Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection

 , and
, and 
Abstract
1. Introduction
2. Results
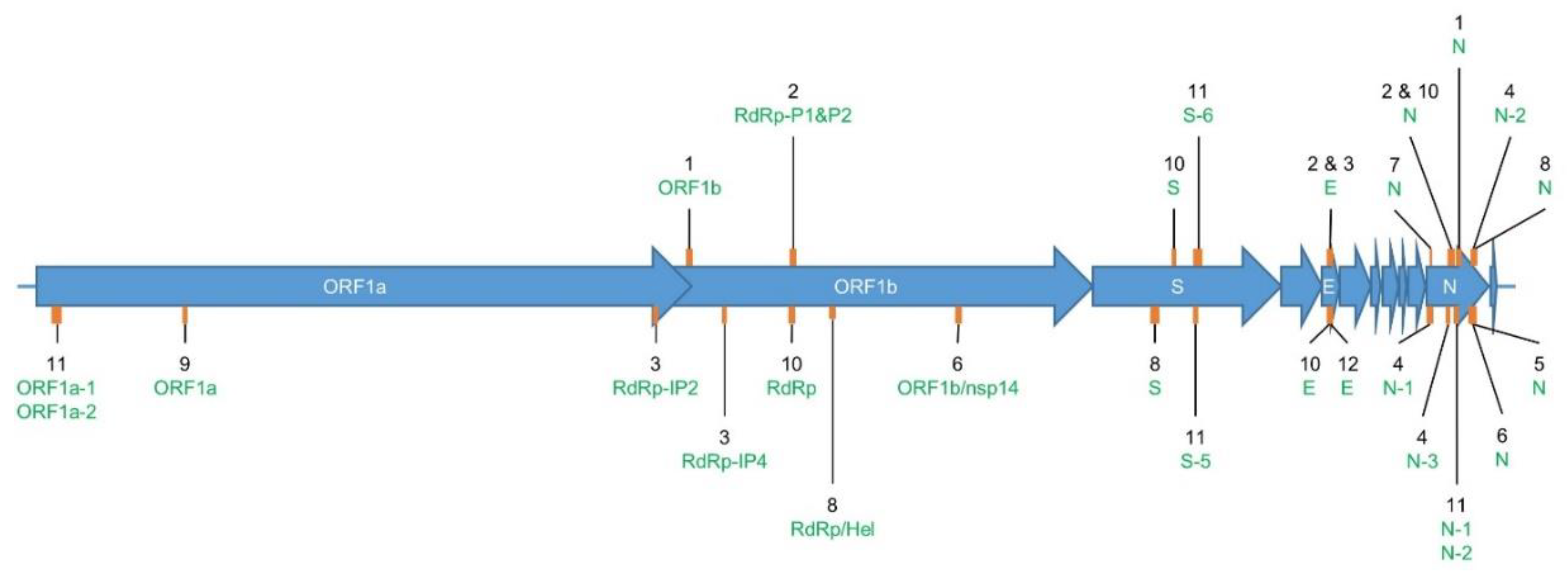
2.1. Overview of RT-qPCR Assays for SARS-CoV-2 Detection
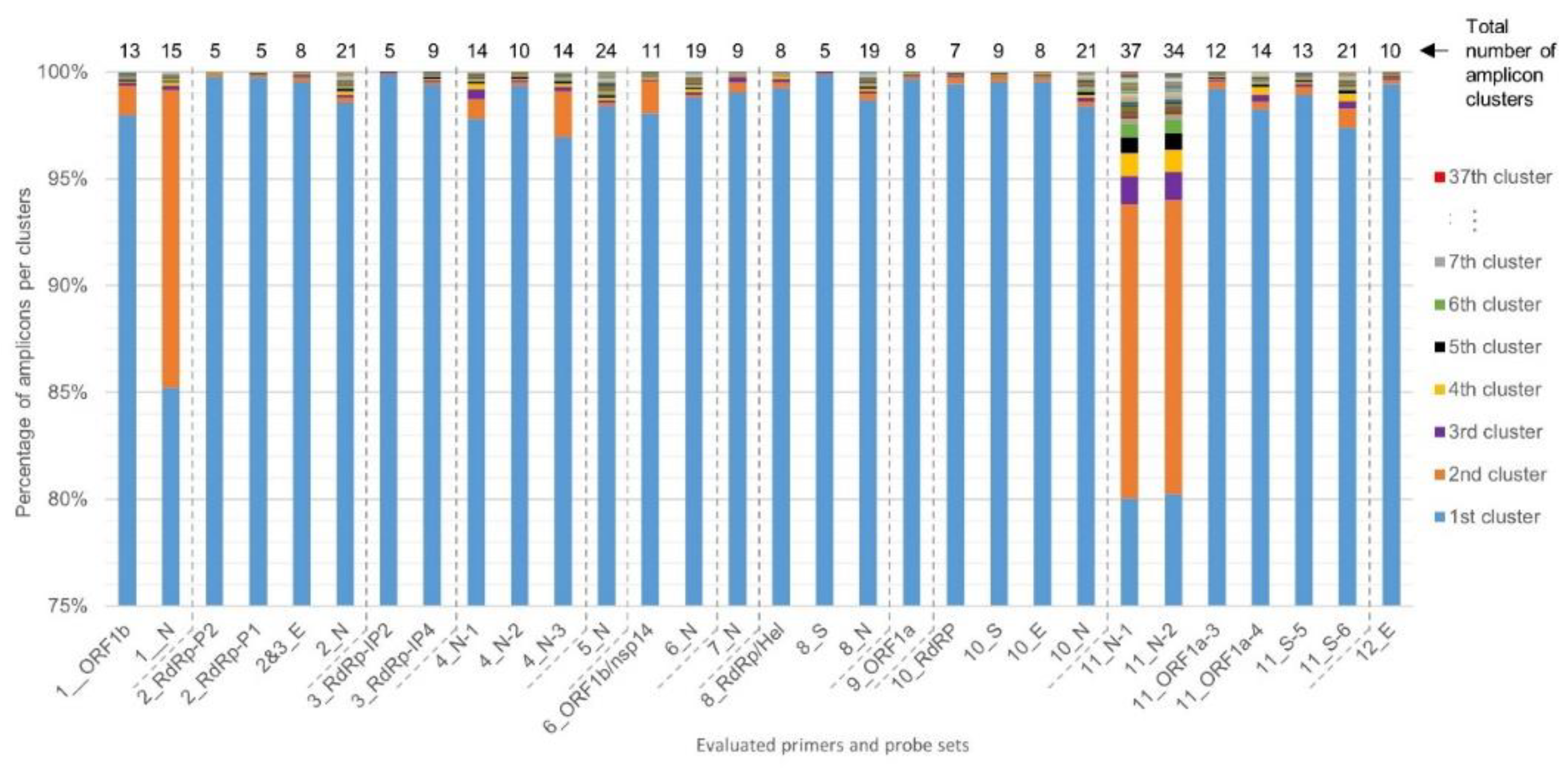
2.2. Determination of RT-qPCR Assay SARS-CoV-2 Inclusivity
2.3. Determination of RT-qPCR Assay Exclusivity
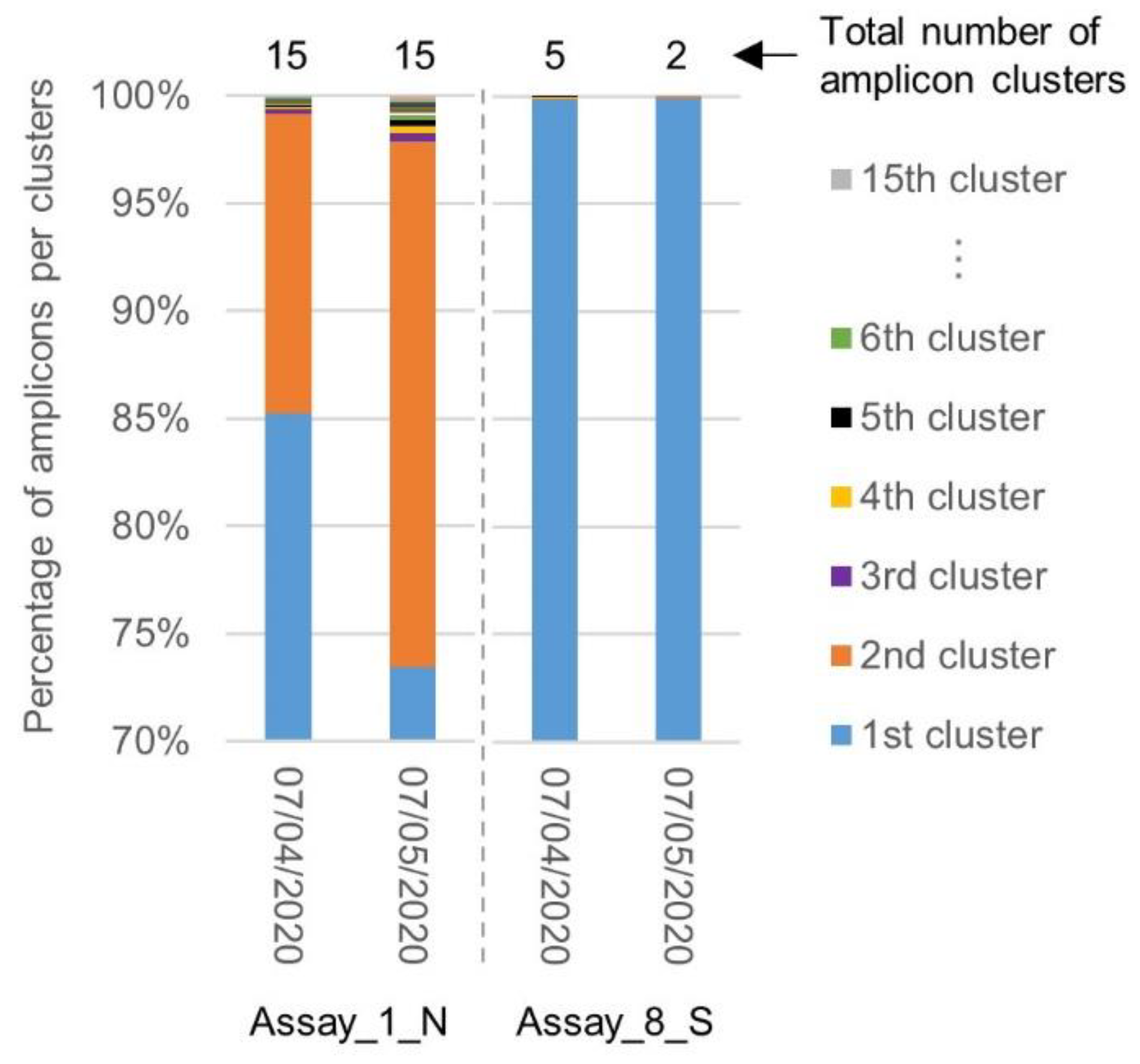
2.4. Evolution of the Inclusivity of Four RT-qPCR Tests after One Month
3. Discussion
4. Material and Methods
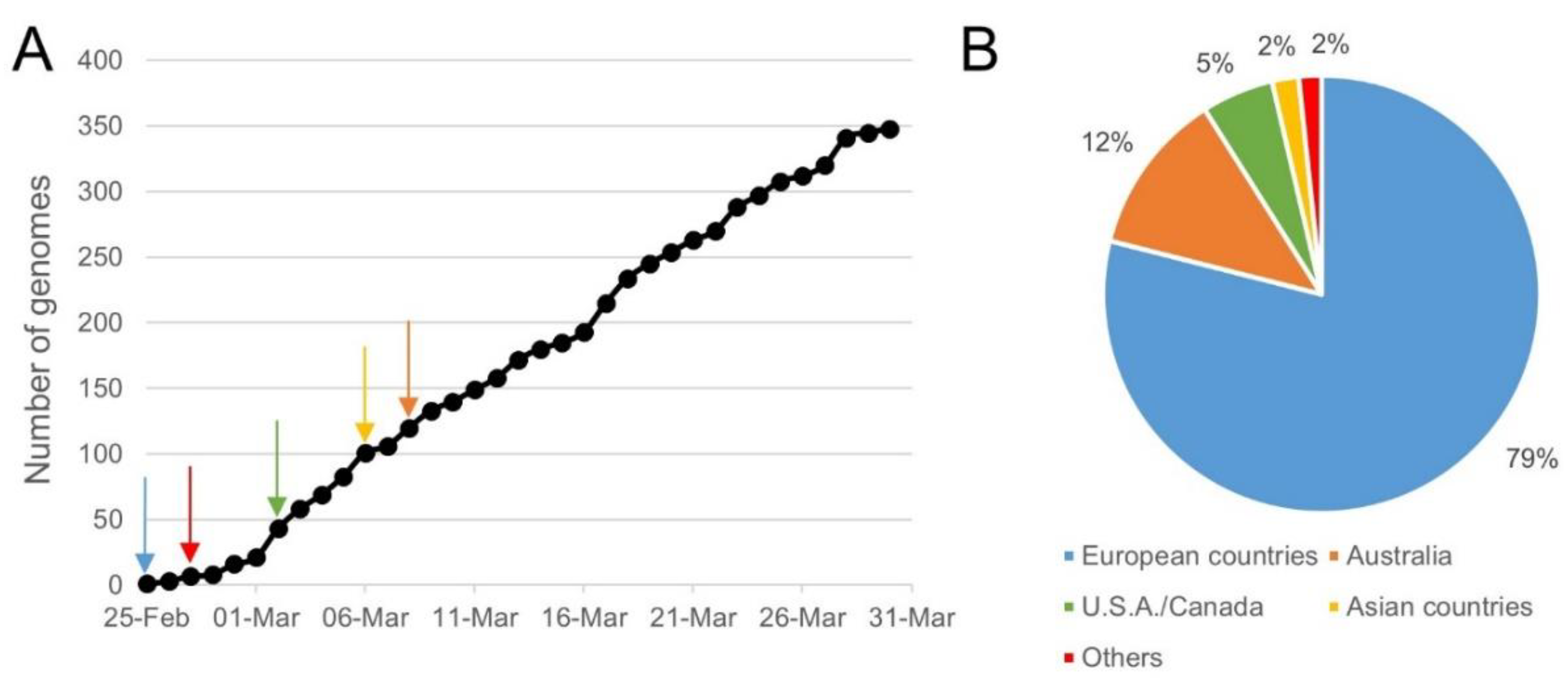
4.1. Collection of WGS Data
4.2. Sequence Identity Clustering to Obtain Unique Representative Genomes
4.3. Settings and Input Files Used in SCREENED
4.4. Determination of the In Silico Analytical Specificity of the Evaluated RT-qPCR Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singhal, T. A Review of Coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 2020, 87, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Valdes, A.M.; Freidin, M.B.; Sudre, C.H.; Nguyen, L.H.; Drew, D.A.; Ganesh, S.; Varsavsky, T.; Cardoso, M.J.; El-Sayed Moustafa, J.S.; et al. Real-time tracking of self-reported symptoms to predict potential COVID-19. Nat. Med. 2020, 26, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Luk, H.K.H.; Wong, A.C.P.; Li, K.S.M.; Zhu, L.; He, Z.; Fung, J.; Chan, T.T.Y.; Fung, K.S.C.; Woo, P.C.Y. Possible Bat Origin of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. J. 2020, 26, 1542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351. [Google Scholar] [CrossRef]
- Li, C.; Yang, Y.; Ren, L. Genetic evolution analysis of 2019 novel coronavirus and coronavirus from other species. Infect. Genet. Evol. 2020, 82, 1–3. [Google Scholar] [CrossRef]
