Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds



Abstract
1. Introduction
2. Results and Discussion
2.1. Comparison of Proteases between SARS-CoV-2 and SARS-CoV
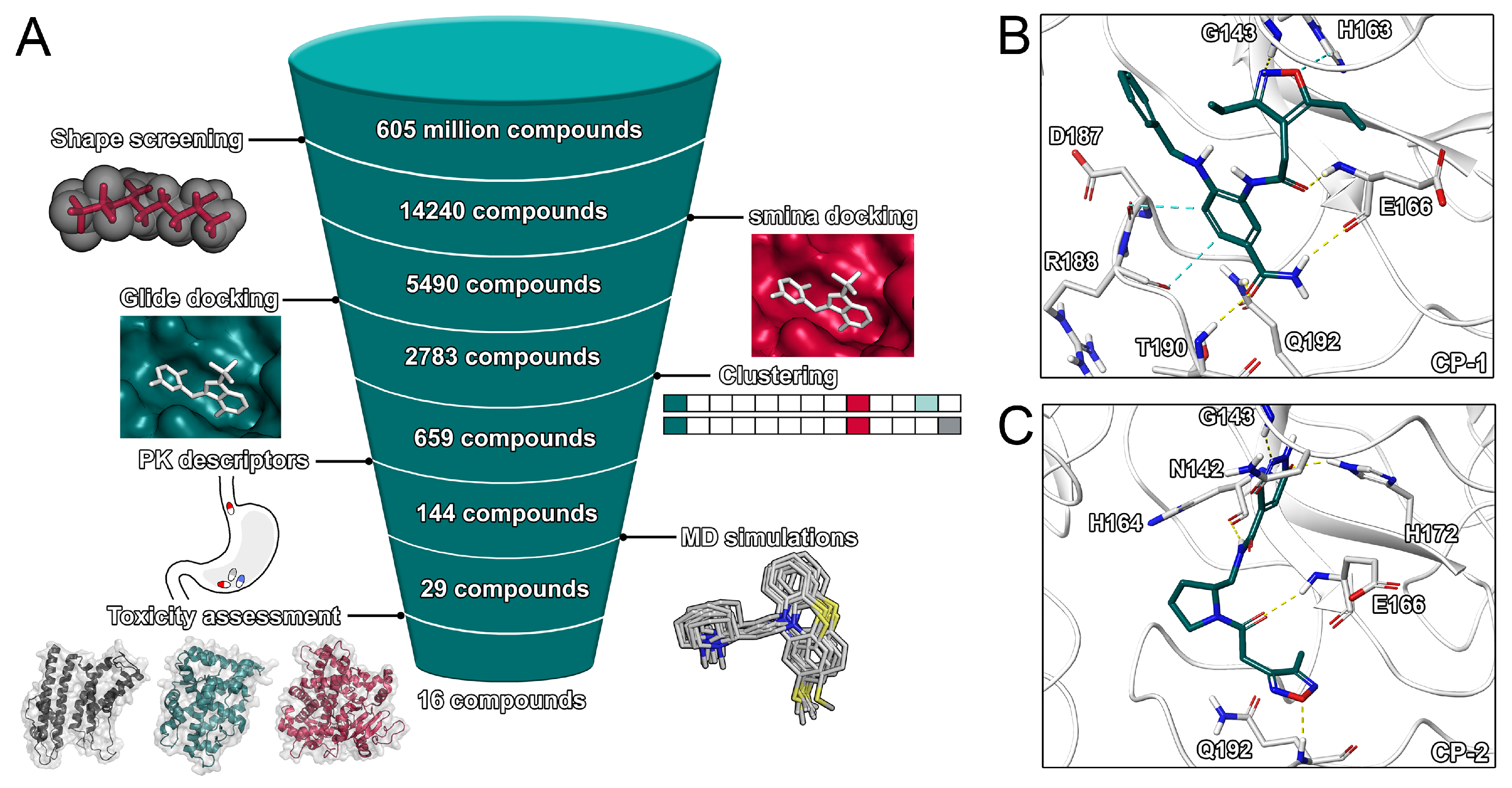
2.2. Virtual Screening Procedures
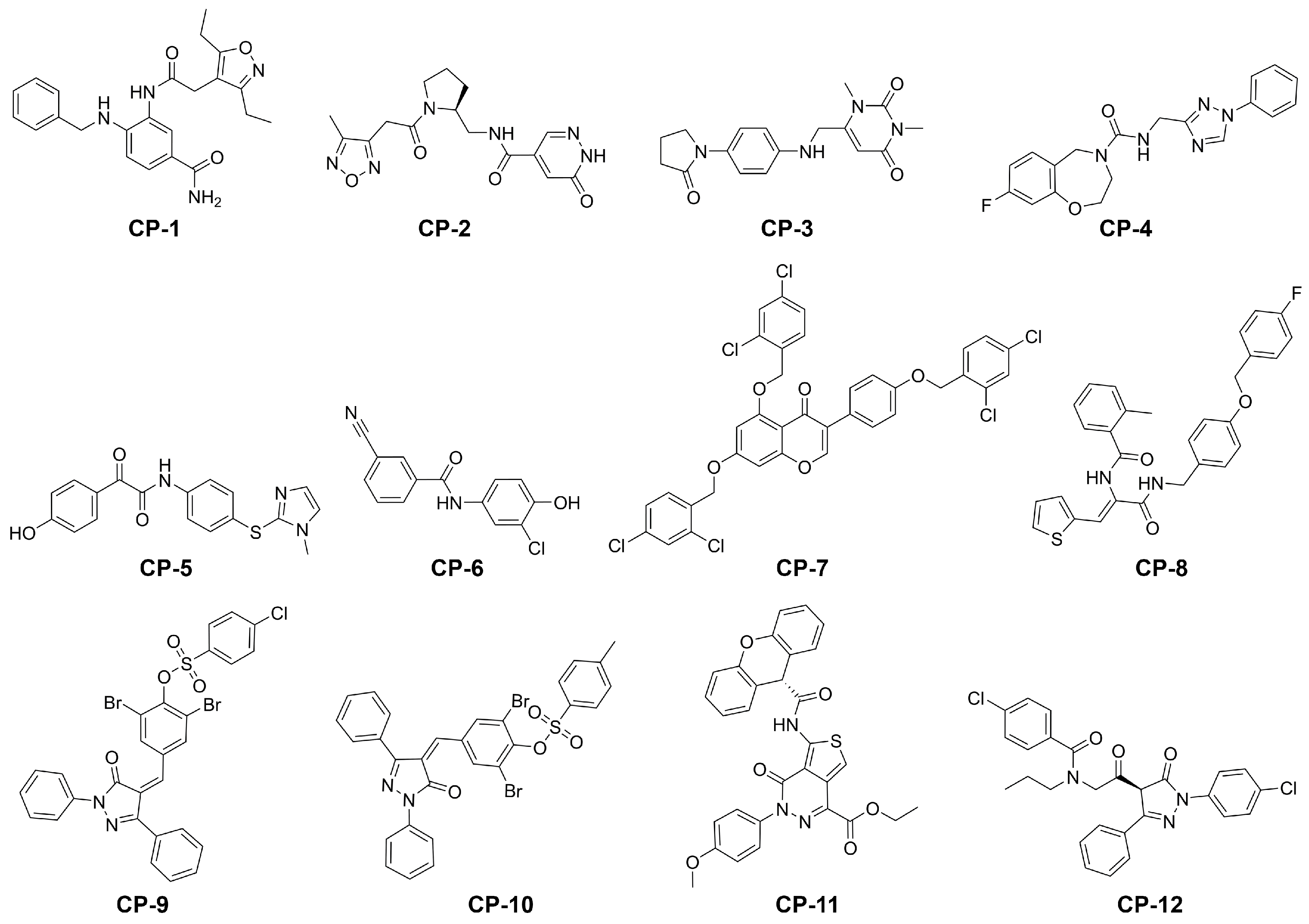
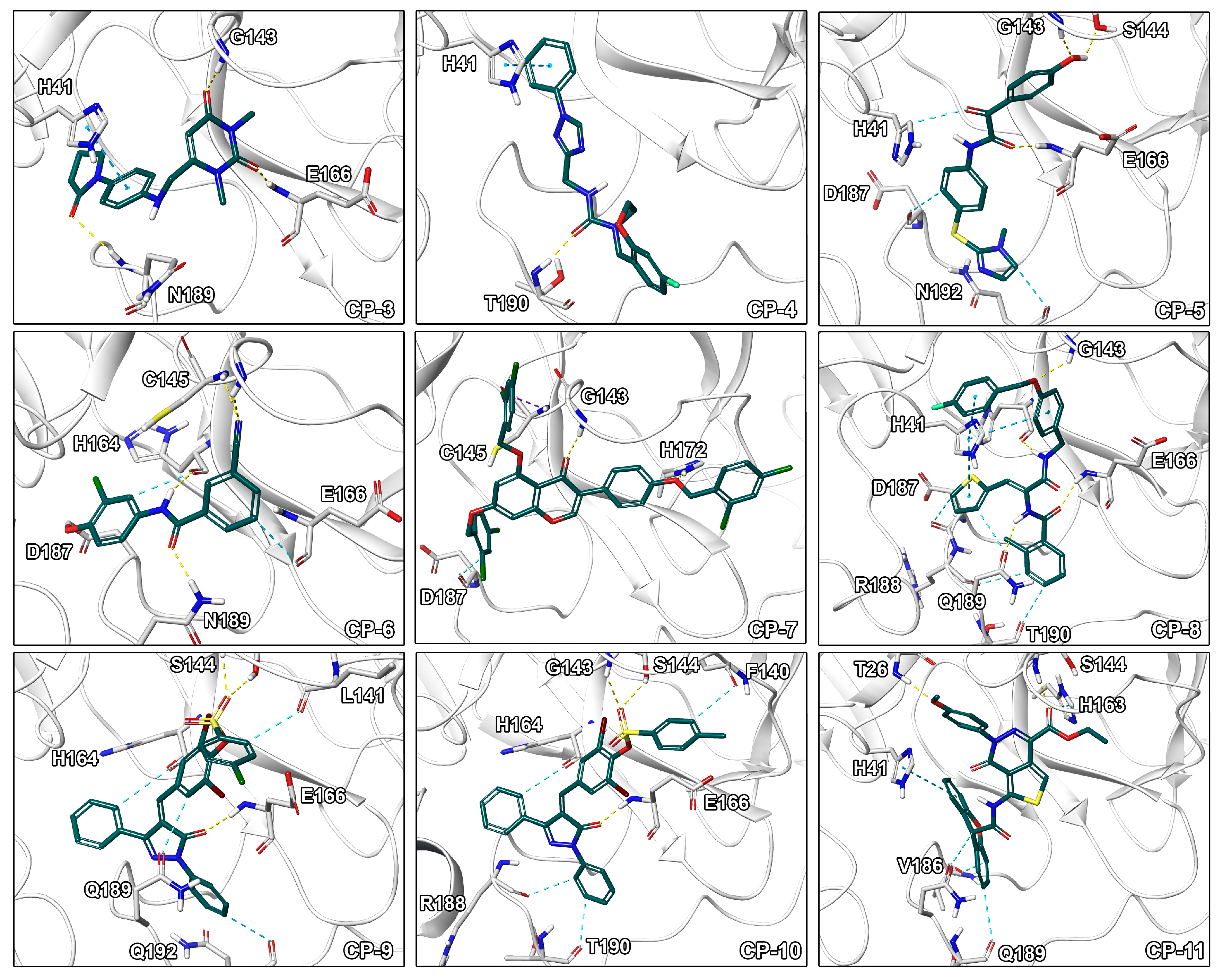
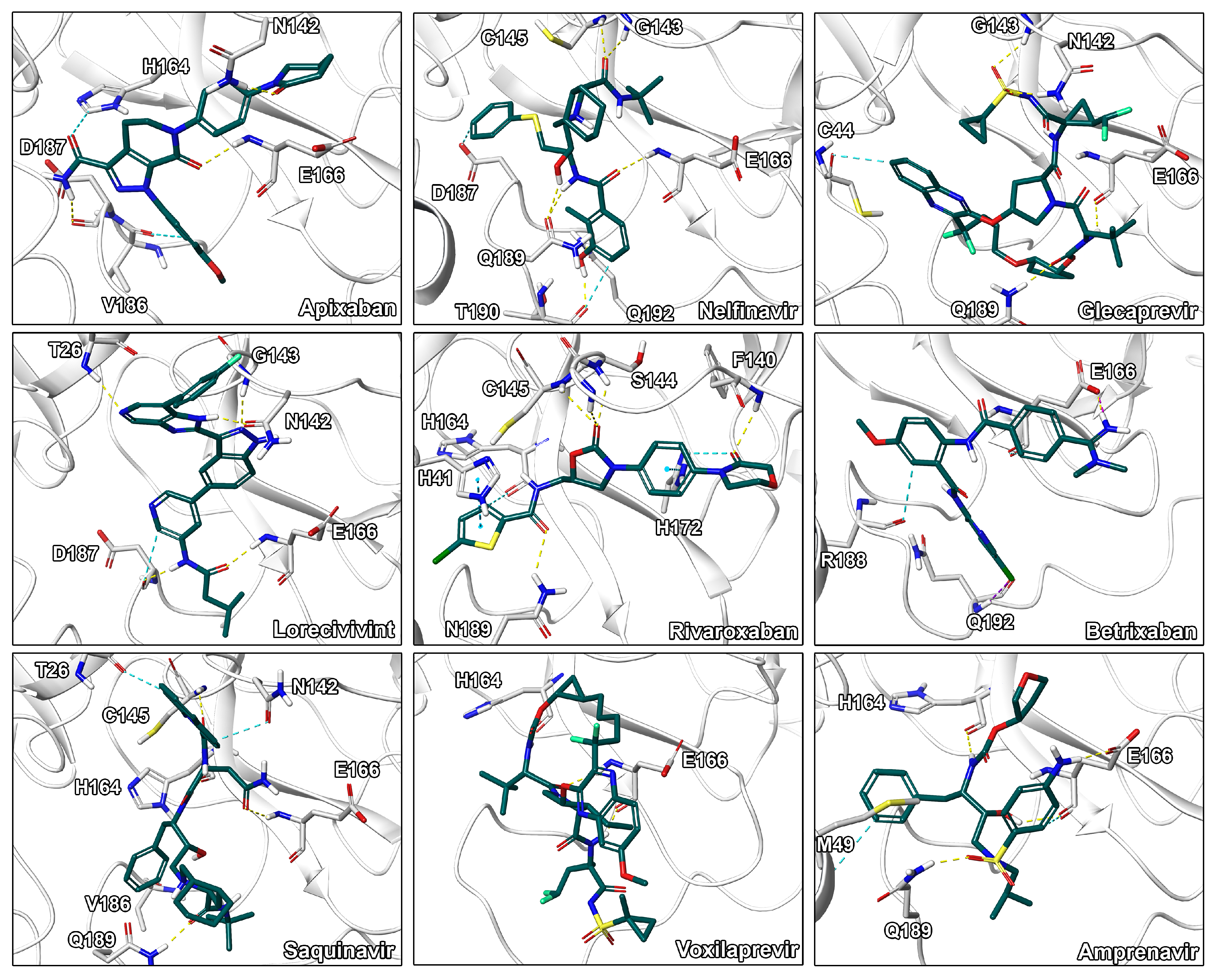
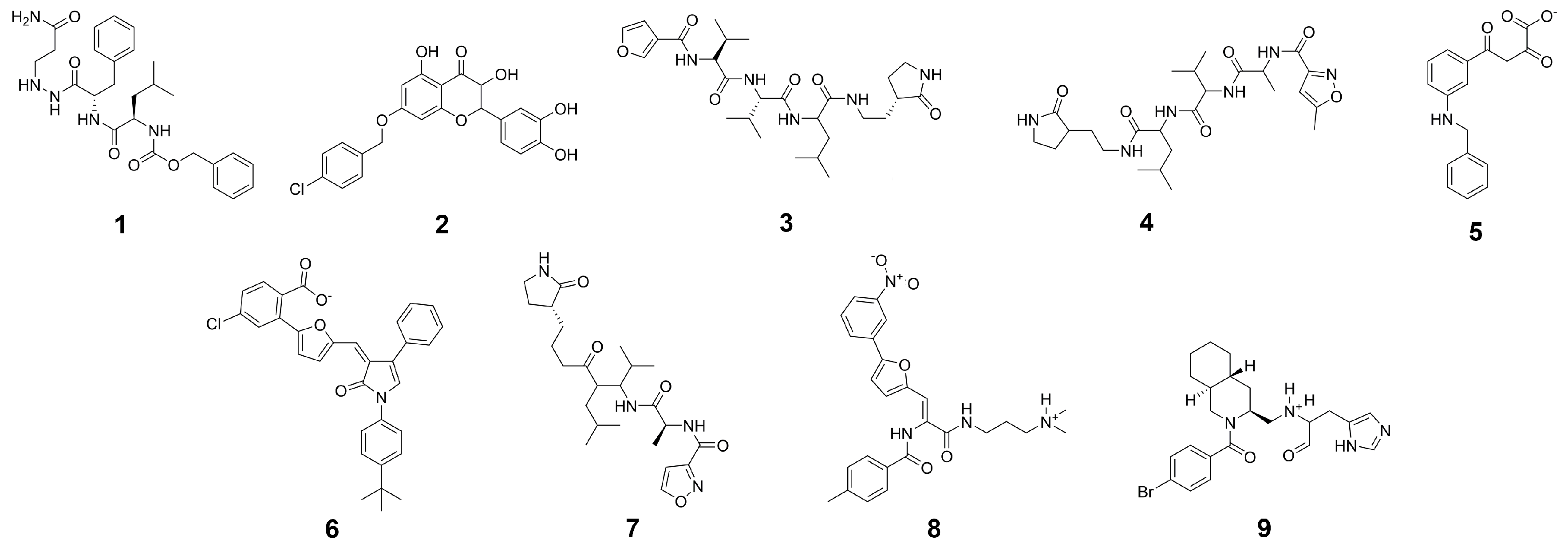
2.3. Final Compound Selection
3. Materials and Methods
3.1. System Preparation and Ensemble Generation
3.2. Pharmacokinetic Descriptors
3.3. Docking and Shape Screening
3.4. MD Simulations and Post-Processing
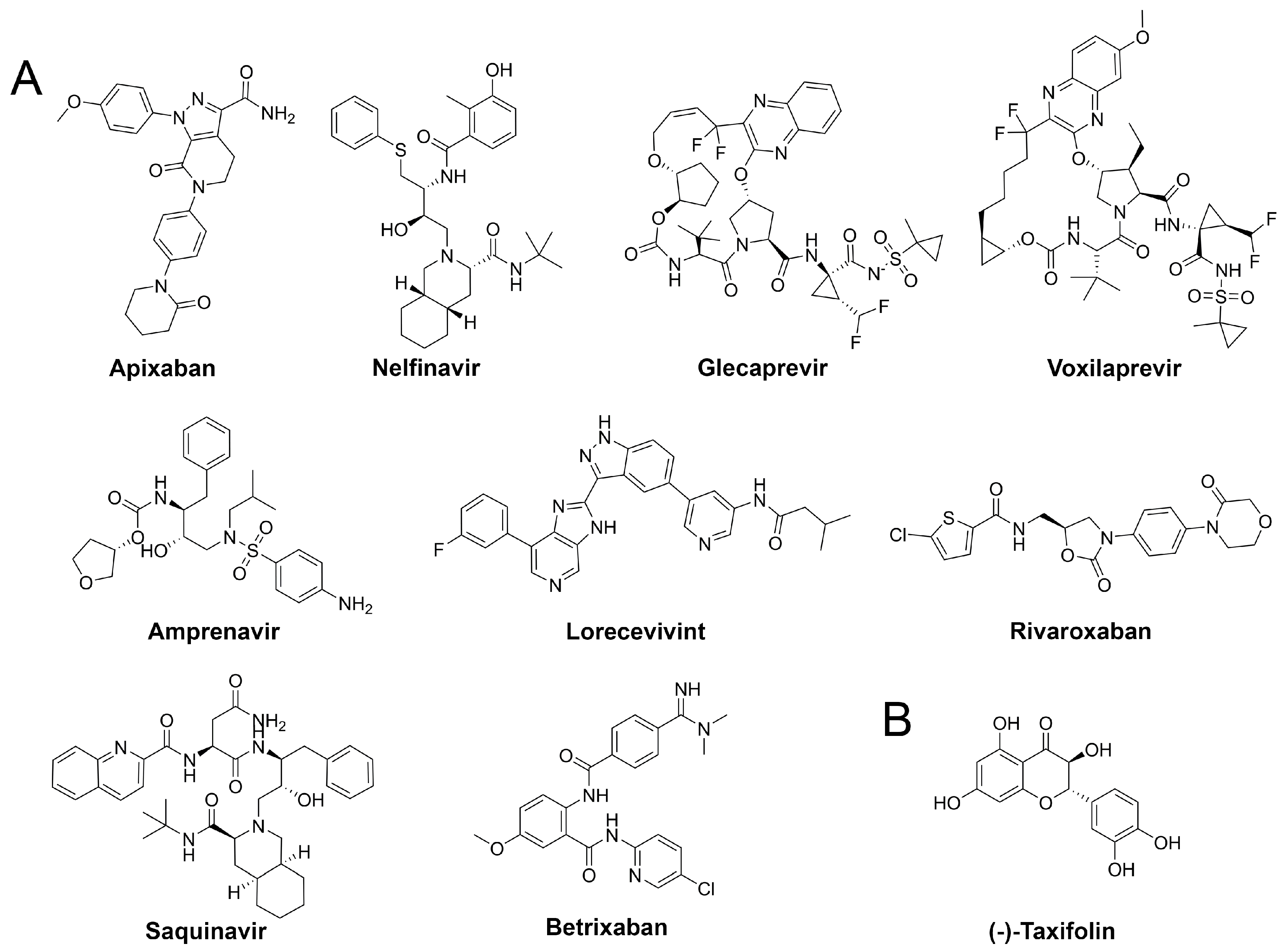
3.5. Drug Repurposing
3.6. Computational Off-Target Profiling and Selection of Final Set of Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Novel Coronavirus (2019-nCoV) Situation Reports; World Health Organisation: Geneva, Switzerland, 2020. [Google Scholar]
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of Main Protease from Human Coronavirus NL63: Insights for Wide Spectrum Anti-Coronavirus Drug Design. Sci. Rep. 2016, 6, 22677. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.M.; Fu, J.F.; Shu, Q.; Chen, Y.H.; Hua, C.Z.; Li, F.B.; Lin, R.; Tang, L.F.; Wang, T.L.; Wang, W.; et al. Diagnosis and treatment recommendations for pediatric respiratory infection caused by the 2019 novel coronavirus. World J. Pediatr. WJP 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. International Health Regulations Emergency Committee on Novel Coronavirus in China; World Health Organisation: Geneva, Switzerland, 2020. [Google Scholar]
