Tobacco, but Not Nicotine and Flavor-Less Electronic Cigarettes, Induces ACE2 and Immune Dysregulation

Abstract
1. Introduction
2. Results
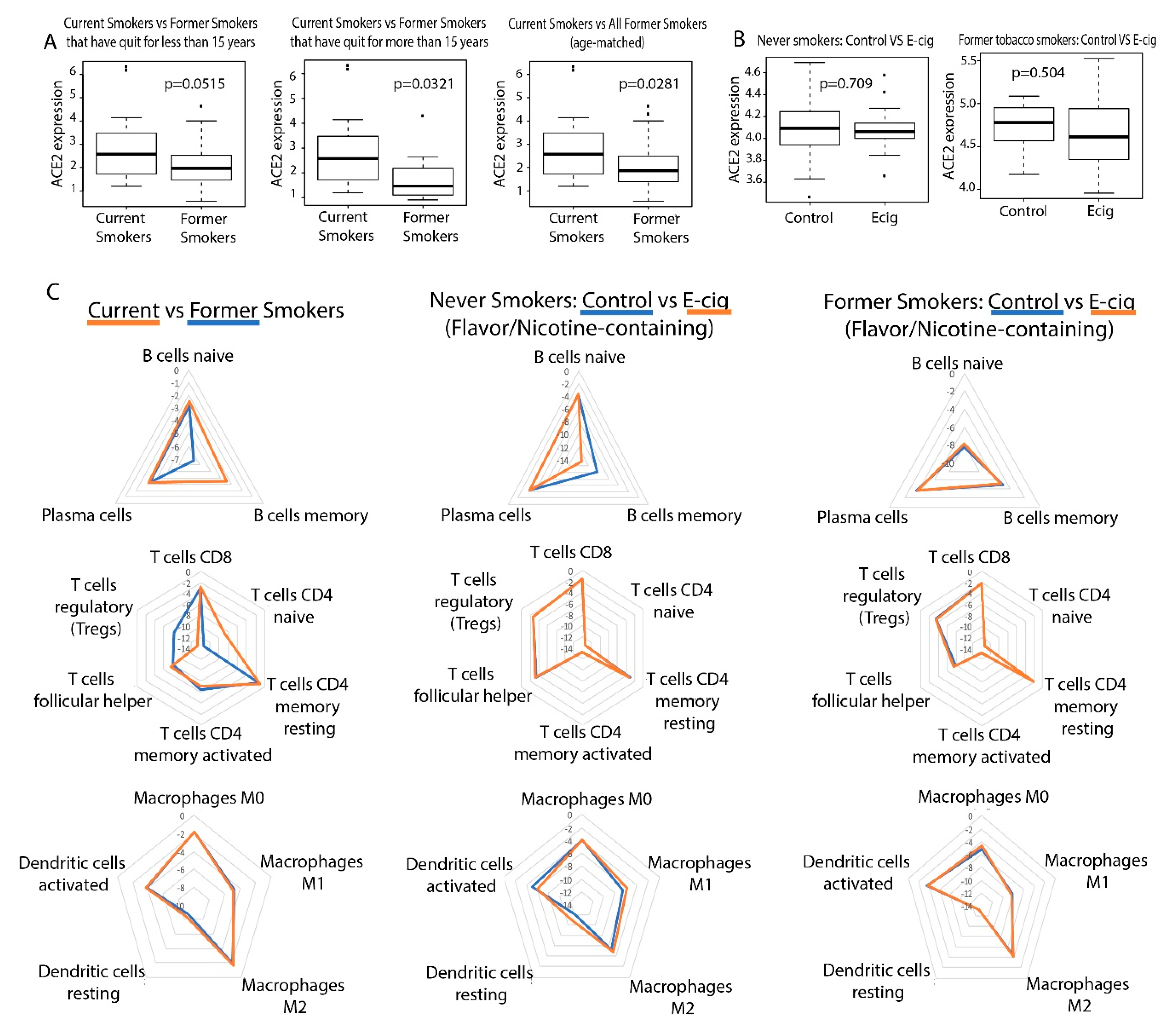
2.1. Correlation between Smoking/Vaping and ACE2 Expression in Bronchial Epithelial Cells
2.2. Correlation between Smoking/Vaping Status and Immune Infiltration
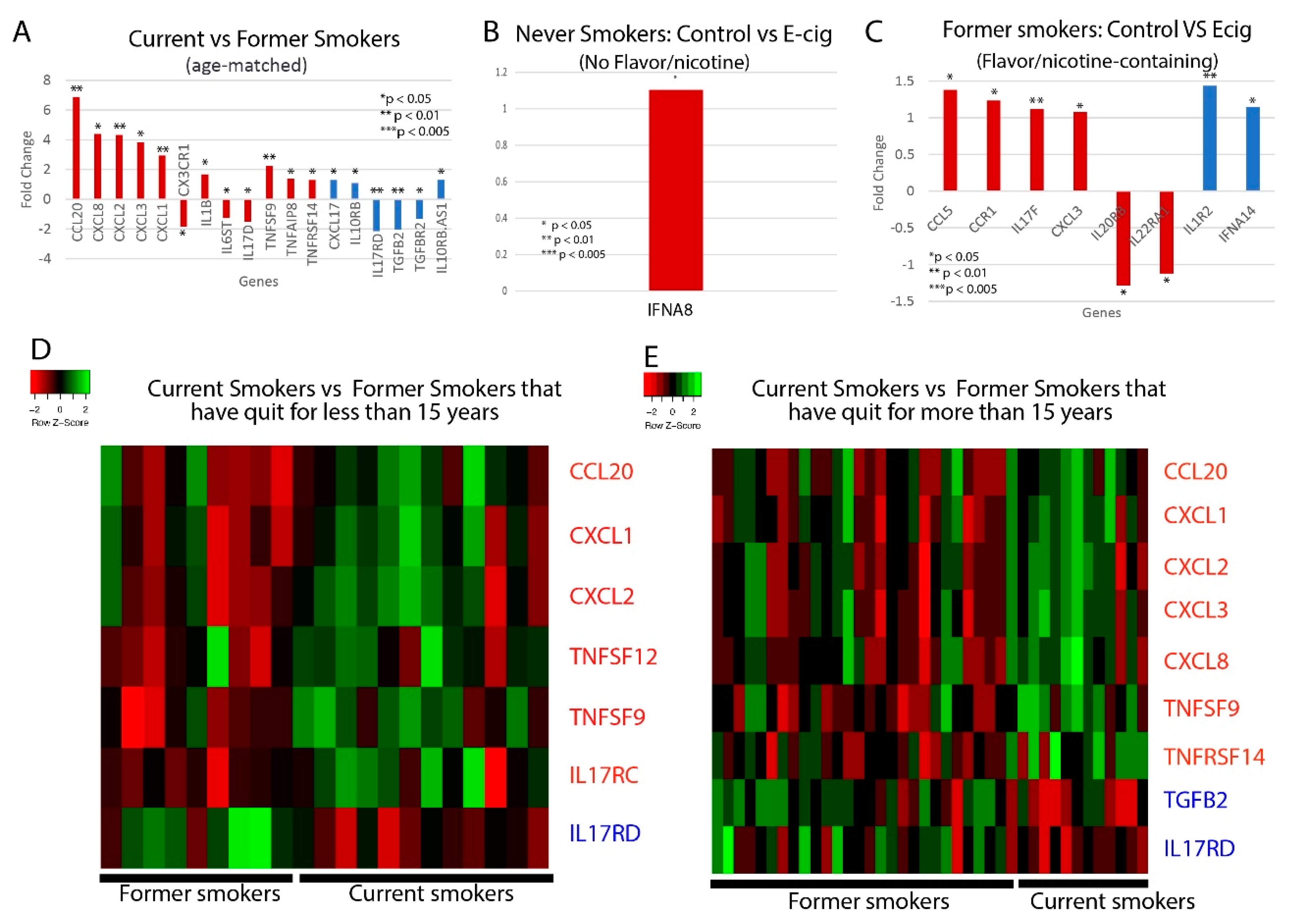
2.3. Correlation between Smoking/Vaping Status and Cytokine Level Dysregulation
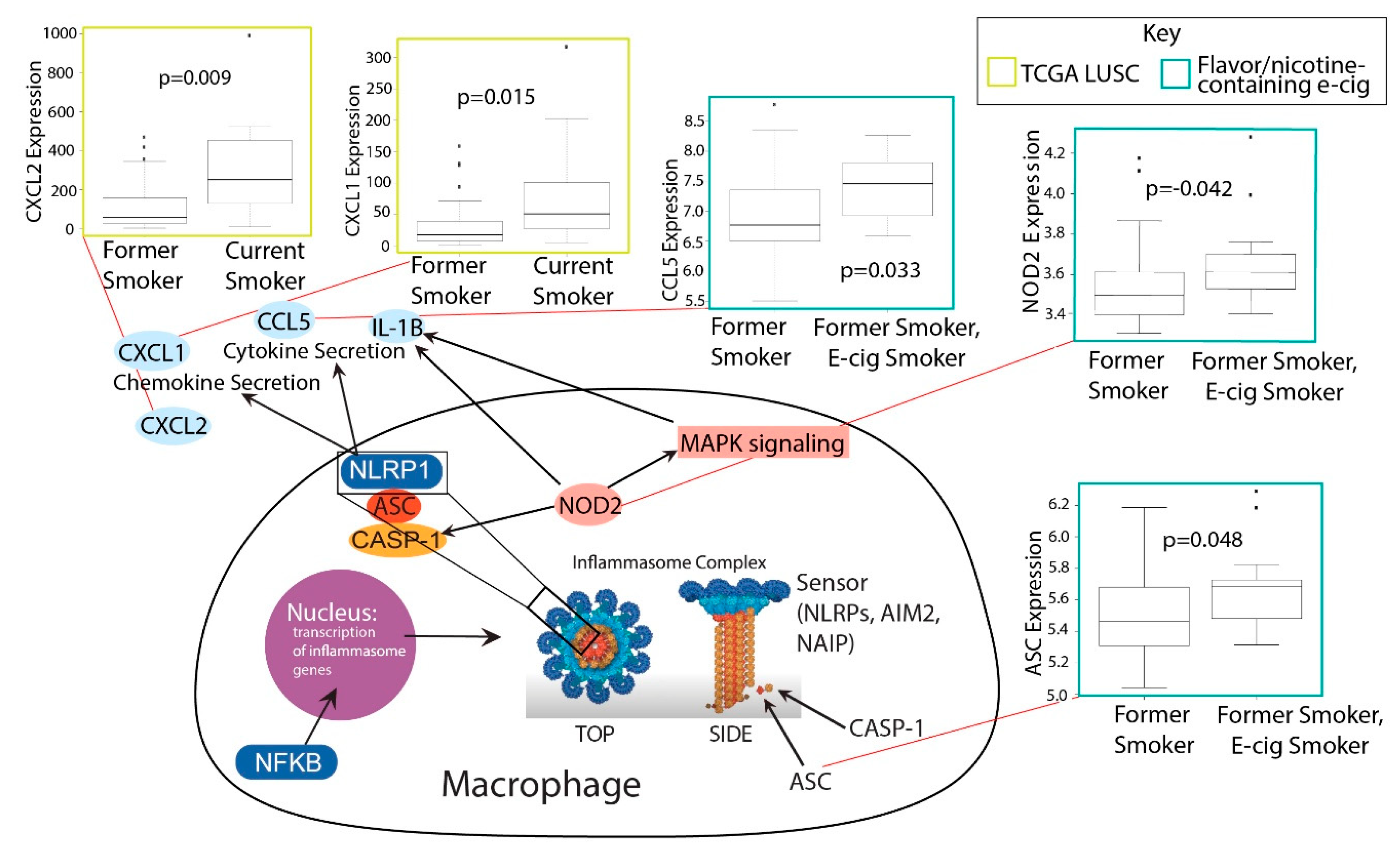
2.4. Investigation of Inflammasome Activation in E-Cig and Tobacco Users
3. Discussion
4. Materials and Methods
4.1. Datasets of Gene Expression from E-Cig and Tobacco Samples
4.2. Differential Expression Analysis
4.3. Inference of Immune Cell Infiltration Populations Using Cibersortx
4.4. Correlation of Smoking Status/E-Cig Vaping Status to Pathways or Signatures Using GSEA
