synapsins regulate alpha-synuclein functions. Proc. Natl. Acad. Sci. USA 2019, 116, 11116–11118. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Longhena, F.; Varanita, T.; Bubacco, L.; Pizzi, M.; Missale, C.; Benfenati, F.; Bjorklund, A.; Spano, P.; Bellucci, A. Synapsin III deficiency hampers alpha-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson’s disease. Acta. Neuropathol. 2018, 136, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Longhena, F.; Bruno, A.; Bono, F.; Grigoletto, J.; La Via, L.; Barbon, A.; Casiraghi, A.; Straniero, V.; Valoti, E.; et al. Alpha-synuclein/synapsin III pathological interplay boosts the motor response to methylphenidate. Neurobiol. Dis. 2020, 138, 104789. [Google Scholar] [CrossRef]
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M.; et al. alpha-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 2015, 128, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- Venda, L.L.; Cragg, S.J.; Buchman, V.L.; Wade-Martins, R. alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 2010, 33, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Aliverti, A.; Barbiroli, A.; Santambrogio, C.; Ragg, E.M.; Casagrande, F.V.M.; Cantele, F.; Beltramone, S.; Marangon, J.; De Gregorio, C.; et al. α-Synuclein is a Novel Microtubule Dynamase. Sci. Rep. 2016, 6, 33289. [Google Scholar] [CrossRef]
- Zhou, R.M.; Huang, Y.X.; Li, X.L.; Chen, C.; Shi, Q.; Wang, G.R.; Tian, C.; Wang, Z.Y.; Jing, Y.Y.; Gao, C.; et al. Molecular interaction of alpha-synuclein with tubulin influences on the polymerization of microtubule in vitro and structure of microtubule in cells. Mol. Biol. Rep. 2010, 37, 3183–3192. [Google Scholar] [CrossRef]
- Sousa, V.L.; Bellani, S.; Giannandrea, M.; Yousuf, M.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. alpha-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol. Biol. Cell 2009, 20, 3725–3739. [Google Scholar] [CrossRef]
- Lee, F.J.; Liu, F.; Pristupa, Z.B.; Niznik, H.B. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 916–926. [Google Scholar] [CrossRef]
- Jeannotte, A.M.; Sidhu, A. Regulation of the norepinephrine transporter by alpha-synuclein-mediated interactions with microtubules. Eur. J. Neurosci. 2007, 26, 1509–1520. [Google Scholar] [CrossRef]
- Wersinger, C.; Rusnak, M.; Sidhu, A. Modulation of the trafficking of the human serotonin transporter by human alpha-synuclein. Eur. J. Neurosci. 2006, 24, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Chadchankar, H.; Ihalainen, J.; Tanila, H.; Yavich, L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res. 2011, 1382, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Bjorklund, A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Chen, A.Q.; Kong, Q.; Zhu, H.; Ma, C.M.; Qin, C. Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell Mol. Neurobiol. 2008, 28, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Specht, C.G.; Schoepfer, R. Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2001, 2, 11. [Google Scholar] [CrossRef]
- Specht, C.G.; Schoepfer, R. Deletion of multimerin-1 in alpha-synuclein-deficient mice. Genomics 2004, 83, 1176–1178. [Google Scholar] [CrossRef]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef]
- Senior, S.L.; Ninkina, N.; Deacon, R.; Bannerman, D.; Buchman, V.L.; Cragg, S.J.; Wade-Martins, R. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur. J. Neurosci. 2008, 27, 947–957. [Google Scholar] [CrossRef]
