Targeting Hypoxia-Driven Metabolic Reprogramming to Constrain Tumor Progression and Metastasis



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
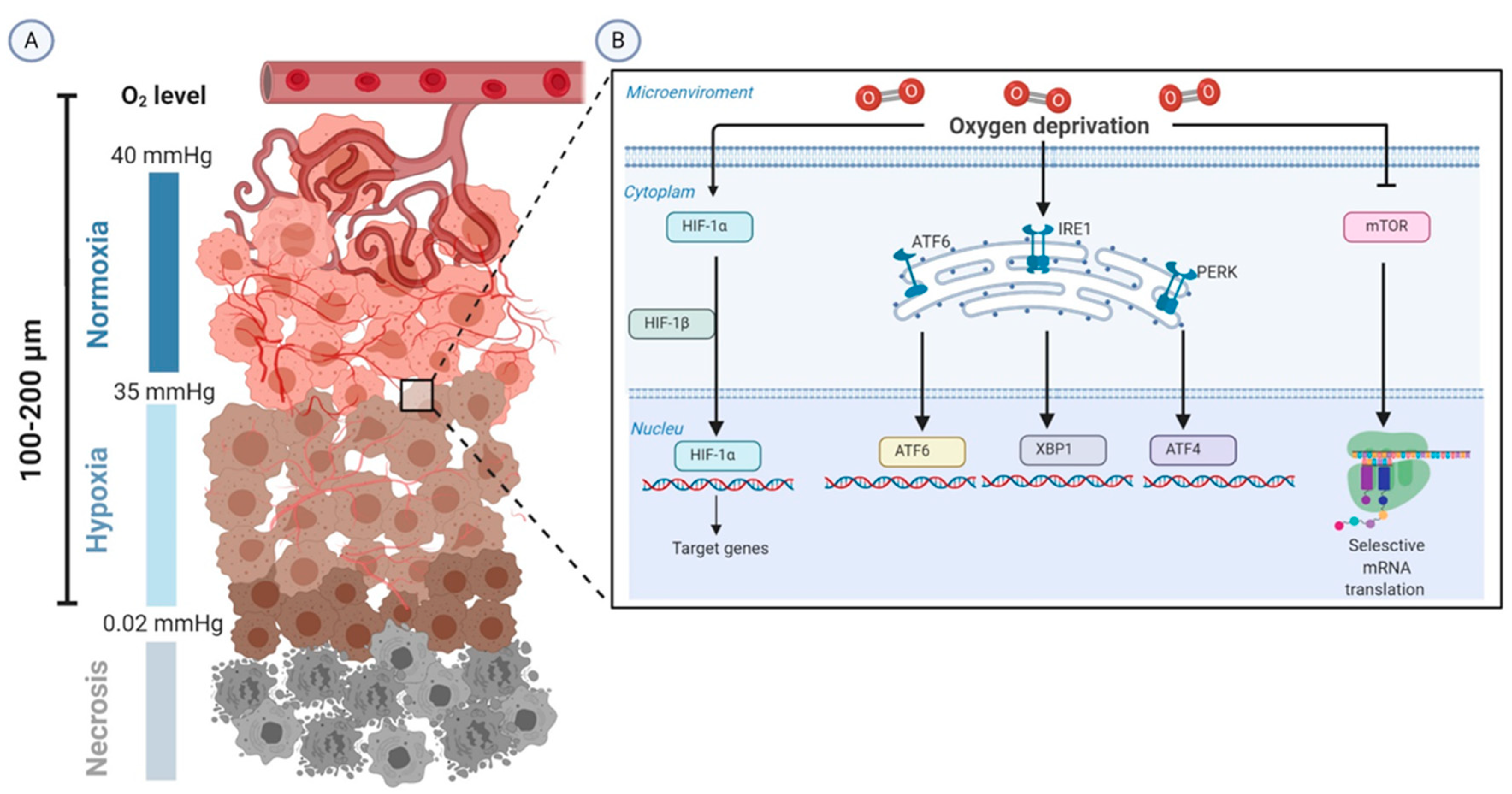
2. Biologic Aspects of Tumor Hypoxia
2.1. Acute Hypoxia
2.2. Cyclic Hypoxia
2.3. Chronic Hypoxia
3. Cellular Adaptation to Hypoxia
3.1. Hypoxia-Inducible Factor 1-Alpha (HIF-1α)
3.2. The mTOR Pathway
3.3. UPR
3.4. NF-κB
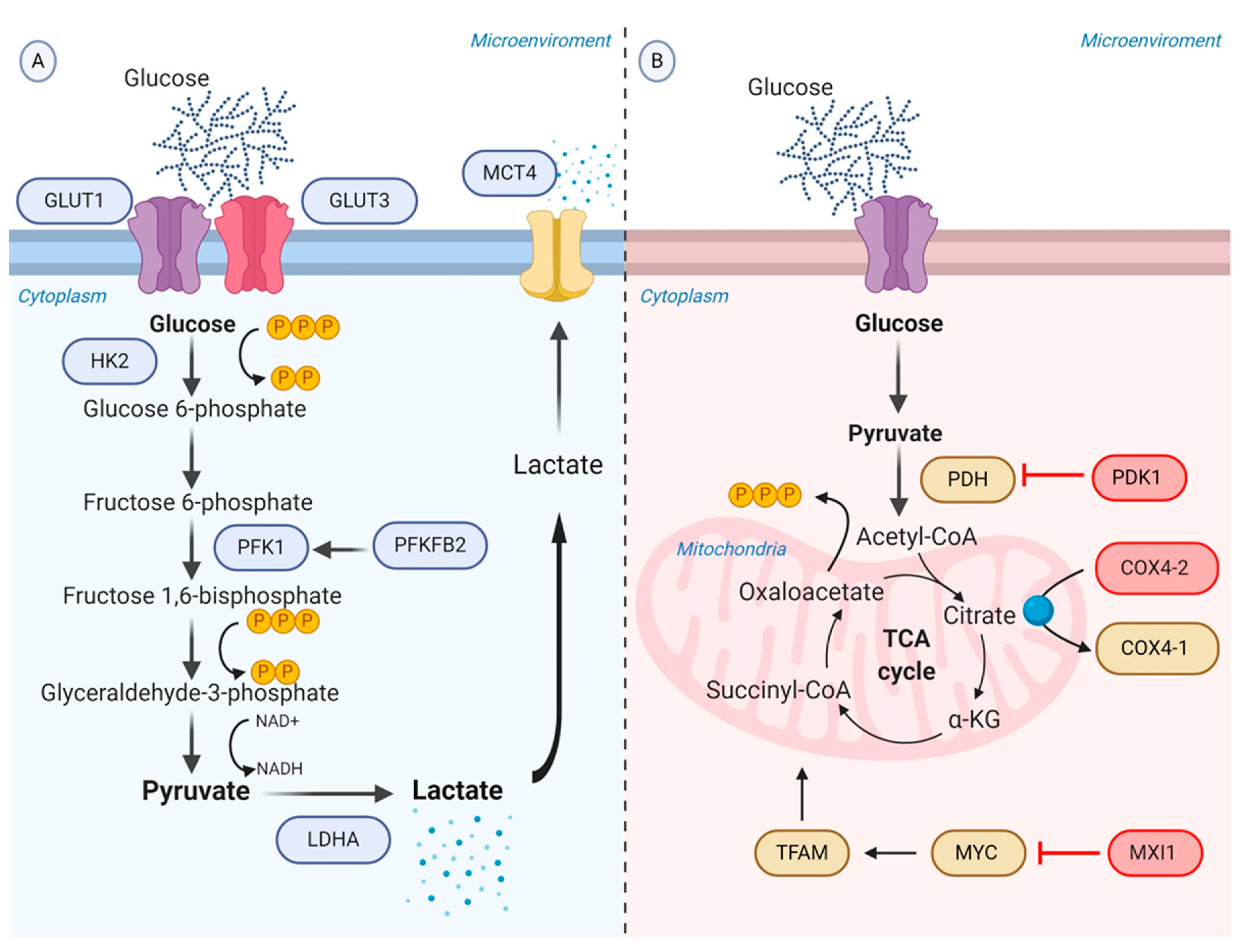
4. Genetic and Metabolic Modifications in the Hypoxic Tumor Microenvironment
4.1. Promotion of Anaerobic Glycolysis
4.2. Repression of Oxidative Phosphorylation
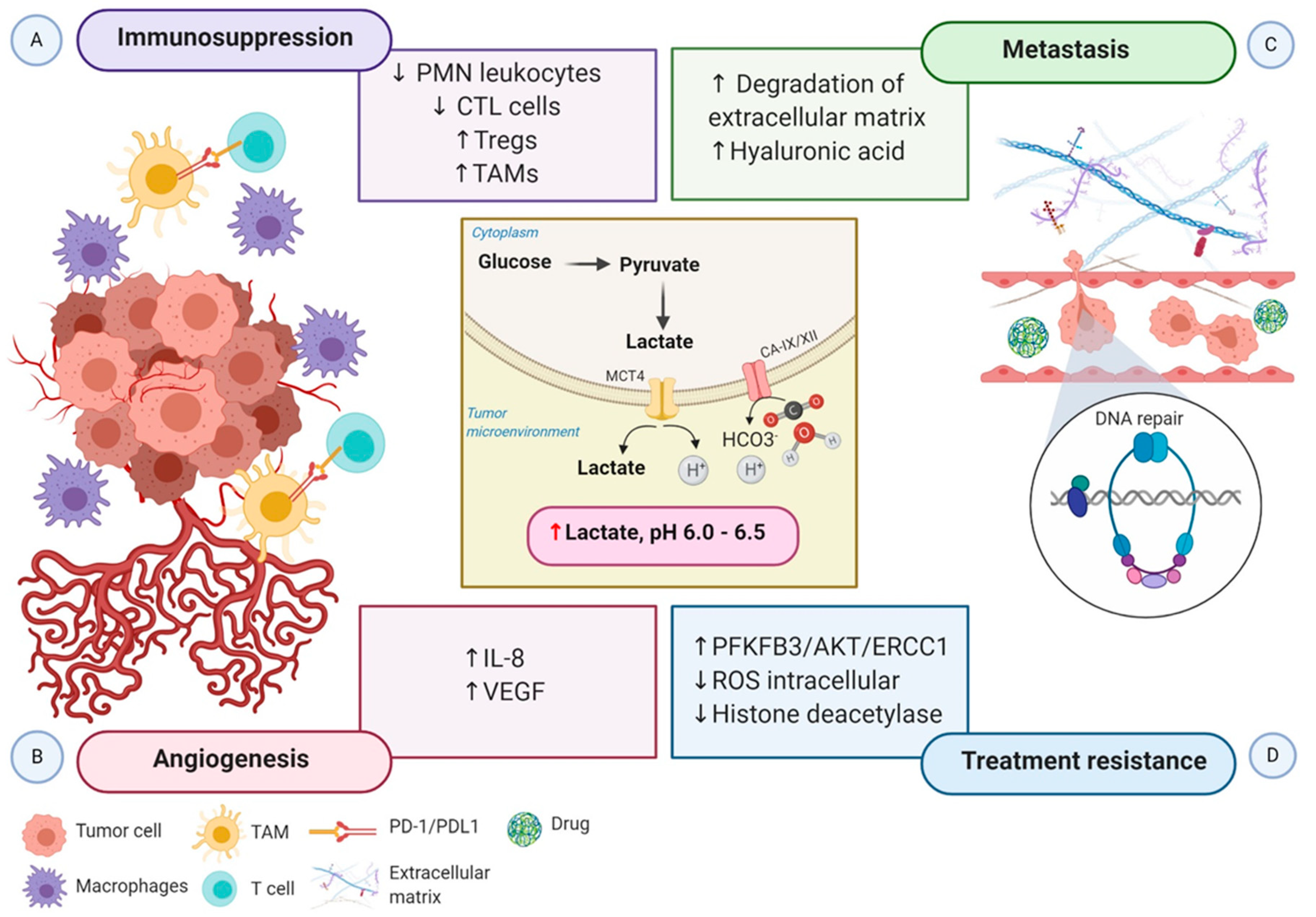
5. Impact of Metabolic Reprogramming Driven-Hypoxia on Tumor Progression
6. Therapeutic Strategies Targeting Hypoxia
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4E-BP1 | Eukaryotic initiation factor 4E binding protein 1 |
| ALDA | Aldolase A |
| AMPK | AMP-activated protein kinase |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| BiP | Immunoglobulin protein |
| BNIP3 | Bcl2/adenovirus E1B 19-kDa protein-interacting protein 3 |
| CA-IX | Carbonic anhydrase IX |
| CA-XII | Carbonic anhydrase XII |
| COX4-2 | Cytochrome oxidase 4-2 |
| CSCs | Cancer stem cells |
| CTLs | Cytotoxic T lymphocytes |
| DCs | Dendritic cells |
| Deptor | DEP-domain-containing mTOR-interacting protein |
| DLK1 | Delta-like 1 Homologue |
| DNA-PKcs | DNA-dependent protein kinase, catalytic subunit |
| ECM | Extracellular matrix |
| EEF2K | Eukaryotic elongation factor 2 kinase |
| EIF2α | Eukaryotic initiation factor 2 alpha |
| EMT | Epithelial to mesenchymal transition |
| ENG | Endoglin |
| EphA2 | Ephrin type-A receptor 2 |
| ER | Endoplasmic reticulum |
| F6P | Fructose 6-phosphate |
| FBW7 | F-box and WD repeat domain-containing 7 |
| FDP | Fructose 1,6-bisphosphate |
| G6P | Glucose 6-phosphate |
| GLUTs | Glucose transporters |
| HA | Hyaluronic acid |
| HDAC | Histone deacetylases |
| HK2 | Hexokinase 2 |
| HIFs | Hypoxia-inducible factors |
| HREs | Hypoxia response elements |
| IGF2 | Insulin-like growth factor |
| IKK | IκB kinase complex |
| IL-8 | Interleukin 8 |
| IRE1 | Inositol-requiring protein 1 |
| LDHA | Lactic dehydrogenase A |
| LEP | Leptin |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