4.5. Selection of Cytokine and Cytokine-Related Genes for Analysis
4.6. Selection of Inflammasome-Related Genes for Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PG | propylene glycol |
| VG | vegetable glycerin |
| LUSC | lung squamous cell carcinoma |
| MSigDB | Molecular Signatures Database |
| TNF | tumor necrosis factor |
| limma | Linear Models for Microarray Analysis |
| Treg | Regulatory T cells |
| PBMCs | Peripheral Blood Mononuclear Cells |
| TCGA | The Cancer Genome Atlas |
References
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in china: Summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Lian, N.; Deng, Y.; Lin, S. The impact of COPD and smoking history on the severity of Covid-19: A systemic review and meta-analysis. J. Med. Virol. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborin, R. Smoking and chronic obstructive pulmonary disease (COPD). Parallel epidemics of the 21 century. Int. J. Environ. Res. Public Health 2009, 6, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Berlin, I.; Thomas, D.; Le Faou, A.L.; Cornuz, J. COVID-19 and smoking. Nicotine Tob. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Farsalinos, K.; Barbouni, A.; Niaura, R. Smoking, vaping and hospitalization for COVID-19. Qeios 2020. [Google Scholar] [CrossRef]
- Lippi, G.; Henry, B.M. Active smoking is not associated with severity of coronavirus disease 2019 (COVID-19). Eur. J. Int. Med. 2020, 75, 107–108. [Google Scholar] [CrossRef]
- Changeux, J.-P.; Amoura, Z.; Rey, F.; Miyara, M. A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications. C. R. Biol. 2020, 343, 33–39. [Google Scholar] [CrossRef]
- Nilsson, P.; Mallet, V. France limits nicotine sales after coronavirus study. Financial Times. 2020. Available online: https://www.ft.com/content/1fe1dda2-bec5-426b-8cfc-a7c2e5a4b139 (accessed on 31 July 2020).