- Chatterjee, P.; Nagi, N.; Agarwal, A.; Das, B.; Banerjee, S.; Sarkar, S.; Gupta, N.; Gangakhedkar, R. The 2019 novel coronavirus disease (COVID-19) pandemic: A review of the current evidence. Indian J. Med. Res. 2020, 151, 147–159. [Google Scholar] [CrossRef]
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 24 June 2020).
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Ren, X.; Liu, Y.; Chen, H.; Liu, W.; Guo, Z.; Zhang, Y.; Chen, C.; Zhou, J.; Xiao, Q.; Jiang, G.-M.; et al. Application and Optimization of RT-PCR in Diagnosis of SARS-CoV-2 Infection. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Zhai, P.; Ding, Y.; Wu, X.; Long, J.; Zhong, Y.; Li, Y. The epidemiology, diagnosis and treatment of COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105955. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, M.; Shen, C.; Wang, F.; Yuan, J.; Li, J.; Zhang, M.; Wang, Z.; Xing, L.; Wei, J.; et al. Evaluating the accuracy of different respiratory specimens in the laboratory diagnosis and monitoring the viral shedding of 2019-nCoV infections. MedRxiv 2020. [Google Scholar] [CrossRef]
- Bai, H.; Cai, X.; Zhang, X. Landscape Coronavirus Disease 2019 test (COVID-19 test) in vitro-A comparison of PCR vs Immunoassay vs Crispr-Based test. OSFpreprints 2020. [Google Scholar] [CrossRef]
- Yong, G.; Yi, Y.; Tuantuan, L.; Xiaowu, W.; Xiuyong, L.; Ang, L.; Mingfeng, H. Evaluation of the auxiliary diagnosis value of antibodies assays for the detection of novel coronavirus (SARS-Cov-2). MedRxiv 2020. [Google Scholar] [CrossRef]
- Li, Z.; Yi, Y.; Luo, X.; Xiong, N.; Liu, Y.; Li, S.; Sun, R.; Wang, Y.; Hu, B.; Chen, W.; et al. Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Kucirka, L.M.; Lauer, S.A.; Laeyendecker, O.; Boon, D.; Lessler, J. Variation in False-Negative Rate of Reverse Transcriptase Polymerase Chain Reaction–Based SARS-CoV-2 Tests by Time Since Exposure. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Lippi, G.; Simundic, A.M.; Plebani, M. Potential preanalytical and analytical vulnerabilities in the laboratory diagnosis of coronavirus disease 2019 (COVID-19). Clin. Chem. Lab. Med. 2020. [Google Scholar] [CrossRef]
- Lescure, F.X.; Bouadma, L.; Nguyen, D.; Parisey, M.; Wicky, P.H.; Behillil, S.; Gaymard, A.; Bouscambert-Duchamp, M.; Donati, F.; Le Hingrat, Q.; et al. Clinical and virological data of the first cases of COVID-19 in Europe: A case series. Lancet Infect. Dis. 2020, 2, 697–706. [Google Scholar] [CrossRef]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of SARS-CoV-2 in Coronavirus Disease 2019 patients. Clin. Infect. Dis. 2020, 71, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.; Kellogg, D.E.; Mckinney, N.; Spasic, D.; Goda, L.; Levenson, C.; Sninsky, J.J. Effects of primer-template mismatches on the polymerase chain reaction: Human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990, 18, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Lefever, S.; Pattyn, F.; Hellemans, J.; Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 2013, 59, 1470–1480. [Google Scholar] [CrossRef]
- Christopherson, C.; Sninsky, J.; Kwok, S. The effects of internal primer-template mismatches on RT-PCR: HIV-1 model studies. Nucleic Acids Res. 1997, 25, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Whiley, D.M.; Sloots, T.P. Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J. Clin. Virol. 2005, 34, 104–107. [Google Scholar] [CrossRef]
- WHO Coronavirus Disease (COVID-19) Technical Guidance: Laboratory Testing for 2019-nCoV in Humans. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/laboratory-guidance (accessed on 30 April 2020).