| MCT1 | Monocarboxylate transporter 1 |
| mLST8 | Mammalian lethal with Sec13 protein 8 |
| mTOR | Mammalian target of rapamycin |
| MXI1 | MAX interactor-1 |
| NAD+ | Nicotinamide adenine dinucleotide |
| NF-κB | Nuclear factor-κB |
| ODDD | Oxygen-dependent degradation domain |
| OXPHOS | Oxidative phosphorylation |
| p70S6K | P70S6 kinase |
| PDGF | Platelet-derived growth factor |
| PDH | Pyruvate dehydrogenase |
| PDK1 | Pyruvate dehydrogenase kinase |
| PERK | Protein kinase RNA-like ER kinase |
| PFK1 | Phosphofructokinase 1 |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| PGK1 | Phosphoglycerate kinase 1 |
| PIKKs | Phosphatidylinositol 3-kinase-related kinases |
| PKM | Pyruvate kinase M |
| PML | Promyelocytic leukemia protein |
| POU5F1 | POU domain, class 5, transcription factor 1 |
| PRAS40 | Proline-rich AKT substrate 40 kDa |
| Raptor | Regulatory-associated protein of mTOR |
| REDD1 | Regulated in development and DNA damage responses 1 |
| ROS | Reactive oxygen species |
| TAD | Transactivation domains |
| TCA | Tricarboxylic acid cycle |
| TAMs | Tumor-associated macrophages |
| TFAM | Transcription factor A, mitochondrial |
| TGF | Transforming growth factor |
| TRAF6 | Tumor necrosis factor receptor associated factor 6 |
| Tregs | T cells regulatory |
| UPR | Uunfolded protein response |
| VEGF-A | Vascular endothelial growth factor-A |
| VHL | Von Hippel-Lindau |
| XBP1 | X-box binding protein |
References
- Giaccia, A.J.; Simon, M.C.; Johnson, R. The biology of hypoxia: The role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004, 18, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef]
- Ortiz-Prado, E.; Dunn, J.F.; Vasconez, J.; Castillo, D.; Viscor, G. Partial pressure of oxygen in the human body: A general review. Am. J. Blood Res. 2019, 9, 1–14. [Google Scholar]
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Palacio-Castañeda, V.; Kooijman, L.; Venzac, B.; Verdurmen, W.P.R.; Le Gac, S. Metabolic Switching of Tumor Cells under Hypoxic Conditions in a Tumor-on-a-chip Model. Micromachines 2020, 11, 382. [Google Scholar] [CrossRef]
- Rehn, M.; Olsson, A.; Reckzeh, K.; Diffner, E.; Carmeliet, P.; Landberg, G.; Cammenga, J. Hypoxic induction of vascular endothelial growth factor regulates murine hematopoietic stem cell function in the low-oxygenic niche. Blood 2011, 118, 1534–1543. [Google Scholar] [CrossRef]