- Bao, W.; Xu, G.; Lu, J.; Snetselaar, L.G.; Wallace, R.B. Changes in electronic cigarette use among adults in the United States, 2014–2016. JAMA 2018, 319, 2039–2041. [Google Scholar] [CrossRef]
- McAlinden, K.D.; Eapen, M.S.; Lu, W.; Chia, C.; Haug, G.; Sohal, S.S. COVID-19 and vaping: Risk for increased susceptibility to SARS-CoV-2 infection? Eur. Respir. J. 2020, 56, 2001645. [Google Scholar] [CrossRef] [PubMed]
- Reinikovaite, V.; Rodriguez, I.E.; Karoor, V.; Rau, A.; Trinh, B.B.; Deleyiannis, F.W.; Taraseviciene-Stewart, L. The effects of electronic cigarette vapour on the lung: Direct comparison to tobacco smoke. Eur. Respir. J. 2018, 51, 1701661. [Google Scholar] [CrossRef] [PubMed]
- Muthumalage, T.; Lamb, T.; Friedman, M.R.; Rahman, I. E-cigarette flavored pods induce inflammation, epithelial barrier dysfunction, and DNA damage in lung epithelial cells and monocytes. Sci. Rep. 2019, 9, 19035. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Vaninov, N. In the eye of the COVID-19 cytokine storm. Nat. Rev. Immunol. 2020, 20, 277. [Google Scholar] [CrossRef]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef]
- Smith, J.C.; Sheltzer, J.M. Cigarette smoke triggers the expansion of a subpopulation of respiratory epithelial cells that express the SARS-CoV-2 receptor ACE2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Scott, A.; Lugg, S.T.; Aldridge, K.; Lewis, K.E.; Bowden, A.; Mahida, R.Y.; Grudzinska, F.S.; Dosanjh, D.; Parekh, D.; Foronjy, R.; et al. Pro-inflammatory effects of e-cigarette vapour condensate on human alveolar macrophages. Thorax 2018, 73, 1161–1169. [Google Scholar] [CrossRef]
- Reidel, B.; Radicioni, G.; Clapp, P.W.; Ford, A.A.; Abdelwahab, S.; Rebuli, M.E.; Haridass, P.; Alexis, N.E.; Jaspers, I.; Kesimer, M. E-cigarette use causes a unique innate immune response in the lung, involving increased neutrophilic activation and altered mucin secretion. Am. J. Respir. Crit. Care Med. 2018, 197, 492–501. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Song, M.A.; Reisinger, S.A.; Freudenheim, J.L.; Brasky, T.M.; Mathe, E.A.; McElroy, J.P.; Nickerson, Q.A.; Weng, D.Y.; Wewers, M.D.; Shields, P.G. Effects of electronic cigarette constituents on the human lung: A pilot clinical trial. Cancer Prev. Res. Phila 2020, 13, 145–152. [Google Scholar] [CrossRef]
- Corbett, S.E.; Nitzberg, M.; Moses, E.; Kleerup, E.; Wang, T.; Perdomo, C.; Perdomo, C.; Liu, G.; Xiao, X.; Liu, H.; et al. Gene expression alterations in the bronchial epithelium of E-Cigarette users. Chest 2019, 156, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Chakladar, J.; Shende, N.; Li, W.T.; Rajasekaran, M.; Chang, E.Y.; Ongkeko, W.M. Smoking-Mediated Upregulation of the Androgen Pathway Leads to Increased SARS-CoV-2 Susceptibility. Int. J. Mol. Sci. 2020, 21, 3627. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Miaw, S.C.; Yu, J.S.; Chen, T.C.; Lin, C.Y.; Lin, Y.C.; Chang, C.S.; He, Y.C.; Chuang, S.H.; Yen, M.I.; et al. Altered T-bet dominance in IFN-gamma-decoupled CD4+ T cells with attenuated cytokine storm and preserved memory in influenza. J. Immunol. 2013, 190, 4205–4214. [Google Scholar] [CrossRef]
- Gogishvili, T.; Langenhorst, D.; Luhder, F.; Elias, F.; Elflein, K.; Dennehy, K.M.; Gold, R.; Hunig, T. Rapid regulatory T-cell response prevents cytokine storm in CD28 superagonist treated mice. PLoS ONE 2009, 4, e4643. [Google Scholar] [CrossRef]