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Catherine Muenker, M.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-COV-2 qRT-PCR assays. Nat. Microbiol. 2020, 1–7. [Google Scholar] [CrossRef]
- Jung, Y.J.; Park, G.-S.; Moon, J.H.; Ku, K.; Beak, S.-H.; Kim, S.; Park, E.C.; Park, D.; Lee, J.-H.; Byeon, C.W.; et al. Comparative analysis of primer-probe sets for the laboratory confirmation of SARS-CoV-2. BioRxiv 2020, 5. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yip, C.C.-Y.; To, K.K.-W.; Tang, T.H.-C.; Wong, S.C.-Y.; Leung, K.-H.; Fung, A.Y.-F.; Ng, A.C.-K.; Zou, Z.; Tsoi, H.-W.; et al. Improved Molecular Diagnosis of COVID-19 by the Novel, Highly Sensitive and Specific COVID-19-RdRp/Hel Real-Time Reverse Transcription-PCR Assay Validated In Vitro and with Clinical Specimens. J. Clin. Microbiol. 2020, 58. [Google Scholar] [CrossRef]
- Won, J.; Lee, S.; Park, M.; Kim, T.Y.; Park, M.G.; Choi, B.Y.; Kim, D.; Chang, H.; Kim, V.N.; Lee, C.J. Development of a Laboratory-safe and Low-cost Detection Protocol for SARS-CoV-2 of the Coronavirus Disease 2019 (COVID-19). Exp. Neurobiol. 2020, 29, 107. [Google Scholar] [CrossRef]
- Sigma-Aldrich Coronavirus qPCR Design Case Study to Support SARS-CoV-2 Research. Available online: https://www.sigmaaldrich.com/technical-documents/protocols/biology/ncov-coronavirus.html (accessed on 5 May 2020).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- European Commission Current Performance of COVID-19 Test Methods and Devices and Proposed Performance Criteria. Available online: https://ec.europa.eu/docsroom/documents/40805 (accessed on 24 June 2020).
- Khan, K.A.; Cheung, P. Presence of mismatches between diagnostic PCR assays and coronavirus SARS-CoV-2 genome. R. Soc. Open Sci. 2020, 7, 200636. [Google Scholar] [CrossRef]
- Toms, D.; Li, J.; Cai, H.Y. Evaluation of WHO listed COVID-19 qPCR primers and probe in silico with 375 SERS-CoV-2 full genome sequences. MedRxiv 2020. [Google Scholar] [CrossRef]
- Phelan, J.; Deelder, W.; Ward, D.; Campino, S.; Hibberd, M.L.; Clark, T.G. Controlling the SARS-CoV-2 outbreak, insights from large scale whole genome sequences generated across the world. BioRxiv 2020. [Google Scholar] [CrossRef]
- Holland, M.; Negrón, D.; Mitchell, S.; Dellinger, N.; Ivancich, M.; Barrus, T.; Thomas, S.; Jennings, K.W.; Goodwin, B.; Sozhamannan, S. BioLaboro: A bioinformatics system for detecting molecular assay signature erosion and designing new assays in response to emerging and reemerging pathogens. BioRxiv 2020. [Google Scholar] [CrossRef]
- Vanneste, K.; Garlant, L.; Broeders, S.; Van Gucht, S.; Roosens, N.H. Application of whole genome data for in silico evaluation of primers and probes routinely employed for the detection of viral species by RT-qPCR using dengue virus as a case study. BMC Bioinform. 2018, 19, 1–18. [Google Scholar] [CrossRef]
- Broeders, S.; Garlant, L.; Fraiture, M.A.; Vandermassen, E.; Suin, V.; Vanhomwegen, J.; Dupont-Rouzeyrol, M.; Rousset, D.; Van Gucht, S.; Roosens, N. A new multiplex RT-qPCR method for the simultaneous detection and discrimination of Zika and chikungunya viruses. Int. J. Infect. Dis. 2020, 92, 160–170. [Google Scholar] [CrossRef]
- Shirato, K.; Nao, N.; Katano, H.; Takayama, I.; Saito, S.; Kato, F.; Katoh, H.; Sakata, M.; Nakatsu, Y.; Mori, Y.; et al. Development of Genetic Diagnostic Methods for Novel Coronavirus 2019 (nCoV-2019) in Japan. Jpn. J. Infect. Dis. 2020, 2019. [Google Scholar] [CrossRef]
- Reusken, C.B.E.M.; Broberg, E.K.; Haagmans, B.; Meijer, A.; Corman, V.M.; Papa, A.; Charrel, R.; Drosten, C.; Koopmans, M.; Leitmeyer, K. Laboratory readiness and response for novel coronavirus (2019-nCoV) in expert laboratories in 30 EU/EEA countries, January 2020. Eurosurveillance 2020, 25, 1–6. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. NextStrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.-P.; Boerwinkle, E.; Fu, Y.-X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21. [Google Scholar] [CrossRef]
- Van Poelvoorde, L.A.E.; Saelens, X.; Thomas, I.; Roosens, N.H. Next-Generation Sequencing: An Eye-Opener for the Surveillance of Antiviral Resistance in Influenza. Trends Biotechnol. 2020, 38, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with 2019 Novel Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 1266. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.; Rodriguez-Morales, A.J.; Jha, R.; Chu, D.K.W.; Gu, H.; Peiris, M.; Bastola, A.; Lal, B.K.; Ojha, H.C.; Rabaan, A.A.; et al. Complete Genome Sequence of a 2019 Novel Coronavirus (SARS-CoV-2) Strain Isolated in Nepal. Microbiol. Resour. Announc. 2020, 9, e00169-20. [Google Scholar] [CrossRef] [PubMed]
- Paden, C.R.; Tao, Y.; Queen, K.; Zhang, J.; Li, Y.; Uehara, A.; Tong, S. Rapid, sensitive, full genome sequencing of Severe Acute Respiratory Syndrome Virus Coronavirus 2 (SARS-CoV-2). BioRxiv 2020. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention Severe Acute Respiratory Syndrome (SARS). Available online: https://www.cdc.gov/sars/index.html (accessed on 27 July 2020).