- Connor-Robson, N.; Peters, O.M.; Millership, S.; Ninkina, N.; Buchman, V.L. Combinational losses of synucleins reveal their differential requirements for compensating age-dependent alterations in motor behavior and dopamine metabolism. Neurobiol. Aging 2016, 46, 107–112. [Google Scholar] [CrossRef]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. alpha-Synuclein knockout mice have cognitive impairments. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-Robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Divane, A.; Goedert, M. Assignment of human alpha-synuclein (SNCA) and beta-synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genomics 1995, 27, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Ninkina, N.N.; Alimova-Kost, M.V.; Paterson, J.W.; Delaney, L.; Cohen, B.B.; Imreh, S.; Gnuchev, N.V.; Davies, A.M.; Buchman, V.L. Organization, expression and polymorphism of the human persyn gene. Hum. Mol. Genet. 1998, 7, 1417–1424. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef]
- Buchman, V.L.; Adu, J.; Pinon, L.G.; Ninkina, N.N.; Davies, A.M. Persyn, a member of the synuclein family, influences neurofilament network integrity. Nat. Neurosci. 1998, 1, 101–103. [Google Scholar] [CrossRef]
- Dunn, T.N.; Akiyama, T.; Lee, H.W.; Kim, J.B.; Knotts, T.A.; Smith, S.R.; Sears, D.D.; Carstens, E.; Adams, S.H. Evaluation of the synuclein-gamma (SNCG) gene as a PPARgamma target in murine adipocytes, dorsal root ganglia somatosensory neurons, and human adipose tissue. PLoS ONE 2015, 10, e0115830. [Google Scholar] [CrossRef]
- Surguchov, A.; Palazzo, R.E.; Surgucheva, I. Gamma synuclein: Subcellular localization in neuronal and non-neuronal cells and effect on signal transduction. Cell Motil. Cytoskelet. 2001, 49, 218–228. [Google Scholar] [CrossRef]
- Robertson, D.C.; Schmidt, O.; Ninkina, N.; Jones, P.A.; Sharkey, J.; Buchman, V.L. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J. Neurochem. 2004, 89, 1126–1136. [Google Scholar] [CrossRef]
- Garcia-Reitboeck, P.; Anichtchik, O.; Dalley, J.W.; Ninkina, N.; Tofaris, G.K.; Buchman, V.L.; Spillantini, M.G. Endogenous alpha-synuclein influences the number of dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 2013, 248, 541–545. [Google Scholar] [CrossRef]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef]
- Xu, J.; Kao, S.Y.; Lee, F.J.; Song, W.; Jin, L.W.; Yankner, B.A. Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 2002, 8, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, Y.; Akiguchi, I.; Nakamura, S.; Honjyo, Y.; Shibasaki, H.; Budka, H. 14-3-3 proteins in Lewy bodies in Parkinson disease and diffuse Lewy body disease brains. J. Neuropathol. Exp. Neurol. 2002, 61, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Cantuti-Castelvetri, I.; Bouzou, B.; Asteris, G.; McLean, P.J.; Hyman, B.T.; Standaert, D.G. Transcriptional dysregulation in a transgenic model of Parkinson disease. Neurobiol. Dis. 2008, 29, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Holzmann, C.; Riess, O. 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 2003, 4, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Ninkina, N.; Papachroni, K.; Robertson, D.C.; Schmidt, O.; Delaney, L.; O’Neill, F.; Court, F.; Rosenthal, A.; Fleetwood-Walker, S.M.; Davies, A.M.; et al. Neurons expressing the highest levels of gamma-synuclein are unaffected by targeted inactivation of the gene. Mol. Cell Biol. 2003, 23, 8233–8245. [Google Scholar] [CrossRef]
- Al-Wandi, A.; Ninkina, N.; Millership, S.; Williamson, S.J.; Jones, P.A.; Buchman, V.L. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 2010, 31, 796–804. [Google Scholar] [CrossRef]
- Kokhan, V.S.; Afanasyeva, M.A.; Vankin, G.I. alpha-Synuclein knockout mice have cognitive impairments. Behav. Brain Res. 2012, 231, 226–230. [Google Scholar] [CrossRef]