- Brandsma, C.A.; Hylkema, M.N.; Geerlings, M.; van Geffen, W.H.; Postma, D.S.; Timens, W.; Kerstjens, H.A. Increased levels of (class switched) memory B cells in peripheral blood of current smokers. Respir. Res. 2009, 10, 108. [Google Scholar] [CrossRef]
- Ng, L.F.; Hibberd, M.L.; Ooi, E.E.; Tang, K.F.; Neo, S.Y.; Tan, J.; Murthy, K.R.; Vega, V.B.; Chia, J.M.; Liu, E.T.; et al. A human in vitro model system for investigating genome-wide host responses to SARS coronavirus infection. BMC Infect. Dis. 2004, 4, 34. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 1. [Google Scholar] [CrossRef]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened innate immune responses in the respiratory Tract of COVID-19 patients. Cell Host Microbe 2020, 27, 883–890. [Google Scholar] [CrossRef]
- Donnelly, S.C.; Strieter, R.M.; Kunkel, S.L.; Walz, A.; Robertson, C.R.; Carter, D.C.; Grant, I.S.; Pollok, A.J.; Haslett, C. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet 1993, 341, 643–647. [Google Scholar] [CrossRef]
- Miller, E.J.; Cohen, A.B.; Nagao, S.; Griffith, D.; Maunder, R.J.; Martin, T.R.; Weiner-Kronish, J.P.; Sticherling, M.; Christophers, E.; Matthay, M.A. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am. Rev. Respir. Dis. 1992, 146, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, A.B.; Raijmakers, P.G.; Hack, C.E.; Thijs, L.G. Interleukin 8-related neutrophil elastase and the severity of the adult respiratory distress syndrome. Cytokine 1995, 7, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.J.; Cohen, A.B.; Matthay, M.A. Increased interleukin-8 concentrations in the pulmonary edema fluid of patients with acute respiratory distress syndrome from sepsis. Crit. Care Med. 1996, 24, 1448–1454. [Google Scholar] [CrossRef]
- Trifilo, M.J.; Bergmann, C.C.; Kuziel, W.A.; Lane, T.E. CC chemokine ligand 3 (CCL3) regulates CD8(+)-T-cell effector function and migration following viral infection. J. Virol. 2003, 77, 4004–4014. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Uchida, O.; Kajiwara, N.; Kim, E.Y.; Patel, A.C.; O’Sullivan, M.P.; Walter, M.J.; Schwendener, R.A.; Cook, D.N.; Danoff, T.M.; et al. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med. 2005, 11, 1180–1187. [Google Scholar] [CrossRef]
- Tekamp-Olson, P.; Gallegos, C.; Bauer, D.; McClain, J.; Sherry, B.; Fabre, M.; van Deventer, S.; Cerami, A. Cloning and characterization of cDNAs for murine macrophage inflammatory protein 2 and its human homologues. J. Exp. Med. 1990, 172, 911–919. [Google Scholar] [CrossRef]
- Lu, A.; Wu, H. Structural mechanisms of inflammasome assembly. FEBS J. 2015, 282, 435–444. [Google Scholar] [CrossRef]
- Boro, M.; Balaji, K.N. CXCL1 and CXCL2 regulate NLRP3 inflammasome activation via G-Protein-Coupled receptor CXCR2. J. Immunol. 2017, 199, 1660–1671. [Google Scholar] [CrossRef]
- Rajamaki, K.; Mayranpaa, M.I.; Risco, A.; Tuimala, J.; Nurmi, K.; Cuenda, A.; Eklund, K.K.; Oorni, K.; Kovanen, P.T. p38delta MAPK: A novel regulator of NLRP3 inflammasome activation with increased expression in coronary atherogenesis. Arter. Thromb. Vasc. Biol. 2016, 36, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.C.; Ali, S.R.; McGillivray, S.; Tseng, P.H.; Mariathasan, S.; Humke, E.W.; Eckmann, L.; Powell, J.J.; Nizet, V.; Dixit, V.M.; et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA 2008, 105, 7803–7808. [Google Scholar] [CrossRef] [PubMed]