- Brown, J.M. Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br. J. Radiol. 1979, 52, 650–656. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.M.; Choyke, P.L. Imaging of angiogenesis: From microscope to clinic. Nat. Med. 2003, 9, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Bayer, C.; Shi, K.; Astner, S.T.; Maftei, C.A.; Vaupel, P. Acute versus chronic hypoxia: Why a simplified classification is simply not enough. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Holmquist-Mengelbier, L.; Fredlund, E.; Lofstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, A.; Gradin, K.; et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.O.; Wang, H. Radiation-modifying effect of oxygen in synchronized cells pre-treated with acute or prolonged hypoxia. Int. J. Radiat. Biol. 1996, 70, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef]
- Bayer, C.; Vaupel, P. Acute versus chronic hypoxia in tumors: Controversial data concerning time frames and biological consequences. Strahlenther. Onkol. 2012, 188, 616–627. [Google Scholar] [CrossRef]
- Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R.K. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat. Med. 1997, 3, 177–182. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Mandl, M.; Depping, R. Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1β): Is it a rare exception? Mol. Med. 2014, 20, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Köditz, J.; Nesper, J.; Wottawa, M.; Stiehl, D.P.; Camenisch, G.; Franke, C.; Myllyharju, J.; Wenger, R.H.; Katschinski, D.M. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 2007, 110, 3610–3617. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- D’Ignazio, L.; Rocha, S. Hypoxia Induced NF-κB. Cells 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, S.F.; Tambuwala, M.M.; Bruning, U.; Schaible, B.; Scholz, C.C.; Byrne, A.; O’Connor, A.; Gallagher, W.M.; Lenihan, C.R.; Garvey, J.F.; et al. An intact canonical NF-κB pathway is required for inflammatory gene expression in response to hypoxia. J. Immunol. 2011, 186, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Bandarra, D.R.S. Tale of two transcription factors: NF-κB and HIF crosstalk. OA Mol. Cell Biol. 2013, 1, 1–7. [Google Scholar] [CrossRef]