- Park, C.; Lee, J.; Hassan, Z.U.; Ku, K.B.; Kim, S.J.; Kim, H.G.; Park, E.C.; Park, G.-S.; Park, D.; Baek, S.-H.; et al. Robust and sensitive detection of SARS-CoV-2 using PCR based methods. BioRxiv 2020. [Google Scholar] [CrossRef]
- Dong, L.; Zhou, J.; Niu, C.; Wang, Q.; Pan, Y.; Sheng, S.; Wang, X.; Zhang, Y.; Yang, J.; Liu, M.; et al. Highly accurate and sensitive diagnostic detection of SARS-CoV-2 by digital PCR. MedRxiv 2020. [Google Scholar] [CrossRef]
- Yu, F.; Yan, L.; Wang, N.; Yang, S.; Wang, L.; Tang, Y.; Gao, G.; Wang, S.; Ma, C.; Xie, R.; et al. Quantitative Detection and Viral Load Analysis of SARS-CoV-2 in Infected Patients. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Suo, T.; Liu, X.; Feng, J.; Guo, M.; Hu, W.; Guo, D.; Ullah, H.; Yang, Y.; Zhang, Q.; Wang, X.; et al. ddPCR: A more sensitive and accurate tool for SARS-CoV-2 detection in low viral load specimens. MedRxiv 2020. [Google Scholar] [CrossRef]
- Hart, O.E.; Halden, R.U. Computational analysis of SARS-CoV-2/COVID-19 surveillance by wastewater-based epidemiology locally and globally: Feasibility, economy, opportunities and challenges. Sci. Total Environ. 2020, 730, 138875. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Luisa Schmidt, M.; et al. Detection of 2019 -nCoV by RT-PCR. Euro Surveill. 2020, 25. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Technology | Target | Primer and Probe Sequences (5′-3′) | Amplicon’s Starting Positio ¶ | Amplicon Length | Annealing T °C | Intended Specificity | Guidelines for Overall Interpretation of the Assay | Source |
|---|---|---|---|---|---|---|---|---|---|
| 1 | TaqMan | ORF1b | Fw CCCTGTGGGTTTTACACTTAA | 13,341 | 119 | NC | NC * | NC1 | China CDC, China [27] |
| Rv ACGATTGTGCATCAGCTGA | |||||||||
| P CCGTCTGCGGTATGTGGAAAGGTTATGG | |||||||||
| 1 | TaqMan | N | Fw GGGGAACTTCTCCTGCTAGAAT | 28,880 | 99 | NC | NC * | ||
| Rv CAGACATTTTGCTCTCAAGCTG | |||||||||
| P TTGCTGCTGCTTGACAGATT | |||||||||
| 2 ** | TaqMan | RdRp-P1 | Fw GTGARATGGTCATGTGTGGCGG | 15,430 | 100 | 58 °C | Sarbeco | The E target can be used for a first screening. Then, positive results must be confirmed by RdRp sets, which must both be positive for the specific detection of SARS-CoV-2. | Charité Hospital, Germany [27] |
| Rv CARATGTTAAASACACTATTAGCATA | |||||||||
| P CCAGGTGGWACRTCATCMGGTGATGC | |||||||||
| 2 ** | TaqMan | RdRp-P2 | Fw GTGARATGGTCATGTGTGGCGG | 15,430 | 100 | 58 °C | SARS- | ||
| Rv CARATGTTAAASACACTATTAGCATA | CoV-2 | ||||||||
| P CAGGTGGAACCTCATCAGGAGATGC | |||||||||
| 2 *** | TaqMan | E | Fw ACAGGTACGTTAATAGTTAATAGCGT | 26,268 | 113 | 58 °C | Sarbeco | ||
| Rv ATATTGCAGCAGTACGCACACA | |||||||||
| P ACACTAGCCATCCTTACTGCGCTTCG | |||||||||
| 2 **** | TaqMan | N | Fw CACATTGGCACCCGCAATC | 28,705 | 128 | 58 °C | NC | ||
| Rv GAGGAACGAGAAGAGGCTTG | |||||||||
| P ACTTCCTCAAGGAACAACATTGCCA | |||||||||
| 3 | TaqMan | RdRp-IP2 | Fw ATGAGCTTAGTCCTGTTG | 12,689 | 108 | 58 °C | SARS- | RdRp-IP2 and RdRp-IP4 must be detected for SARS-CoV-2 determination. The E target can be used as a confirmatory result. | Institut Pasteur, France [27] |
| Rv CTCCCTTTGTTGTGTTGT | CoV-2 | ||||||||
| P AGATGTCTTGTGCTGCCGGTA | |||||||||
| 3 | TaqMan | RdRp-IP4 | Fw GGTAACTGGTATGATTTCG | 14,079 | 107 | 58 °C | SARS- | ||
| Rv CTGGTCAAGGTTAATATAGG | CoV-2 | ||||||||
| P TCATACAAACCACGCCAGG | |||||||||
| 3 *** | TaqMan | E | Fw ACAGGTACGTTAATAGTTAATAGCGT | 26,268 | 113 | 58 °C | Sarbeco | ||
| Rv ATATTGCAGCAGTACGCACACA | |||||||||
| P ACACTAGCCATCCTTACTGCGCTTCG | |||||||||
| 4 | TaqMan | N-1 | Fw GACCCCAAAATCAGCGAAAT | 28,286 | 72 | 55 °C | SARS- | Both N-1 and N-2 tests must be positive to confirm the detection of SARS-CoV-2. If only one target is detected, the result of the test is inconclusive. | US CDC, USA [27] |