- Kokhan, V.S.; Van’kin, G.I.; Ninkina, N.N.; Shelkovnikova, T.A.; Bachurin, S.O. Impaired spatial and working memory in ageing mice with targeted inactivation of alpha-synuclein gene. Dokl. Biol. Sci.: Proc. Acad. Sci. Ussrbiol. Sci. Sect. 2011, 441, 354–356. [Google Scholar] [CrossRef]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A.; et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar] [CrossRef]
- Thomas, B.; Mandir, A.S.; West, N.; Liu, Y.; Andrabi, S.A.; Stirling, W.; Dawson, V.L.; Dawson, T.M.; Lee, M.K. Resistance to MPTP-neurotoxicity in alpha-synuclein knockout mice is complemented by human alpha-synuclein and associated with increased beta-synuclein and Akt activation. PLoS ONE 2011, 6, e16706. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Tabarean, I.; Tang, Y.; Cartier, A.; Masliah, E.; Roy, S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 8083–8095. [Google Scholar] [CrossRef]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef]
- Gaugler, M.N.; Genc, O.; Bobela, W.; Mohanna, S.; Ardah, M.T.; El-Agnaf, O.M.; Cantoni, M.; Bensadoun, J.C.; Schneggenburger, R.; Knott, G.W.; et al. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta. Neuropathol. 2012, 123, 653–669. [Google Scholar] [CrossRef]
- Barzilai, A.; Melamed, E. Molecular mechanisms of selective dopaminergic neuronal death in Parkinson’s disease. Trends Mol. Med. 2003, 9, 126–132. [Google Scholar] [CrossRef]
- von Linstow, C.U.; DeLano-Taylor, M.; Kordower, J.H.; Brundin, P. Does Developmental Variability in the Number of Midbrain Dopamine Neurons Affect Individual Risk for Sporadic Parkinson’s Disease? J. Parkinsons Dis. 2020, 10, 405–411. [Google Scholar] [CrossRef]
- Alegre-Abarrategui, J.; Brimblecombe, K.R.; Roberts, R.F.; Velentza-Almpani, E.; Tilley, B.S.; Bengoa-Vergniory, N.; Proukakis, C. Selective vulnerability in alpha-synucleinopathies. Acta. Neuropathol. 2019, 138, 681–704. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta. Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef]
- Stefanis, L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Xia, Y.; Wan, F.; Ma, K.; Guo, X.; Kou, L.; Yin, S.; Han, C.; Liu, L.; Huang, J.; et al. New Perspectives on Roles of Alpha-Synuclein in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.S.; Morrison, J.P.; Southwell, M.F.; Foley, J.F.; Bolon, B.; Elmore, S.A. Histology Atlas of the Developing Prenatal and Postnatal Mouse Central Nervous System, with Emphasis on Prenatal Days E7.5 to E18.5. Toxicol. Pathol. 2017, 45, 705–744. [Google Scholar] [CrossRef] [PubMed]
- Gale, E.; Li, M. Midbrain dopaminergic neuron fate specification: Of mice and embryonic stem cells. Mol. Brain 2008, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.C.; Luo, X.; Chen, X.S.; Cai, Q.Y.; Liu, J.; Chen, X.H.; Yao, Z.X. Expression and subcellular location of alpha-synuclein during mouse-embryonic development. Cell Mol. Neurobiol. 2010, 30, 469–482. [Google Scholar] [CrossRef]
- Jakowec, M.W.; Donaldson, D.M.; Barba, J.; Petzinger, G.M. Postnatal expression of alpha-synuclein protein in the rodent substantia nigra and striatum. Dev. Neurosci. 2001, 23, 96. [Google Scholar] [CrossRef]
- Hong, L.; Ko, H.W.; Gwag, B.J.; Joe, E.; Lee, S.; Kim, Y.T.; Suh, Y.H. The cDNA cloning and ontogeny of mouse alpha-synuclein. Neuroreport 1998, 9, 1239–1243. [Google Scholar] [CrossRef]
- Smidt, M.P.; Smits, S.M.; Bouwmeester, H.; Hamers, F.P.; van der Linden, A.J.; Hellemons, A.J.; Graw, J.; Burbach, J.P. Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development 2004, 131, 1145–1155. [Google Scholar] [CrossRef]
- Michell, A.W.; Tofaris, G.K.; Gossage, H.; Tyers, P.; Spillantini, M.G.; Barker, R.A. The effect of truncated human alpha-synuclein (1-120) on dopaminergic cells in a transgenic mouse model of Parkinson’s disease. Cell Transpl. 2007, 16, 461–474. [Google Scholar] [CrossRef]