- van Uden, P.; Kenneth, N.S.; Webster, R.; Müller, H.A.; Mudie, S.; Rocha, S. Evolutionary conserved regulation of HIF-1β by NF-κB. PLoS Genet. 2011, 7, e1001285. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Schito, L.; Pellegrini, L.; Villanova, L.; Marfe, G.; Anwar, T.; Rosa, R.; Indelicato, M.; Fini, M.; Pucci, B.; et al. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-κB. Carcinogenesis 2011, 32, 1167–1175. [Google Scholar] [CrossRef]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; et al. Modulators of HIF1α and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front. Pharm. 2013, 4, 13. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e359. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [PubMed]
- Gleadle, J.M.; Ratcliffe, P.J. Induction of hypoxia-inducible factor-1, erythropoietin, vascular endothelial growth factor, and glucose transporter-1 by hypoxia: Evidence against a regulatory role for Src kinase. Blood 1997, 89, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yang, D.; Hou, Y.; Liu, S.; Zhao, M.; Qin, Y.; Chen, R.; Teng, Y.; Liu, M. Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis. Cell Death Dis. 2019, 10, 228. [Google Scholar] [CrossRef] [PubMed]
- Lang, L.; Chemmalakuzhy, R.; Shay, C.; Teng, Y. PFKP Signaling at a Glance: An Emerging Mediator of Cancer Cell Metabolism. Adv. Exp. Med. Biol. 2019, 1134, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Bai, R.; Yang, K.; Tian, Z. MiR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1α regulation. Oncogenesis 2016, 5, e224. [Google Scholar] [CrossRef]
- Minchenko, O.; Opentanova, I.; Caro, J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003, 554, 264–270. [Google Scholar] [CrossRef]
- Cui, X.-G.; Han, Z.-T.; He, S.-H.; Wu, X.-D.; Chen, T.-R.; Shao, C.-H.; Chen, D.-L.; Su, N.; Chen, Y.-M.; Wang, T.; et al. HIF1/2α mediates hypoxia-induced LDHA expression in human pancreatic cancer cells. Oncotarget 2017, 8. [Google Scholar] [CrossRef]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J. Biol. Chem. 1995, 270, 21021–21027. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and Beyond: Multiple Functions of the Human Mitochondrial Transcription Machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar]