- Williford, J.; Zablotsky, B.; Zelaya, C. QuickStats: Percentage of adults aged 18–24 years who currently smoke cigarettes* or who currently use electronic cigarettes, (dagger) by Year—National Health Interview Survey, United States, 2014–2018 (section sign). Morb. Mortal. Wkly. Rep. 2019, 68, 870. [Google Scholar] [CrossRef]
- Bagaitkar, J.; Demuth, D.R.; Scott, D.A. Tobacco use increases susceptibility to bacterial infection. Tob. Induc. Dis. 2008, 4, 12. [Google Scholar] [CrossRef]
- Van Zyl-Smit, R.N.; Richards, G.; Leone, F.T. Tobacco smoking and COVID-19 infection. Lancet Respir. Med. 2020, 8, 664–665. [Google Scholar] [CrossRef]
- Cullen, K.A.; Ambrose, B.K.; Gentzke, A.S.; Apelberg, B.J.; Jamal, A.; King, B.A. Notes from the Field: Use of electronic cigarettes and any tobacco product among middle and high school students—United States, 2011–2018. Morb. Mortal. Wkly. Rep. 2018, 67, 1276–1277. [Google Scholar] [CrossRef]
- Chaffee, B.W. Electronic cigarettes: Trends, health effects and advising patients amid uncertainty. J. Calif Dent. Assoc. 2019, 47, 85–92. [Google Scholar]
- Yu, V.; Rahimy, M.; Korrapati, A.; Xuan, Y.; Zou, A.E.; Krishnan, A.R.; Tsui, T.; Aguilera, J.A.; Advani, S.; Crotty Alexander, L.E.; et al. Electronic cigarettes induce DNA strand breaks and cell death independently of nicotine in cell lines. Oral Oncol. 2016, 52, 58–65. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Leung, J.M.; Yang, C.X.; Tam, A.; Shaipanich, T.; Hackett, T.L.; Singhera, G.K.; Dorscheid, D.R.; Sin, D.D. ACE-2 Expression in the small airway epithelia of smokers and COPD patients: Implications for COVID-19. Eur. Respir. J. 2020, 55, 2000688. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Bosse, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco smoking increases the lung gene Expression of ACE2, the receptor of SARS-CoV-2. Am. J. Respir Crit Care Med. 2020, 201, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sundar, I.K.; Li, D.; Lucas, J.H.; Muthumalage, T.; McDonough, S.R.; Rahman, I. E-cigarette-induced pulmonary inflammation and dysregulated repair are mediated by nAChR alpha7 receptor: Role of nAChR alpha7 in SARS-CoV-2 Covid-19 ACE2 receptor regulation. Respir. Res. 2020, 21, 154. [Google Scholar] [CrossRef]
- Tsai, M.; Song, M.A.; McAndrew, C.; Brasky, T.M.; Freudenheim, J.L.; Mathe, E.; McElroy, J.; Reisinger, S.A.; Shields, P.G.; Wewers, M.D. Electronic versus combustible cigarette effects on inflammasome component release into human lung. Am. J. Respir. Crit. Care Med. 2019, 199, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Lerner, C.A.; Sundar, I.K.; Yao, H.; Gerloff, J.; Ossip, D.J.; McIntosh, S.; Rahman, I. Vapors produced by electronic cigarettes and e-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS ONE 2015, 10, e0116732. [Google Scholar] [CrossRef] [PubMed]
- Chaumont, M.; van de Borne, P.; Bernard, A.; Van Muylem, A.; Deprez, G.; Ullmo, J.; Debbas, N. Fourth generation e-cigarette vaping induces transient lung inflammation and gas exchange disturbances: Results from two randomized clinical trials. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Rowell, T.R.; Tarran, R. Will chronic e-cigarette use cause lung disease? Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, 1398–1409. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, Y.; Duan, Y. Breath biomarkers in diagnosis of pulmonary diseases. Clin. Chim. Acta 2012, 413, 1770–1780. [Google Scholar] [CrossRef]