- Budday, S.; Steinmann, P.; Kuhl, E. Physical biology of human brain development. Front. Cell Neurosci. 2015, 9, 257. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.K. Chapter 28 Development of the Nervous System. In Basic Neurochemistry (Eighth Edition); Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: New York, NY, USA, 2012; pp. 533–545. [Google Scholar] [CrossRef]
- Lenroot, R.K.; Giedd, J.N. Brain development in children and adolescents: Insights from anatomical magnetic resonance imaging. Neurosci. Biobehav. Rev. 2006, 30, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, R.; Kruijff, L.; Sterrenburg, M.D.; Rogers, B.B.; Hladik, C.L.; White, C.L., 3rd. Alpha-synuclein expression in the developing human brain. Pediatr. Dev. Pathol. 2004, 7, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Schuck, T.M.; Lee, V.M.; Trojanowski, J.Q. Differential expression and distribution of alpha-, beta-, and gamma-synuclein in the developing human substantia nigra. Exp. Neurol. 2001, 168, 347–355. [Google Scholar] [CrossRef]
- Gundersen, C.B. The Structure of the Synaptic Vesicle-Plasma Membrane Interface Constrains SNARE Models of Rapid, Synchronous Exocytosis at Nerve Terminals. Front. Mol. Neurosci. 2017, 10, 48. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Crews, L.; Pathel, P.; Morvinski-Friedmann, D.; Kosberg, K.; Roberts, S.; Patrick, C.; Winner, B.; Winkler, J.; et al. alpha-Synuclein induces alterations in adult neurogenesis in Parkinson disease models via p53-mediated repression of Notch1. J. Biol. Chem. 2012, 287, 31691–31702. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Lim, D.A.; Alvarez-Buylla, A. The Adult Ventricular-Subventricular Zone (V-SVZ) and Olfactory Bulb (OB) Neurogenesis. Cold Spring Harb. Perspect Biol. 2016, 8. [Google Scholar] [CrossRef]
- Lledo, P.M.; Alonso, M.; Grubb, M.S. Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 2006, 7, 179–193. [Google Scholar] [CrossRef]
- Toda, T.; Gage, F.H. Review: Adult neurogenesis contributes to hippocampal plasticity. Cell Tissue Res. 2018, 373, 693–709. [Google Scholar] [CrossRef]
- Abbott, L.C.; Nigussie, F. Adult neurogenesis in the mammalian dentate gyrus. Anat. Histol. Embryol. 2020, 49, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Danzer, S.C. Adult Neurogenesis in the Human Brain: Paradise Lost? Epilepsy Curr. 2018, 18, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef]
- Ernst, A.; Alkass, K.; Bernard, S.; Salehpour, M.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; Frisen, J. Neurogenesis in the striatum of the adult human brain. Cell 2014, 156, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.A.; Belnoue, L.; Song, H.; Simon, A. Neurotransmitter-mediated control of neurogenesis in the adult vertebrate brain. Development 2013, 140, 2548–2561. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, C.S.; Apostolopoulou, M.; Tian, Y.; Temple, S. It takes a village: Constructing the neurogenic niche. Dev. Cell 2015, 32, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Man, H.Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef]
- Packer, A. Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci. Biobehav. Rev. 2016, 64, 185–195. [Google Scholar] [CrossRef]
- Meechan, D.W.; Tucker, E.S.; Maynard, T.M.; LaMantia, A.S. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 16434–16445. [Google Scholar] [CrossRef]