| Rv TCTGGTTACTGCCAGTTGAATCTG | CoV-2 | ||||||||
| P ACCCCGCATTACGTTTGGTGGACC | |||||||||
| 4 | TaqMan | N-2 | Fw TTACAAACATTGGCCGCAAA | 29,163 | 67 | 55 °C | SARS- | ||
| Rv GCGCGACATTCCGAAGAA | CoV-2 | ||||||||
| P ACAATTTGCCCCCAGCGCTTCAG | |||||||||
| 4 † | TaqMan | N-3 | Fw GGGAGCCTTGAATACACCAAAA | 28,680 | 72 | 55 °C | SARS- | ||
| Rv ACAATTTGCCCCCAGCGCTTCAG | CoV-2 | ||||||||
| P AYCACATTGGCACCCGCAATCCTG | |||||||||
| 5 | TaqMan | N | Fw AAATTTTGGGGACCAGGAAC | 29,124 | 158 | 60 °C | SARS- | NA | NIID, Japan [27] |
| Rv TGGCAGCTGTGTAGGTCAAC | CoV-2 § | ||||||||
| P ATGTCGCGCATTGGCATGGA | |||||||||
| 6 | TaqMan | ORF1b/ | Fw TGGGGYTTTACRGGTAACCT | 18,777 | 132 | 60 °C | Sarbeco‡ | The N gene detection is recommended for a first screening and the Orf1b/nsp14 detection as a confirmatory test. Mixed positive/negative results between the 2 targets should be regarded as undetermined. | HKU Med, Hong-Kong [27] |
| nsp14 | Rv AACRCGCTTAACAAAGCACTC | ||||||||
| P TAGTTGTGATGCWATCATGACTAG | |||||||||
| 6 | TaqMan | N | Fw TAATCAGACAAGGAACTGATTA | 29,144 | 110 | 60 °C | Sarbeco‡ | ||
| Rv CGAAGGTGTGACTTCCATG | |||||||||
| P GCAAATTGTGCAATTTGCGG | |||||||||
| 7 | TaqMan | N | Fw CGTTTGGTGGACCCTCAGAT | 28,319 | 57 | 55 °C | SARS- | NA | NIH, Thailand [27] |
| Rv CCCCACTGCGTTCTCCATT | CoV-2 § | ||||||||
| P CAACTGGCAGTAACCA | |||||||||
| 8 | TaqMan | RdRp/ | Fw CGCATACAGTCTTRCAGGCT | 16,219 | 134 | 55 °C | SARS- | The primers and probe set targeting RdRp/Hel is the most sensitive of Assay 8 and can be used alone for the specific detection of SARS-CoV-2 with no cross-reactivity with other human coronaviruses. | Chan et al. [30] |
| Hel | Rv GTGTGATGTTGAWATGACATGGTC | CoV-2 | |||||||
| P TTAAGATGTGGTGCTTGCATACGTAGAC | |||||||||
| 8 | TaqMan | S | Fw CCTACTAAATTAAATGATCTCTGCTTTACT | 22,711 | 158 | 55 °C | SARS- | ||
| Rv CAAGCTATAACGCAGCCTGTA | CoV-2 | ||||||||
| P CGCTCCAGGGCAAACTGGAAAG | |||||||||
| 8 | TaqMan | N | Fw GCGTTCTTCGGAATGTCG | 29,209 | 97 | 55 °C | SARS- | ||
| Rv TTGGATCTTTGTCATCCAATTTG | CoV-2 | ||||||||
| P AACGTGGTTGACCTACACAGST | |||||||||
| 9 | TaqMan | ORF1a | Fw AGAAGATTGGTTAGATGATGATAGT | 3192 | 118 | 58 °C | SARS- | NA | Lu et al. [3] |
| Rv TTCCATCTCTAATTGAGGTTGAACC | CoV-2 | ||||||||
| P TCCTCACTGCCGTCTTGTTGACCA | |||||||||
| 10 | SYBR | RdRP | Fw CATGTGTGGCGGTTCACTAT | 15,440 | 118 | NC | SARS- | The main objective of this assay was the determination of patients negative for SARS-CoV-2. Thus, for a negative result, the 4 targets must remain undetected. Presence of SARS-CoV-2 is suspected if at least one target is detected, but further investigations need to confirm this. | Won et al. [31] |
| Green | Rv TGCATTAACATTGGCCGTGA | CoV-2 | |||||||
| 10 | SYBR | S | Fw CTACATGCACCAGCAACTGT | 23,113 | 100 | NC | SARS- | ||
| Green | Rv CACCTGTGCCTGTTAAACCA | CoV-2 | |||||||
| 10 | SYBR | E | Fw TTCGGAAGAGACAGGTACGTT | 26,258 | 107 | NC | SARS- | ||
| Green | Rv CACACAATCGATGCGCAGTA | CoV-2 | |||||||
| 10 | SYBR | N | Fw CAATGCTGCAATCGTGCTAC | 28,731 | 118 | NC | SARS- | ||
| Green | Rv GTTGCGACTACGTGATGAGG | CoV-2 | |||||||
| 11 ∥ | SYBR | N-1 | Fw GCCTCTTCTCGTTCCTCATCAC | 28,816 | 111 | NC | SARS | N and ORF1a targets can be detected in SARS-CoV-2 and possibly in SARS-CoV even if there are 2 SNPs of difference. S targets should be detected in SARS-CoV-2 only, but some variants can be missed. Consequently, a combination of these targets must be used for test development. | Sigma-Aldrich [32] |
| Green | Rv AGCAGCATCACCGCCATTG | ||||||||
| 11 ∥ | SYBR | N-2 | Fw AGCCTCTTCTCGTTCCTCATCAC | 28,815 | 102 | NC | SARS | ||
| Green | Rv CCGCCATTGCCAGCCATTC | ||||||||
| 11 ∥ | TaqMan | ORF1a-3 | Fw CCGCAAGGTTCTTCTTCGTAAG | 618 | 146 | NC | SARS | ||