- Stoermer, M.J. Homology Models of the Papain-Like Protease PLpro from Coronavirus 2019-nCoV. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Keogh-Brown, M.R.; Smith, R.D. The economic impact of SARS: How does the reality match the predictions? Health Policy 2008, 88, 110–120. [Google Scholar] [CrossRef]
- Xu, Z.; Peng, C.; Shi, Y.; Zhu, Z.; Mu, K.; Wang, X.; Zhu, W. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. BioRxiv 2020, 1201, 1–20. [Google Scholar]
- Zumla, A.; Hui, D.S.; Azhar, E.I.; Memish, Z.A.; Maeurer, M. Reducing mortality from 2019-nCoV: Host-directed therapies should be an option. Lancet 2020, 395, e35–e36. [Google Scholar] [CrossRef]
- Zhang, H.; Saravanan, K.M.; Yang, Y.; Hossain, T. Deep learning based drug screening for novel coronavirus 2019-nCov. Preprints 2020, 19, 1–17. [Google Scholar] [CrossRef][Green Version]
- Yang, S.; Chen, S.J.; Hsu, M.F.; Wu, J.D.; Tseng, C.T.K.; Liu, Y.F.; Chen, H.C.; Kuo, C.W.; Wu, C.S.; Chang, L.W.; et al. Synthesis, Crystal Structure, Structure-Activity Relationships, and Antiviral Activity of a Potent SARS Coronavirus 3CL Protease Inhibitor. J. Med. Chem. 2006, 49, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, B.; Jin, Z.; Yang, H.; Rao, Z. The Crystal Structure of COVID-19 Main Protease in Complex with an Inhibitor N3; Protein DataBank: New York, NY, USA, 2020. [Google Scholar]