- Pisinger, C.; Døssing, M. A systematic review of health effects of electronic cigarettes. Prev. Med. 2014, 69, 248–260. [Google Scholar] [CrossRef]
- Rom, O.; Pecorelli, A.; Valacchi, G.; Reznick, A.Z. Are E-cigarettes a safe and good alternative to cigarette smoking? Ann. N. Y. Acad. Sci. 2015, 1340, 65–74. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.; Hafezi, M.; Chia, A.; Chia, W.N. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020. [Google Scholar] [CrossRef]
- Levet, S.; Charvet, B.; Bertin, A.; Deschaumes, A.; Perron, H.; Hober, D. Human endogenous retroviruses and Type 1 Diabetes. Curr. Diab. Rep. 2019, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Matousková, M.; Blazková, J.; Pajer, P.; Pavlícek, A.; Hejnar, J. CpG methylation suppresses transcriptional activity of human syncytin-1 in non-placental tissues. Exp. Cell Res. 2006, 312, 1011–1020. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qin, C. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Sakata, M. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [PubMed]
- Caliri, A.W.; Caceres, A.; Tommasi, S.; Besaratinia, A. Hypomethylation of LINE-1 repeat elements and global loss of DNA hydroxymethylation in vapers and smokers. Epigenetics 2020, 15, 1–14. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Geo2R | Kruskal-Wallis | Cohort | ||
|---|---|---|---|---|---|
| p-Value | logFC | p-Value | logFC | ||
| CCL20 | 0.5055 | 0.1778 | 0.7851 | 0.0019 | E-cig vs. Former Smokers |
| 0.3871 | −0.1674 | 0.2907 | −0.0177 | E-cig vs. None | |
| 0.0076 | 2.7834 | Current vs. Former Tobacco Smokers | |||
| CXCL17 | 0.6345 | 0.0701 | 0.5315 | 0.0007 | E-cig vs. Former Smokers |
| 0.5831 | −0.0391 | 0.7968 | −0.0010 | E-cig vs. None | |
| 0.0353 | 0.3798 | Current vs. Former Tobacco Smokers | |||
| CXCL3 | 0.0491 | 0.1098 | 0.0650 | 0.0122 | E-cig vs. Former Smokers |
| 0.9108 | 0.0071 | 0.2106 | 0.0099 | E-cig vs. None | |
| 0.0103 | 1.9380 | Current vs. Former Tobacco Smokers | |||
| CXCL8 | 0.5096 | 0.0407 | 0.4508 | 0.0093 | E-cig vs. Former Smokers |
| 0.0137 | 2.1347 | Current vs. Former Tobacco Smokers | |||
| IL10RB | 0.1907 | −0.1347 | 0.1051 | 0.0074 | E-cig vs. Former Smokers |
| 0.0742 | −0.0526 | 0.0439 | −0.0021 | E-cig vs. None | |
| 0.0481 | 0.1260 | Current vs. Former Tobacco Smokers | |||
| IL10RB-AS1 | 0.3466 | −0.0597 | 0.5055 | −0.0040 | E-cig vs. None |
| 0.0426 | 0.3871 | Current vs. Former Tobacco Smokers | |||
| IL17D | 0.1372 | 0.1445 | 0.1197 | 0.0152 | E-cig vs. Former Smokers |
| 0.9680 | 0.0021 | 0.5639 | −0.0017 | E-cig vs. None | |
| 0.0182 | −0.6035 | Current vs. Former Tobacco Smokers | |||
| IL17RD | 0.9196 | −0.0084 | 0.7362 | 0.0029 | E-cig vs. Former Smokers |
| 0.3403 | 0.0438 | 0.4942 | 0.0004 | E-cig vs. None | |
| 0.0017 | −1.1050 | Current vs. Former Tobacco Smokers | |||
| IL1B | 0.5137 | 0.1273 | 0.6649 | −0.0052 | E-cig vs. Former Smokers |
| 0.3701 | −0.1931 | 0.7968 | −0.0020 | E-cig vs. None | |
| 0.0376 | 0.7497 | Current vs. Former Tobacco Smokers | |||
| IL6ST | 0.1507 | 0.0736 | 0.4132 | −0.0010 | E-cig vs. Former Smokers |
| 0.5503 | 0.0511 | 0.8660 | 0.0016 | E-cig vs. None | |
| 0.0353 | −0.3333 | Current vs. Former Tobacco Smokers | |||
| TGFB2 | 0.2557 | 0.0795 | 0.9361 | −0.0134 | E-cig vs. Former Smokers |
| 0.0231 | 0.2371 | 0.1800 | 0.0022 | E-cig vs. None | |