- Domblides, C.; Lartigue, L.; Faustin, B. Control of the Antitumor Immune Response by Cancer Metabolism. Cells 2019, 8, 104. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Elia, A.R.; Cappello, P.; Puppo, M.; Fraone, T.; Vanni, C.; Eva, A.; Musso, T.; Novelli, F.; Varesio, L.; Giovarelli, M. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J. Leukoc. Biol. 2008, 84, 1472–1482. [Google Scholar] [CrossRef]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.-P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.X.; Kazlauskas, A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J. Biol. Chem. 2013, 288, 21161–21172. [Google Scholar] [CrossRef] [PubMed]
- Rudrabhatla, S.R.; Mahaffey, C.L.; Mummert, M.E. Tumor microenvironment modulates hyaluronan expression: The lactate effect. J. Investig. Derm. 2006, 126, 1378–1387. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Rothberg, J.M.; Kumar, V.; Schramm, K.J.; Haller, E.; Proemsey, J.B.; Lloyd, M.C.; Sloane, B.F.; Gillies, R.J. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 2012, 72, 3938–3947. [Google Scholar] [CrossRef]
- Shi, W.K.; Zhu, X.D.; Wang, C.H.; Zhang, Y.Y.; Cai, H.; Li, X.L.; Cao, M.Q.; Zhang, S.Z.; Li, K.S.; Sun, H.C. PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT. Cell Death Dis. 2018, 9, 428. [Google Scholar] [CrossRef]
- Wagner, W.; Ciszewski, W.M.; Kania, K.D. L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun. Signal. 2015, 13, 36. [Google Scholar] [CrossRef]
- Sattler, U.G.A.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lin, Q.; Zelterman, D.; Yun, Z. Hypoxia-regulated delta-like 1 homologue enhances cancer cell stemness and tumorigenicity. Cancer Res. 2009, 69, 9271–9280. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Bandopadhyay, G.; Coyle, B.; Grabowska, A. A HIF-independent, CD133-mediated mechanism of cisplatin resistance in glioblastoma cells. Cell. Oncol. 2018, 41, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Harding, M.A.; Smith, S.C.; Overdevest, J.B.; Nitz, M.D.; Frierson, H.F.; Tomlins, S.A.; Kristiansen, G.; Theodorescu, D. CD24 Is an Effector of HIF-1–Driven Primary Tumor Growth and Metastasis. Cancer Res. 2012, 72, 5600–5612. [Google Scholar] [CrossRef] [PubMed]