- Li, Y.; Zhang, J.; Wang, N.; Li, H.; Shi, Y.; Guo, G.; Liu, K.; Zeng, H.; Zou, Q. Therapeutic Drugs Targeting 2019-nCoV Main Protease by High-Throughput Screening. bioRxiv 2020, 922922. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Raugi, D.N.; Smith, R.A.; Gottlieb, G.S. Four Amino Acid Changes in HIV-2 Protease Confer Class-Wide Sensitivity to Protease Inhibitors. J. Virol. 2016, 90, 1062–1069. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.P.; Wong, K.Y. Prediction of the 2019-nCoV 3C-like protease (3CLpro) structure_ virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates.pdf.crdownload. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2014, 31, 405–412. [Google Scholar] [CrossRef]
- Vedani, A.; Dobler, M.; Hu, Z.; Smieško, M. OpenVirtualToxLab-A platform for generating and exchanging in silico toxicity data. Toxicol. Lett. 2015, 232, 519–532. [Google Scholar] [CrossRef]
- He, L.; Zhu, J. Computational tools for epitope vaccine design and evaluation. Curr. Opin. Virol. 2015, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Alisi, M.A.; Cazzolla, N.; Coletta, I.; Furlotti, G.; Garofalo, B.; Mangano, G.; Mancini, F.; Vitiello, M.; Ombrato, R. A Novel Multi-step Virtual Screening for the Identification of Human and Mouse mPGES-1 Inhibitors. Mol. Inform. 2016, 91045543, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Sun, H.; Xi, L.; Li, J.; Yang, Y.; Liu, H.; Yao, X. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM-GBSA calculation. J. Comput. Chem. 2011, 32, 2800–2809. [Google Scholar] [CrossRef]