- Meechan, D.W.; Maynard, T.M.; Tucker, E.S.; LaMantia, A.S. Three phases of DiGeorge/22q11 deletion syndrome pathogenesis during brain development: Patterning, proliferation, and mitochondrial functions of 22q11 genes. Int. J. Dev. Neurosci. 2011, 29, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Lie, D.C.; Rockenstein, E.; Aigner, R.; Aigner, L.; Masliah, E.; Kuhn, H.G.; Winkler, J. Human wild-type alpha-synuclein impairs neurogenesis. J. Neuropathol. Exp. Neurol. 2004, 63, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- May, V.E.; Nuber, S.; Marxreiter, F.; Riess, O.; Winner, B.; Winkler, J. Impaired olfactory bulb neurogenesis depends on the presence of human wild-type alpha-synuclein. Neuroscience 2012, 222, 343–355. [Google Scholar] [CrossRef]
- Kohl, Z.; Ben Abdallah, N.; Vogelgsang, J.; Tischer, L.; Deusser, J.; Amato, D.; Anderson, S.; Muller, C.P.; Riess, O.; Masliah, E.; et al. Severely impaired hippocampal neurogenesis associates with an early serotonergic deficit in a BAC alpha-synuclein transgenic rat model of Parkinson’s disease. Neurobiol. Dis. 2016, 85, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Rockenstein, E.; Lie, D.C.; Aigner, R.; Mante, M.; Bogdahn, U.; Couillard-Despres, S.; Masliah, E.; Winkler, J. Mutant alpha-synuclein exacerbates age-related decrease of neurogenesis. Neurobiol. Aging 2008, 29, 913–925. [Google Scholar] [CrossRef]
- Zhang, X.M.; Anwar, S.; Kim, Y.; Brown, J.; Comte, I.; Cai, H.; Cai, N.N.; Wade-Martins, R.; Szele, F.G. The A30P alpha-synuclein mutation decreases subventricular zone proliferation. Hum. Mol. Genet. 2019, 28, 2283–2294. [Google Scholar] [CrossRef]
- Lim, Y.; Kehm, V.M.; Li, C.; Trojanowski, J.Q.; Lee, V.M. Forebrain overexpression of alpha-synuclein leads to early postnatal hippocampal neuron loss and synaptic disruption. Exp. Neurol. 2010, 221, 86–97. [Google Scholar] [CrossRef]
- Regensburger, M.; Schreglmann, S.R.; Stoll, S.; Rockenstein, E.; Loskarn, S.; Xiang, W.; Masliah, E.; Winner, B. Oligomer-prone E57K-mutant alpha-synuclein exacerbates integration deficit of adult hippocampal newborn neurons in transgenic mice. Brain Struct. Funct. 2018, 223, 1357–1368. [Google Scholar] [CrossRef]
- Winner, B.; Desplats, P.; Hagl, C.; Klucken, J.; Aigner, R.; Ploetz, S.; Laemke, J.; Karl, A.; Aigner, L.; Masliah, E.; et al. Dopamine receptor activation promotes adult neurogenesis in an acute Parkinson model. Exp. Neurol. 2009, 219, 543–552. [Google Scholar] [CrossRef]
- Salvi, R.; Steigleder, T.; Schlachetzki, J.C.; Waldmann, E.; Schwab, S.; Winner, B.; Winkler, J.; Kohl, Z. Distinct Effects of Chronic Dopaminergic Stimulation on Hippocampal Neurogenesis and Striatal Doublecortin Expression in Adult Mice. Front. Neurosci. 2016, 10, 77. [Google Scholar] [CrossRef]
- Greenberg, D.A.; Jin, K. Turning neurogenesis up a Notch. Nat. Med. 2006, 12, 884–885. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.F.; Gao, X.; Ding, X.C.; Fan, M.; Chen, J. Postnatal dysregulation of Notch signal disrupts dendrite development of adult-born neurons in the hippocampus and contributes to memory impairment. Sci. Rep. 2016, 6, 25780. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Bjornevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schule, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef]
- Schneider, B.L.; Seehus, C.R.; Capowski, E.E.; Aebischer, P.; Zhang, S.C.; Svendsen, C.N. Over-expression of alpha-synuclein in human neural progenitors leads to specific changes in fate and differentiation. Hum. Mol. Genet. 2007, 16, 651–666. [Google Scholar] [CrossRef]
- Flierl, A.; Oliveira, L.M.; Falomir-Lockhart, L.J.; Mak, S.K.; Hesley, J.; Soldner, F.; Arndt-Jovin, D.J.; Jaenisch, R.; Langston, J.W.; Jovin, T.M.; et al. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS ONE 2014, 9, e112413. [Google Scholar] [CrossRef]
- Oliveira, L.M.; Falomir-Lockhart, L.J.; Botelho, M.G.; Lin, K.H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1994. [Google Scholar] [CrossRef]