- Nascimento-Filho, C.H.V.; Webber, L.P.; Borgato, G.B.; Goloni-Bertollo, E.M.; Squarize, C.H.; Castilho, R.M. Hypoxic niches are endowed with a protumorigenic mechanism that supersedes the protective function of PTEN. FASEB J. 2019, 33, 13435–13449. [Google Scholar] [CrossRef]
- Santos, J.C.; Lima, N.D.S.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L.; Derchain, S.F.M. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci. Rep. 2018, 8, 829. [Google Scholar] [CrossRef]
- Onnis, B.; Rapisarda, A.; Melillo, G. Development of HIF-1 inhibitors for cancer therapy. J. Cell Mol. Med. 2009, 13, 2780–2786. [Google Scholar] [CrossRef]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Köhler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin alpha/beta pathway. Biochim. Biophys. Acta 2008, 1783, 394–404. [Google Scholar] [CrossRef]
- Kosyna, F.K.; Nagel, M.; Kluxen, L.; Kraushaar, K.; Depping, R. The importin α/β-specific inhibitor Ivermectin affects HIF-dependent hypoxia response pathways. Biol. Chem. 2015, 396, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Kosyna, F.K.; Depping, R. Controlling the Gatekeeper: Therapeutic Targeting of Nuclear Transport. Cells 2018, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Chafe, S.C.; Dedhar, S. Overcoming Hypoxia-Mediated Tumor Progression: Combinatorial Approaches Targeting pH Regulation, Angiogenesis and Immune Dysfunction. Front. Cell Dev. Biol. 2016, 4, 27. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Galvis, M.; Teng, Y. Targeting Hypoxia-Driven Metabolic Reprogramming to Constrain Tumor Progression and Metastasis. Int. J. Mol. Sci. 2020, 21, 5487. https://doi.org/10.3390/ijms21155487
Miranda-Galvis M, Teng Y. Targeting Hypoxia-Driven Metabolic Reprogramming to Constrain Tumor Progression and Metastasis. International Journal of Molecular Sciences. 2020; 21(15):5487. https://doi.org/10.3390/ijms21155487
Chicago/Turabian StyleMiranda-Galvis, Marisol, and Yong Teng. 2020. "Targeting Hypoxia-Driven Metabolic Reprogramming to Constrain Tumor Progression and Metastasis" International Journal of Molecular Sciences 21, no. 15: 5487. https://doi.org/10.3390/ijms21155487
APA StyleMiranda-Galvis, M., & Teng, Y. (2020). Targeting Hypoxia-Driven Metabolic Reprogramming to Constrain Tumor Progression and Metastasis. International Journal of Molecular Sciences, 21(15), 5487. https://doi.org/10.3390/ijms21155487