- Ye, W.L.; Yang, S.Q.; Zhang, L.X.; Deng, Z.K.; Li, W.Q.; Zhang, J.W.; Zhang, L.; Yun, Y.H.; Chen, A.F.; Cao, D.S. Multistep virtual screening for rapid identification of G Protein-Coupled Receptors Kinase 2 inhibitors for heart failure treatment. Chemom. Intell. Lab. Syst. 2019, 185, 32–40. [Google Scholar] [CrossRef]
- Kotowski, K.; Supplitt, S.; Wiczew, D.; Przystupski, D.; Bartosik, W.; Saczko, J.; Rossowska, J.; Dra̧g-Zalesińska, M.; Michel, O.; Kulbacka, J. 3PO as a Selective Inhibitor of 6-Phosphofructo-2-Kinase/ Fructose-2,6-Biphosphatase 3 in A375 Human Melanoma Cells. Anticancer Res. 2020, 40, 2613–2625. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Nguyen, D.D.; Wang, R.; Wei, G.W. Machine intelligence design of 2019-nCoV drugs. bioRxiv 2020, 927889. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Shekfeh, S.; Çalışkan, B.; Fischer, K.; Yalçın, T.; Garscha, U.; Werz, O.; Banoglu, E. A Multi-step Virtual Screening Protocol for the Identification of Novel Non-acidic Microsomal Prostaglandin E 2 Synthase-1 (mPGES-1) Inhibitors. ChemMedChem 2019, 14, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cao, S.; Hevener, K.E.; Truong, L.; Gatuz, J.L.; Patel, K.; Ghosh, A.K.; Johnson, M.E. Synergistic inhibitor binding to the papain-like protease of human SARS coronavirus: Mechanistic and inhibitor design implications. ChemMedChem 2013, 8, 1361–1372. [Google Scholar] [CrossRef]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Forssén, P.; Multia, E.; Samuelsson, J.; Andersson, M.; Aastrup, T.; Altun, S.; Wallinder, D.; Wallbing, L.; Liangsupree, T.; Riekkola, M.L.; et al. Reliable Strategy for Analysis of Complex Biosensor Data. Anal. Chem. 2018, 90, 5366–5374. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P. Going further than Lipinski’s rule in drug design. Expert Opin. Drug Discov. 2012, 7, 99–107. [Google Scholar] [CrossRef]
- Nguyen, T.T.H.; Woo, H.J.; Kang, H.K.; Nguyen, V.D.; Kim, Y.M.; Kim, D.W.; Ahn, S.A.; Xia, Y.; Kim, D. Flavonoid-mediated inhibition of SARS coronavirus 3C-like protease expressed in Pichia pastoris. Biotechnol. Lett. 2012, 34, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Slámová, K.; Kapešová, J.; Valentová, K. “Sweet Flavonoids”: Glycosidase-Catalyzed Modifications. Int. J. Mol. Sci. 2018, 19, 2126. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J.; Dupont, M.S.; Ridley, S.; Rhodes, M.; Rhodes, M.J.; Morgan, M.R.; Williamson, G. Deglycosylation of flavonoid and isoflavonoid glycosides by human small intestine and liver β-glucosidase activity. FEBS Lett. 1998, 436, 71–75. [Google Scholar] [CrossRef]
- Han, H.; Ma, Z.; Wang, W.; Xu, M.; Zhou, S.; Li, L.; Jiang, H. Deglycosylation and absorption of marein, flavanomarein and taxifolin-7-O-β-D-glucopyranoside from capitula of Coreopsis tinctoria in rats and humans. J. Funct. Foods 2016, 27, 178–188. [Google Scholar] [CrossRef]
- Pozharitskaya, O.N.; Karlina, M.V.; Shikov, A.N.; Kosman, V.M.; Makarova, M.N.; Makarov, V.G. Determination and pharmacokinetic study of taxifolin in rabbit plasma by high-performance liquid chromatography. Phytomedicine 2009, 16, 244–251. [Google Scholar] [CrossRef]