| Rv TGCTATGTTTAGTGTTCCAGTTTTC | |||||||||
| P AAGGATCAGTGCCAAGCTCGTCGCC | |||||||||
| 11 ∥ | TaqMan | ORF1a-4 | Fw GGCTTACCGCAAGGTTCTTC | 612 | 152 | NC | SARS | ||
| Rv TGCTATGTTTAGTGTTCCAGTTTTC | |||||||||
| P AAGGATCAGTGCCAAGCTCGTCGCC | |||||||||
| 11 ∥ | TaqMan | S-5 | Fw CAGGTATATGCGCTAGTTATCAGAC | 23,564 | 97 | NC | SARS- | ||
| Rv CCAAGTGACATAGTGTAGGCAATG | CoV-2 | ||||||||
| P AGACTAATTCTCCTCGGCGGGCACG | |||||||||
| 11 ∥ | TaqMan | S-6 | Fw GCAGGTATATGCGCTAGTTATCAG | 23,563 | 187 | NC | SARS- | ||
| Rv ACACTGGTAGAATTTCTGTGGTAAC | CoV-2 | ||||||||
| P AGACTAATTCTCCTCGGCGGGCACG | |||||||||
| 12 | TaqMan | E | Fw ACTTCTTTTTCTTGCTTTCGTGGT | 26,294 | 82 | 60 °C | SARS- | NA | Huang et al. [33] |
| Rv GCAGCAGTACGCACACAATC | CoV-2 | ||||||||
| P CTAGTTACACTAGCCATCCTTACTGC |
| Assay | Target | Genomes with Mismatches in the First Five Nucleotides of the Primer’s 3′ End | Genomes with >10% Mismatches in the Annealing Sites of Primers and Probes | False Negative Results *** | Inclusivity | ||
|---|---|---|---|---|---|---|---|
| Number * | Modifications ** | Number * | Modifications ** | ||||
| 1 | ORF1b | 1 | Fw GTGGGTTTTACATTTAA | 0 | - | 1 | 99.96% |
| 1 | N | 1 | Rv CAGACATTTTGCTCTCAAACTG | 358 | Fw AACGAACTTCTCCTGCTAGAAT | 359 | 86.03% |
| 2 | RdRp-P1 | 0 | - | 1 | Pb CCAGGTGGGACCTCATCAGGAGATGC | 1 | 99.96% |
| 2 | RdRp-P2 | 0 | - | 0 | - | 0 | 100% |
| 2 | E | 0 | - | 0 | - | 0 | 100% |
| 2 | N | 4 | Fw CACATTGGCACCCGTAATC | 0 | - | 5 | 99.81% |
| 1 | Rv GAGGAACGAGAAGAGACTTG | ||||||
| 3 | RdRp-IP2 | 3 | Rv CTCCCTTTGTTGTGTTAT | 0 | - | 3 | 99.88% |
| 3 | RdRp-IP4 | 0 | - | 0 | - | 0 | 100% |
| 3 | E | 0 | - | 0 | - | 0 | 100% |
| 4 | N-1 | 7 | Rv TCTGGTTACTGCCAGTTGAACCTG | 0 | - | 7 | 99.73% |
| 4 | N-2 | 1 | Fw TTACAAACATTGGCCTCAAA | 0 | - | 1 | 99.96% |
| 4 | N-3 | 0 | - | 0 | - | 0 | 100% |
| 5 | N | 3 | Fw AAATTTTGGGGACCATGAAC | 0 | - | 8 | 99.69% |
| 4 | Fw AAATTTTGGGGACCAGGAAT | ||||||
| 1 | Rv TGGCACCTGTGTAGGTAAAC | ||||||
| 6 | ORF1b/nsp14 | 0 | - | 0 | - | 0 | 100% |
| 6 | N | 2 | Rv CGAAGGTGTGACTTCAATG | 0 | - | 2 | 99.92% |
| 7 | N | 7 | Fw CGTTTGGTGGACCCTCAGGT | 0 | - | 7 | 99.73% |
| 8 | RdRp/Hel | 0 | - | 0 | - | 0 | 100% |
| 8 | S | 0 | - | 0 | - | 0 | 100% |
| 8 | N | 1 | Fw GCGTTCTTCGGAATGTCT | 0 | - | 1 | 99.96% |
| 9 | ORF1a | 0 | - | 0 | - | 0 | 100% |
| 10 | RdRp | 8 | Rv TGCATTAACATTGGCCGTAA | 0 | - | 8 | 99.69% |
| 10 | S | 0 | - | 0 | - | 0 | 100% |
| 10 | E | 0 | - | 0 | - | 0 | 100% |
| 10 | N | 1 | Rv GTTGCGACTACGTGATGAGT | 0 | - | 1 | 99.96% |
| 11 | N-1 | 3 | Fw GCCTCTTCTCGTTCCTCACCAC | 0 | - | 8 | 99.69% |
| 5 | FW GCCTCTTCTCGTTCCTCATTAC | ||||||
| 11 | N-2 | 3 | Fw AGCCTCTTCTCGTTCCTCACCAC | 0 | - | 8 | 99.69% |
| 5 | Fw AGCCTCTTCTCGTTCCTCATTAC | ||||||
| 11 | ORF1a-3 | 0 | - | 0 | - | 0 | 100% |
| 11 | ORF1a-4 | 0 | - | 0 | - | 0 | 100% |
| 11 | S-5 | 9 | Fw CAGGTATATGTGCTAGTTATCACAC | 0 | - | 11 | 99.57% |
| 1 | Fw CAGGTATATGCGCTAGTTATCATAC | ||||||
| 1 | Fw CAGGTATATGCGCTAGTTATCGGAC | ||||||
| 11 | S-6 | 9 | Fw GCAGGTATATGTGCTAGTTATCAC | 0 | - | 11 | 99.57% |
| 1 | Fw GCAGGTATATGCGCTAGTTATCGT | ||||||
| 1 | Fw GCAGGTATATGCGCTAGTTATCGG | ||||||
| 12 | E | 0 | - | 0 | - | 0 | 100% |
| Assay | Target | Modifications * | Number of Genomes with the Modification(s) |
|---|---|---|---|
| 2 | RdRp-P1 | Pb CCAGGTGGAACCTCATCAGGAGATGC | 2566 (99.88%) |
| 2 | RdRp-P1&P2 | Rv CAAATGTTAAAAACACTATTAGCATA | 2569 (100%) |
| 5 | N | Rv TGGCACCTGTGTAGGTCAAC † | 2569 (100%) |
| 8 | RdRp/Hel | Rv ATGTGATGTTGATATGACATGGTC | 2569 (100%) |
| 10 | E | Rv CACACAATCGAAGCGCAGTA | 2567 (99.92%) |
| Assay | Target | Genomes Giving a Positive Signal | False Positive Results * | Exclusivity |
|---|---|---|---|---|
| 1 | ORF1b | 0 | 0 | 100% |
