Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy


 ,
,  and
and 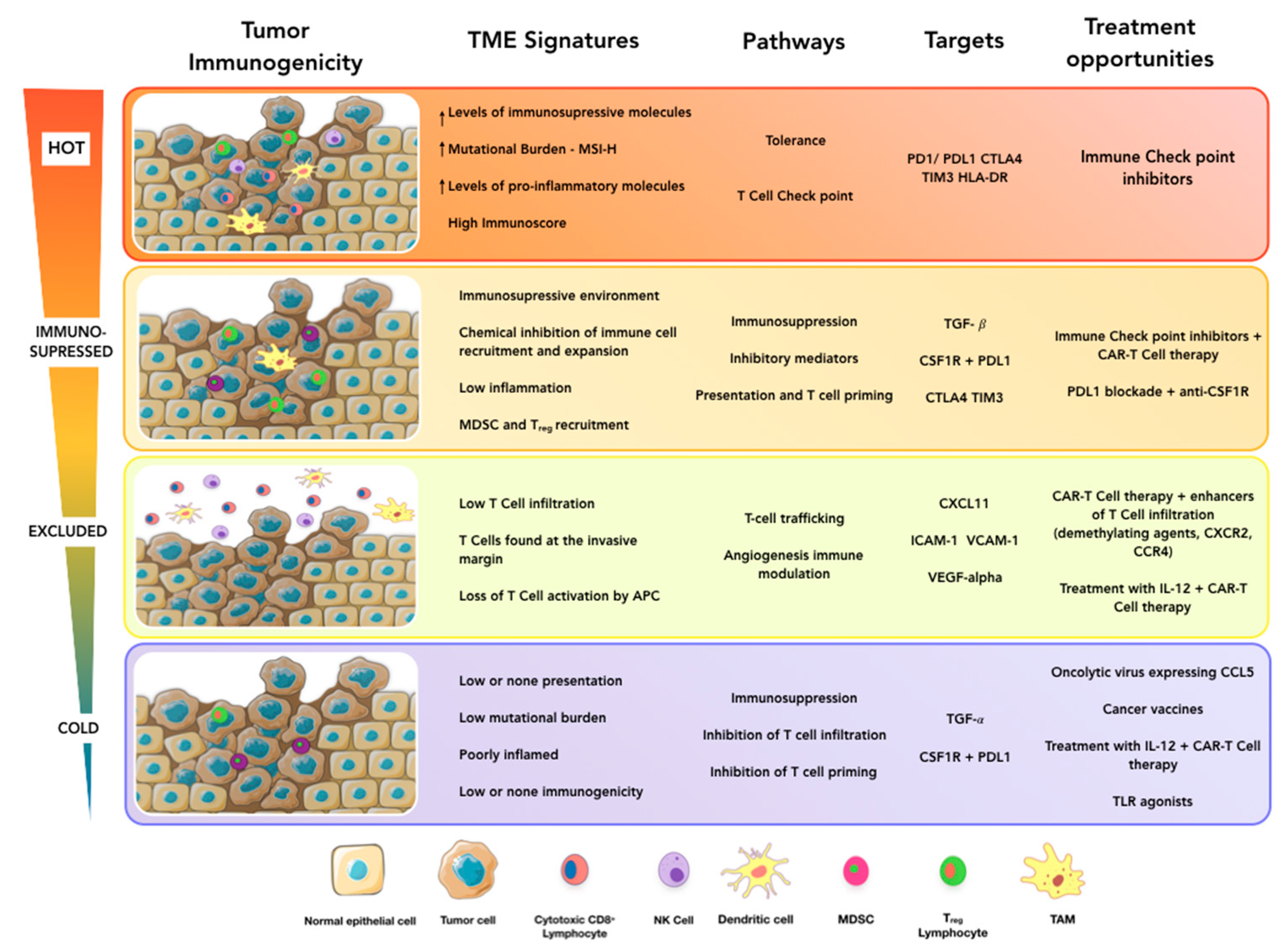{kind=link}
Abstract
1. Introduction
2. Immunological Assessment of the TME for Cancer Prognosis
3. Evaluation of the Tumor Immune Ecosystem to Refine Current Immunotherapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cells |
| CAR | chimeric antigen receptor |
| CCL | C-C motif chemokine ligand |
| CD | cluster of differentiation |
| CRC | colorectal cancer |
| CSF1R | colony-stimulating factor 1 receptor |
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL | C-X-C motif chemokine ligand |
| DC | dendritic cells |
| DNA | deoxyribonucleic acid |
| EGF | epidermal growth factor |
| Erα | estrogen receptor alpha |
| FASL | FAS ligand |
| GMCSF | granulocyte macrophage colony-stimulating factor |
| HCC | hepatocellular carcinoma |
| HLA | human leukocyte antigen |
| HLADR | human leukocyte antigen DR- isotype |
| ICAM | intracellular cell adhesion molecule |
| IFNγ | interferon gamma |
| IL | interleukin |
| KLRK1 | killer cell lectin-like receptor K1 |
| LAMP | lysosome-associated membrane glycoprotein |
| MCSF | macrophage colony-stimulating factor |
| MDSC | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MPE: | molecular pathological epidemiology |
| MSI | microsatellite instability |
| NK | natural killer cell |
| NKT | natural killer T cell |
| PD1 | programmed cell death protein 1 |
| PDL1 | programmed cell death ligand 1 |
| RNA | ribonucleic acid |
| TAM | tumor-associated macrophages |
| TGFβ | transforming growth factor-beta |
| Th | T helper cell |
| TIM3 | T cell immunoglobulin-mucin domain 3 |
| TLR | Toll-like receptor |
| TLS | tertiary lymphoid structures |
| TME | tumor microenvironment |
| TNFα | tumor necrosis factor-alpha |
| Treg | regulatory T cell |
| VCAM | vascular cell adhesion molecule |
| VEGF | vascular endothelial growth factor |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Galon, J.; Fridman, W.-H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Invest. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed]
- RHOADS, C.P. Paul ehrlich and the cancer problem. Ann. N. Y. Acad. Sci. 1954, 59, 190–197. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Linette, G.P.; Becker-Hapak, M.; Skidmore, Z.L.; Baroja, M.L.; Xu, C.; Hundal, J.; Spencer, D.H.; Fu, W.; Cummins, C.; Robnett, M.; et al. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc. Natl. Acad. Sci. USA 2019, 116, 23662–23670. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of Tumors by the Innate Immune System and Natural Killer Cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar]
- Tiwary, S.; Berzofsky, J.A.; Terabe, M. Altered Lipid Tumor Environment and Its Potential Effects on NKT Cell Function in Tumor Immunity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Hamada, T.; Nowak, J.A.; Milner, D.A.; Song, M.; Ogino, S. Integration of microbiology, molecular pathology, and epidemiology: A new paradigm to explore the pathogenesis of microbiome-driven neoplasms. J. Pathol. 2019, 247, 615–628. [Google Scholar] [CrossRef]