- Winner, B.; Regensburger, M.; Schreglmann, S.; Boyer, L.; Prots, I.; Rockenstein, E.; Mante, M.; Zhao, C.; Winkler, J.; Masliah, E.; et al. Role of alpha-synuclein in adult neurogenesis and neuronal maturation in the dentate gyrus. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 16906–16916. [Google Scholar] [CrossRef]
- Perez-Villalba, A.; Sirerol-Piquer, M.S.; Belenguer, G.; Soriano-Canton, R.; Munoz-Manchado, A.B.; Villadiego, J.; Alarcon-Aris, D.; Soria, F.N.; Dehay, B.; Bezard, E.; et al. Synaptic Regulator alpha-Synuclein in Dopaminergic Fibers Is Essentially Required for the Maintenance of Subependymal Neural Stem Cells. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 814–825. [Google Scholar] [CrossRef]
- Taoufik, E.; Kouroupi, G.; Zygogianni, O.; Matsas, R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: An overview of induced pluripotent stem-cell-based disease models. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef]
- Schirinzi, T.; Madeo, G.; Martella, G.; Maltese, M.; Picconi, B.; Calabresi, P.; Pisani, A. Early synaptic dysfunction in Parkinson’s disease: Insights from animal models. Mov. Disord. 2016, 31, 802–813. [Google Scholar] [CrossRef]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef]
- Riley, B.E.; Gardai, S.J.; Emig-Agius, D.; Bessarabova, M.; Ivliev, A.E.; Schüle, B.; Alexander, J.; Wallace, W.; Halliday, G.M.; Langston, J.W.; et al. Systems-based analyses of brain regions functionally impacted in Parkinson’s disease reveals underlying causal mechanisms. PLoS ONE 2014, 9, e102909. [Google Scholar] [CrossRef]
- Burre, J. The Synaptic Function of alpha-Synuclein. J. Parkinsons Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef]
- Bridi, J.C.; Hirth, F. Mechanisms of alpha-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef]
- Bereczki, E.; Branca, R.M.; Francis, P.T.; Pereira, J.B.; Baek, J.H.; Hortobagyi, T.; Winblad, B.; Ballard, C.; Lehtio, J.; Aarsland, D. Synaptic markers of cognitive decline in neurodegenerative diseases: A proteomic approach. Brain 2018, 141, 582–595. [Google Scholar] [CrossRef]
- Bellucci, A.; Mercuri, N.B.; Venneri, A.; Faustini, G.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P. Review: Parkinson’s disease: From synaptic loss to connectome dysfunction. Neuropathol. Appl. Neurobiol. 2016, 42, 77–94. [Google Scholar] [CrossRef]
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Pique, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morato Torres, C.A.; Wassouf, Z.; Zafar, F.; Sastre, D.; Outeiro, T.F.; Schüle, B. The Role of Alpha-Synuclein and Other Parkinson’s Genes in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 5724. https://doi.org/10.3390/ijms21165724
Morato Torres CA, Wassouf Z, Zafar F, Sastre D, Outeiro TF, Schüle B. The Role of Alpha-Synuclein and Other Parkinson’s Genes in Neurodevelopmental and Neurodegenerative Disorders. International Journal of Molecular Sciences. 2020; 21(16):5724. https://doi.org/10.3390/ijms21165724
Chicago/Turabian StyleMorato Torres, C. Alejandra, Zinah Wassouf, Faria Zafar, Danuta Sastre, Tiago Fleming Outeiro, and Birgitt Schüle. 2020. "The Role of Alpha-Synuclein and Other Parkinson’s Genes in Neurodevelopmental and Neurodegenerative Disorders" International Journal of Molecular Sciences 21, no. 16: 5724. https://doi.org/10.3390/ijms21165724
APA StyleMorato Torres, C. A., Wassouf, Z., Zafar, F., Sastre, D., Outeiro, T. F., & Schüle, B. (2020). The Role of Alpha-Synuclein and Other Parkinson’s Genes in Neurodevelopmental and Neurodegenerative Disorders. International Journal of Molecular Sciences, 21(16), 5724. https://doi.org/10.3390/ijms21165724