| 1 | N | 0 | 0 | 100% |
| 2 | RdRp-P1 | 172 SARS-related coronavirus | 0 | 100% |
| 2 | RdRp-P2 | 5 SARS-related coronavirus and 2 unclassified bat coronavirus | 7 | 99.75% |
| 2 | E | 179 SARS-related coronavirus and 6 unclassified bat coronavirus | 0 | 100% |
| 2 | N | 162 SARS-related coronavirus and 5 unclassified bat coronavirus | 0 | 100% |
| 3 | RdRp-IP2 | 0 | 0 | 100% |
| 3 | RdRp-IP4 | 0 | 0 | 100% |
| 3 | E | 179 SARS-related coronavirus and 6 unclassified bat coronavirus | 0 | 100% |
| 4 | N-1 | 0 | 0 | 100% |
| 4 | N-2 | 0 | 0 | 100% |
| 4 | N-3 | 13 SARS-related coronavirus and 1 unclassified bat coronavirus | 0 | 100% |
| 5 | N | 0 | 0 | 100% |
| 6 | ORF1b/nsp14 | 170 SARS-related coronavirus and 5 unclassified bat coronavirus | 0 | 100% |
| 6 | N | 179 SARS-related coronavirus and 5 unclassified bat coronavirus | 0 | 100% |
| 7 | N | 0 | 0 | 100% |
| 8 | RdRp/Hel | 0 | 0 | 100% |
| 8 | S | 0 | 0 | 100% |
| 8 | N | 0 | 0 | 100% |
| 9 | ORF1a | 0 | 0 | 100% |
| 10 | RdRP | 0 | 0 | 100% |
| 10 | S | 0 | 0 | 100% |
| 10 | E | 181 SARS-related coronavirus and 5 unclassified bat coronavirus | 186 | 92.32% |
| 10 | N | 2 SARS-related coronavirus | 2 | 99.92% |
| 11 | N-1 | 0 | 0 | 100% |
| 11 | N-2 | 0 | 0 | 100% |
| 11 | ORF1a-3 | 2 SARS-related coronavirus | 2 | 100% |
| 11 | ORF1a-4 | 0 | 0 | 100% |
| 11 | S-5 | 0 | 0 | 100% |
| 11 | S-6 | 0 | 0 | 100% |
| 12 | E | 178 SARS-related coronavirus and 6 unclassified bat coronavirus | 184 | 92.41% |
| Assay | Target | Genomes with Mismatches in the First Five Nucleotides of the Primer’s 3′ End | Genomes with too Many Mismatches in the Annealing Sites of Primers and Probes | False Negative Results *** | Inclusivity | ||
|---|---|---|---|---|---|---|---|
| Number * | Modifications ** | Number * | Modifications ** | ||||
| 1 | ORF1b | 0 | - | 0 | - | 0 | 100% |
| 1 | N | 1 | Fw GGGGAACTTCTCCTGCTAAAAT | 241 | Fw AACGAACTTCTCCTGCTAGAAT | 247 | 74.54% |
| 4 | Fw GGGGAACTTCTCCTGCTACAAT | 1 | Fw AACGAACTTCTCCTTCTAGAAT | ||||
| 2 **** | RdRp-P1 | 1 | Fw GTGAAATGGTCATGTGTAGCGG | 0 | - | 2 | 99.79% |
| 1 | Fw GTGAAATGGTCATGTGTGGTGG | 0 | - | ||||
| 2 **** | RdRp-P2 | 1 | Fw GTGAAATGGTCATGTGTAGCGG | 0 | - | 2 | 99.79% |
| 1 | Fw GTGAAATGGTCATGTGTGGTGG | 0 | - | ||||
| 2 | E | 0 | - | 0 | - | 0 | 100% |
| 2 | N | 0 | - | 0 | - | 0 | 100% |
| 4 | N-1 | 0 | - | 0 | - | 0 | 100% |
| 4 | N-2 | 0 | - | 0 | - | 0 | 100% |
| 4 | N-3 | 0 | - | 0 | - | 0 | 100% |
| 8 | RdRp/Hel | 0 | - | 0 | - | 0 | 100% |
| 8 | S | 0 | - | 0 | - | 0 | 100% |
| 8 | N | 1 | Rv TTGGATCTTTGTCATCCAATTTA | 0 | - | 1 | 99.90% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gand, M.; Vanneste, K.; Thomas, I.; Van Gucht, S.; Capron, A.; Herman, P.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection. Int. J. Mol. Sci. 2020, 21, 5585. https://doi.org/10.3390/ijms21155585
Gand M, Vanneste K, Thomas I, Van Gucht S, Capron A, Herman P, Roosens NHC, De Keersmaecker SCJ. Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection. International Journal of Molecular Sciences. 2020; 21(15):5585. https://doi.org/10.3390/ijms21155585
Chicago/Turabian StyleGand, Mathieu, Kevin Vanneste, Isabelle Thomas, Steven Van Gucht, Arnaud Capron, Philippe Herman, Nancy H. C. Roosens, and Sigrid C. J. De Keersmaecker. 2020. "Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection" International Journal of Molecular Sciences 21, no. 15: 5585. https://doi.org/10.3390/ijms21155585
APA StyleGand, M., Vanneste, K., Thomas, I., Van Gucht, S., Capron, A., Herman, P., Roosens, N. H. C., & De Keersmaecker, S. C. J. (2020). Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection. International Journal of Molecular Sciences, 21(15), 5585. https://doi.org/10.3390/ijms21155585