- Abd El-Hack, M.E.; Alagawany, M.; Elrys, A.S.; Desoky, E.S.M.; Tolba, H.M.; Elnahal, A.S.; Elnesr, S.S.; Swelum, A.A. Effect of forage moringa oleifera l. (moringa) on animal health and nutrition and its beneficial applications in soil, plants and water purification. Agriculture 2018, 8, 145. [Google Scholar] [CrossRef]
- Novo Belchor, M.; Hessel Gaeta, H.; Fabri Bittencourt Rodrigues, C.; Ramos da Cruz Costa, C.; de Oliveira Toyama, D.; Domingues Passero, L.F.; Dalastra Laurenti, M.; Hikari Toyama, M. Evaluation of Rhamnetin as an Inhibitor of the Pharmacological Effect of Secretory Phospholipase A2. Molecules 2017, 22, 1441. [Google Scholar] [CrossRef]
- Lee, S.; Shin, S.Y.; Lee, Y.; Park, Y.; Kim, B.G.; Ahn, J.H.; Chong, Y.; Lee, Y.H.; Lim, Y. Rhamnetin production based on the rational design of the poplar O-methyltransferase enzyme and its biological activities. Bioorg. Med. Chem. Lett. 2011, 21, 3866–3870. [Google Scholar] [CrossRef]
- Moon, Y.J.; Wang, L.; DiCenzo, R.; Morris, M.E. Quercetin pharmacokinetics in humans. Biopharm. Drug Dispos. 2008, 29, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.; Hoppensteadt, D.; Jeske, W.; Iqbal, O.; Tafur, A.; Fareed, J. Factor Xa Inhibitory Profile of Apixaban, Betrixaban, Edoxaban, and Rivaroxaban Does Not Fully Reflect Their Biologic Spectrum. Clin. Appl. Thromb. 2019, 25, 1076029619847524. [Google Scholar] [CrossRef] [PubMed]