- Koh, H.; Hamada, T.; Song, M.; Liu, L.; Cao, Y.; Nowak, J.A.; da Silva, A.; Twombly, T.; Morikawa, T.; Kim, S.A.; et al. Physical Activity and Colorectal Cancer Prognosis According to Tumor-Infiltrating T Cells. JNCI Cancer Spectr. 2018, 2, pky058. [Google Scholar] [CrossRef]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Milner, D.A.; Nishihara, R. Insights into Pathogenic Interactions Among Environment, Host, and Tumor at the Crossroads of Molecular Pathology and Epidemiology. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 83–103. [Google Scholar] [CrossRef]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Phipps, A.I.; Peters, U.; Milner, D.A.; Giovannucci, E.L.; Nishihara, R.; Giannakis, M.; Garrett, W.S.; et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut 2018, 67, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector Memory T Cells, Early Metastasis, and Survival in Colorectal Cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Galon, J. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pages, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of Intratumoral Immune Reaction and Human Colorectal Cancer Recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In Situ Cytotoxic and Memory T Cells Predict Outcome in Patients With Early-Stage Colorectal Cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.-H.; Pagès, F.; et al. Histopathologic-Based Prognostic Factors of Colorectal Cancers Are Associated With the State of the Local Immune Reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Martinet, L.; Garrido, I.; Filleron, T.; Le Guellec, S.; Bellard, E.; Fournie, J.-J.; Rochaix, P.; Girard, J.-P. Human Solid Tumors Contain High Endothelial Venules: Association with T- and B-Lymphocyte Infiltration and Favorable Prognosis in Breast Cancer. Cancer Res. 2011, 71, 5678–5687. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B Cells in Tertiary Lymphoid Structures Is Associated with a Protective Immunity in Patients with Lung Cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Møller, H.J.; Høyer, M.; Maniecki, M.B.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Macrophage Markers in Serum and Tumor Have Prognostic Impact in American Joint Committee on Cancer Stage I/II Melanoma. J. Clin. Oncol. 2009, 27, 3330–3337. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Chao, Y.-J.; Tung, H.-L.; Wang, H.-C.; Shan, Y.-S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef]
- Hirayama, S.; Ishii, G.; Nagai, K.; Ono, S.; Kojima, M.; Yamauchi, C.; Aokage, K.; Hishida, T.; Yoshida, J.; Suzuki, K.; et al. Prognostic Impact of CD204-Positive Macrophages in Lung Squamous Cell Carcinoma: Possible Contribution of Cd204-Positive Macrophages to the Tumor-Promoting Microenvironment. J. Thorac. Oncol. 2012, 7, 1790–1797. [Google Scholar] [CrossRef]