| 0.0040 | −1.0251 | Current vs. Former Tobacco Smokers | |||
| TGFBR2 | 0.2268 | 0.0852 | 0.2671 | 0.0032 | E-cig vs. None |
| 0.0137 | −0.3959 | Current vs. Former Tobacco Smokers | |||
| TNFAIP8 | 0.5235 | 0.1650 | 0.1938 | −0.0106 | E-cig vs. Former Smokers |
| 0.9072 | 0.0040 | 0.6253 | 0.0003 | E-cig vs. None | |
| 0.0453 | 0.4613 | Current vs. Former Tobacco Smokers | |||
| TNFRSF14 | 0.2259 | −0.1559 | 0.9361 | −0.0009 | E-cig vs. Former Smokers |
| 0.3154 | 0.0407 | 0.1289 | 0.0028 | E-cig vs. None | |
| 0.0376 | 0.3800 | Current vs. Former Tobacco Smokers | |||
| TNFSF9 | 0.9974 | −0.0002 | 0.4132 | 0.0010 | E-cig vs. Former Smokers |
| 0.4118 | −0.0497 | 0.3990 | −0.0016 | E-cig vs. None | |
| 0.0016 | 1.1718 | Current vs. Former Tobacco Smokers | |||
| Gene | Geo2R | Kruskal-Wallis | Cohort | ||
|---|---|---|---|---|---|
| p-Value | logFC | p-Value | logFC | ||
| CCL5 | 0.85415 | 0.024121 | 0.273192 | 0.009959 | E-cig vs. None |
| 0.049094 | 0.463617 | 0.032858 | 0.0421 | E-cig vs. Former Smokers | |
| 0.897886 | 0.077011 | Current vs. Former Tobacco Smokers | |||
| CXCL1 | 0.622626 | 0.083064 | 0.241121 | 0.016747 | E-cig vs. None |
| 0.274418 | 0.177215 | 0.395149 | 0.00718 | E-cig vs. Former Smokers | |
| 0.008846 | 1.553637 | Current vs. Former Tobacco Smokers | |||
| CXCL2 | 0.802924 | −0.03393 | 0.429555 | 0.004948 | E-cig vs. None |
| 0.359789 | 0.122607 | 0.413227 | 0.003556 | E-cig vs. Former Smokers | |
| 0.008202 | 2.113189 | Current vs. Former Tobacco Smokers | |||
| NLRP3 | 0.995982 | 0.000195 | 0.918813 | 0.002349 | E-cig vs. None |
| 0.183356 | 0.07004 | 0.056236 | 0.013534 | E-cig vs. Former Smokers | |
| 0.758085 | 0.158417 | Current vs. Former Tobacco Smokers | |||
| NOD2 | 0.317884 | 0.034773 | 0.459914 | 0.001984 | E-cig vs. None |
| 0.207633 | 0.099462 | 0.041595 | 0.014107 | E-cig vs. Former Smokers | |
| 0.777691 | 0.16816 | Current vs. Former Tobacco Smokers | |||
| PYCARD | 0.659102 | −0.0204 | 0.721277 | −0.00158 | E-cig vs. None |
| 0.048781 | 0.187701 | 0.065034 | 0.017268 | E-cig vs. Former Smokers | |
| 0.100456 | 0.272032 | Current vs. Former Tobacco Smokers | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, A.C.; Chakladar, J.; Li, W.T.; Chen, C.; Chang, E.Y.; Wang-Rodriguez, J.; Ongkeko, W.M. Tobacco, but Not Nicotine and Flavor-Less Electronic Cigarettes, Induces ACE2 and Immune Dysregulation. Int. J. Mol. Sci. 2020, 21, 5513. https://doi.org/10.3390/ijms21155513
Lee AC, Chakladar J, Li WT, Chen C, Chang EY, Wang-Rodriguez J, Ongkeko WM. Tobacco, but Not Nicotine and Flavor-Less Electronic Cigarettes, Induces ACE2 and Immune Dysregulation. International Journal of Molecular Sciences. 2020; 21(15):5513. https://doi.org/10.3390/ijms21155513
Chicago/Turabian StyleLee, Abby C., Jaideep Chakladar, Wei Tse Li, Chengyu Chen, Eric Y. Chang, Jessica Wang-Rodriguez, and Weg M. Ongkeko. 2020. "Tobacco, but Not Nicotine and Flavor-Less Electronic Cigarettes, Induces ACE2 and Immune Dysregulation" International Journal of Molecular Sciences 21, no. 15: 5513. https://doi.org/10.3390/ijms21155513
APA StyleLee, A. C., Chakladar, J., Li, W. T., Chen, C., Chang, E. Y., Wang-Rodriguez, J., & Ongkeko, W. M. (2020). Tobacco, but Not Nicotine and Flavor-Less Electronic Cigarettes, Induces ACE2 and Immune Dysregulation. International Journal of Molecular Sciences, 21(15), 5513. https://doi.org/10.3390/ijms21155513