- De Leuw, P.; Stephan, C. Protease inhibitor therapy for hepatitis C virus-infection. Expert Opin. Pharmacother. 2018, 19, 577–587. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS Res. Palliat. Care 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. JTH 2020, 1094–1099. [Google Scholar] [CrossRef]
- Xu, Z.; Yao, H.; Shen, J.; Wu, N.; Xu, Y.; Lu, X.; Zhu, W.; Li, L.J. Nelfinavir Is Active Against SARS-CoV-2 in Vero E6 Cells. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Deshmukh, V.; O’Green, A.L.; Bossard, C.; Seo, T.; Lamangan, L.; Ibanez, M.; Ghias, A.; Lai, C.; Do, L.; Cho, S.; et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr. Cartil. 2019, 27, 1347–1360. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Kenny, P.W. The nature of ligand efficiency. J. Cheminform. 2019, 11, 1–18. [Google Scholar] [CrossRef]
- Waszkowycz, B. Towards improving compound selection in structure-based virtual screening. Drug Discov. Today 2008, 13, 219–226. [Google Scholar] [CrossRef]
- Chen, S.; Chen, L.L.; Luo, H.B.; Sun, T.; Chen, J.; Ye, F.; Cai, J.H.; Shen, J.K.; Shen, X.; Jiang, H.L. Enzymatic activity characterization of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer technique. Acta Pharmacol. Sin. 2005, 26, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger LCC. Maestro Small-Molecular Drug Discovery Suite 2019-4; Schrodinger: New York, NY, USA, 2019. [Google Scholar]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, SC, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar] [CrossRef]
- ChemAxon. Marvin (v.20.4.0). 2020. Available online: http://www.chemaxon.com (accessed on 19 May 2020).