- Shimizu, K.; Nakata, M.; Hirami, Y.; Yukawa, T.; Maeda, A.; Tanemoto, K. Tumor-Infiltrating Foxp3+ Regulatory T Cells are Correlated with Cyclooxygenase-2 Expression and are Associated with Recurrence in Resected Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 585–590. [Google Scholar] [CrossRef]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of Regulatory T Cells Enables the Identification of High-Risk Breast Cancer Patients and Those at Risk of Late Relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef]
- Ling, A.; Edin, S.; Wikberg, M.L.; Öberg, Å.; Palmqvist, R. The intratumoural subsite and relation of CD8+ and FOXP3+ T lymphocytes in colorectal cancer provide important prognostic clues. Br. J. Cancer 2014, 110, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-Infiltrating FOXP3 + T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Lee, K.S.; Cho, H.J.; Kim, Y.H.; Yang, H.K.; Kim, W.H.; Kang, G.H. Prognostic implications of tumor-infiltrating FoxP3+ regulatory T cells and CD8+ cytotoxic T cells in microsatellite-unstable gastric cancers. Hum. Pathol. 2014, 45, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Niccolai, E.; Ricci, F.; Russo, E.; Nannini, G.; Emmi, G.; Taddei, A.; Ringressi, M.N.; Melli, F.; Miloeva, M.; Cianchi, F.; et al. The Different Functional Distribution of “Not Effector” T Cells (Treg/Tnull) in Colorectal Cancer. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Badoual, C. Prognostic Value of Tumor-Infiltrating CD4+ T-Cell Subpopulations in Head and Neck Cancers. Clin. Cancer Res. 2006, 12, 465–472. [Google Scholar] [CrossRef]
- Sun, D.; Zhao, M.; Xia, M.; Li, L.; Jiang, Y. The correlation between tumor-infiltrating Foxp3+ regulatory T cells and cyclooxygenase-2 expression and their association with recurrence in resected head and neck cancers. Med. Oncol. 2012, 29, 707–713. [Google Scholar] [CrossRef]
- Kitz, A.; Dominguez-Villar, M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell. Mol. Life Sci. 2017, 74, 4059–4075. [Google Scholar] [CrossRef]
- Sawant, D.V.; Vignali, D.A.A. Once a Treg, always a Treg? Immunol. Rev. 2014, 259, 173–191. [Google Scholar] [CrossRef]
- Bhela, S.; Varanasi, S.K.; Jaggi, U.; Sloan, S.S.; Rajasagi, N.K.; Rouse, B.T. The Plasticity and Stability of Regulatory T Cells during Viral-Induced Inflammatory Lesions. J. Immunol. 2017, 199, 1342–1352. [Google Scholar] [CrossRef]
- Voo, K.S.; Wang, Y.H.; Santori, F.R.; Boggiano, C.; Wang, Y.H.; Arima, K.; Bover, L.; Hanabuchi, S.; Khalili, J.; Marinova, E.; et al. Identification of IL-17-producing FOXP3 + regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 2009, 12, 4793–4798. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Mu, S.; Wang, Y.; Wang, H.; Cai, L.; Li, W.; Hu, Y. Prognostic role of myeloid-derived suppressor cells in cancers: A systematic review and meta-analysis. BMC Cancer 2018, 18, 1220. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Niccolai, E.; Taddei, A.; Ricci, F.; Rolla, S.; D’Elios, M.M.; Benagiano, M.; Bechi, P.; Bencini, L.; Ringressi, M.N.; Pini, A.; et al. Intra-tumoral IFN-γ-producing Th22 cells correlate with TNM staging and the worst outcomes in pancreatic cancer. Clin. Sci. 2016, 130, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Friedman, C.F.; Wolchok, J.D. Combinatorial Cancer Immunotherapies. Adv. Immunol. 2016, 130, 251–277. [Google Scholar]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 2014, 124, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Magen, A.; Nie, J.; Ciucci, T.; Tamoutounour, S.; Zhao, Y.; Mehta, M.; Tran, B.; McGavern, D.B.; Hannenhalli, S.; Bosselut, R. Single-Cell Profiling Defines Transcriptomic Signatures Specific to Tumor-Reactive versus Virus-Responsive CD4+ T Cells. Cell Rep. 2019, 29, 3019–3032.e6. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Sung, L.; Pollard, J.A.; Aplenc, R.; Loken, M.R.; Gamis, A.S.; et al. Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2016, 22, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting Migration of T Cells to Chemokine Secreted from Tumors by Genetic Modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Bodey, B.; Bodey, B., Jr.; Siegel, S.E.; Kaiser, H.E. Failure of cancer vaccines: The significant limitations of this approach to immunotherapy. Anticancer. Res. 2000, 20, 2665–2676. [Google Scholar] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-Delivered Transforming Growth Factor-β siRNA Enhances Vaccination against Advanced Melanoma by Modifying Tumor Microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Braunstein, M.J.; Kucharczyk, J.; Adams, S. Targeting Toll-Like Receptors for Cancer Therapy. Target. Oncol. 2018, 13, 583–598. [Google Scholar] [CrossRef]
- Van Herpen, C.M.L. Intratumoral Recombinant Human Interleukin-12 Administration in Head and Neck Squamous Cell Carcinoma Patients Modifies Locoregional Lymph Node Architecture and Induces Natural Killer Cell Infiltration in the Primary Tumor. Clin. Cancer Res. 2005, 11, 1899–1909. [Google Scholar] [CrossRef]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor-Modified T Cells in Solid Tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Romero, K.; Rodríguez, R.M.; Amedei, A.; Barceló-Coblijn, G.; Lopez, D.H. Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5521. https://doi.org/10.3390/ijms21155521
Pérez-Romero K, Rodríguez RM, Amedei A, Barceló-Coblijn G, Lopez DH. Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy. International Journal of Molecular Sciences. 2020; 21(15):5521. https://doi.org/10.3390/ijms21155521
Chicago/Turabian StylePérez-Romero, Karim, Ramón M. Rodríguez, Amedeo Amedei, Gwendolyn Barceló-Coblijn, and Daniel H. Lopez. 2020. "Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy" International Journal of Molecular Sciences 21, no. 15: 5521. https://doi.org/10.3390/ijms21155521
APA StylePérez-Romero, K., Rodríguez, R. M., Amedei, A., Barceló-Coblijn, G., & Lopez, D. H. (2020). Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy. International Journal of Molecular Sciences, 21(15), 5521. https://doi.org/10.3390/ijms21155521