- Bhal, S.K.; Kassam, K.; Peirson, I.G.; Pearl, G.M. The rule of five revisited: Applying log D in place of log P in drug-likeness filters. Mol. Pharm. 2007, 4, 556–560. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.M.; Lee, N.R.; Jin, B.S.; Jang, K.J.; Kim, D.E.; Jeong, Y.J.; Chong, Y. Aryl diketoacids (ADK) selectively inhibit duplex DNA-unwinding activity of SARS coronavirus NTPase/helicase. Bioorg Med. Chem. Lett. 2009, 19, 1636–1638. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Lee, J.M.; Lee, N.R.; Kim, D.E.; Jeong, Y.J.; Chong, Y. Investigation of the pharmacophore space of Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) NTPase/helicase by dihydroxychromone derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 4538–4541. [Google Scholar] [CrossRef]
- Hanh Nguyen, T.T.; Ryu, H.J.; Lee, S.H.; Hwang, S.; Breton, V.; Rhee, J.H.; Kim, D. Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorg. Med. Chem. Lett. 2011, 21, 3088–3091. [Google Scholar] [CrossRef]
- Kumar, V.; Tan, K.P.; Wang, Y.M.; Lin, S.W.; Liang, P.H. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2016, 24, 3035–3042. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Diamond. Main Protease Structure and XChem Fragment Screen; Diamond Light Source Ltd.: Oxfordshire, UK, 2020. [Google Scholar]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R., III; Mackerell, J.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; Caflisch, A.; et al. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Morris, G.; Huey, R. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Tungol, A.; Rademacher, K.; Schafer, J.A. Formulary management of the protease inhibitors boceprevir and telaprevir for chronic hepatitis C virus. J. Manag. Care Pharm. JMCP 2011, 17, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Kiser, J.J.; Burton, J.R.; Anderson, P.L.; Everson, G.T. Review and management of drug interactions with boceprevir and telaprevir. Hepatology 2012, 55, 1620–1628. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Vedani, A.; Dobler, M.; Smieško, M. VirtualToxLab—A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 2012, 261, 142–153. [Google Scholar] [CrossRef]
- Goldinger, D.M.; Demierre, A.L.; Zoller, O.; Rupp, H.; Reinhard, H.; Magnin, R.; Becker, T.W.; Bourqui-Pittet, M. Endocrine activity of alternatives to BPA found in thermal paper in Switzerland. Regul. Toxicol. Pharmacol. 2015, 71, 453–462. [Google Scholar] [CrossRef]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | G a (kcal/mol) | Score b (kcal/mol) | LigEff c | logD | logS | ToxPot d | Visual e |
|---|---|---|---|---|---|---|---|
| CP-1 | −78.2 ± 5.2 | −16.7 | −2.6 | 3.0 | −4.1 | 0.226 | *** |
| CP-2 | −75.1 ± 4.1 | −15.3 | −3.0 | −1.9 | −1.8 | 0.409 | **** |
| CP-3 | −70.6 ± 3.9 | −14.5 | −2.9 | 0.0 | −2.3 | 0.352 | **** |
| CP-4 | −70.3 ± 5.0 | −15.3 | −2.6 | 2.5 | −4.2 | 0.365 | ** |
| CP-5 | −69.8 ± 4.2 | −15.3 | −2.8 | 3.3 | −4.7 | 0.291 | ** |
| CP-6 | −69.6 ± 3.1 | −14.9 | −3.7 | 3.1 | −4.0 | 0.493 | **** |
| CP-7 | −91.8 ± 4.1 | −17.4 | −2.0 | 11.7 | −12.6 | 0.439 | ** |
| CP-8 | −91.4 ± 5.1 | −15.4 | −2.5 | 5.9 | −8.0 | 0.454 | **** |
| CP-9 | −84.8 ± 5.1 | −17.1 | −2.2 | 8.4 | −9.4 | 0.331 | *** |
| CP-10 | −82.7 ± 4.6 | −16.5 | −2.2 | 8.3 | −9.3 | 0.291 | *** |
| CP-11 | −81.7 ± 4.8 | −17.0 | −2.0 | 5.5 | −7.7 | 0.304 | *** |
| CP-12 | −74.5 ± 4.7 | −14.6 | −2.1 | 6.0 | −7.4 | 0.387 | *** |
| N3 | −59.3 ± 7.6 | −17.6 | −1.6 | 2.3 | −6.2 | 0.299 | **** |
| (−)-taxifolin | −53.3 ± 5.1 | −16.0 | −2.4 | 1.7 | −2.1 | 0.289 | **** |
| rhamnetin | −52.4 ± 3.5 | −16.8 | −2.3 | 0.0 | −2.1 | n/a | * |
| Compound | G (kcal/mol) a | LigEff b | Indicationc | Approval d | Visual e |
|---|---|---|---|---|---|
| Apixaban | −84.0 ± 5.5 | −2.5 | Anticoagulant | approved | **** |
| Nelfinavir | −80.6 ± 8.2 | −2.0 | Antiviral | approved | **** |
| Glecaprevir | −80.3 ± 5.2 | −1.4 | Antiviral | approved | *** |
| Lorecivivint | −79.7 ± 3.6 | −1.4 | Inflammation | experimental | **** |
| Rivaroxaban | −77.2 ± 4.8 | −2.7 | Anticoagulant | approved | **** |
| Betrixaban | −73.3 ± 6.0 | −2.3 | Anticoagulant | approved | *** |
| Saquinavir | −71.5 ± 6.2 | −1.5 | Antiviral | approved | ** |
| Voxilaprevir | −66.5 ± 5.7 | −1.1 | Antiviral | approved | ** |
| Amprenavir | −66.5 ± 4.6 | −1.9 | Antiviral | approved | **** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. https://doi.org/10.3390/ijms21103626
Fischer A, Sellner M, Neranjan S, Smieško M, Lill MA. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. International Journal of Molecular Sciences. 2020; 21(10):3626. https://doi.org/10.3390/ijms21103626
Chicago/Turabian StyleFischer, André, Manuel Sellner, Santhosh Neranjan, Martin Smieško, and Markus A. Lill. 2020. "Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds" International Journal of Molecular Sciences 21, no. 10: 3626. https://doi.org/10.3390/ijms21103626
APA StyleFischer, A., Sellner, M., Neranjan, S., Smieško, M., & Lill, M. A. (2020). Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. International Journal of Molecular Sciences, 21(10), 3626. https://doi.org/10.3390/ijms21103626