Non-Melanoma Skin Cancers: Biological and Clinical Features

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Normal Skin and the Mechanisms of Cancerogenesis
3. Basal Cell Carcinoma
3.1. The Genetic Landscape of BCC
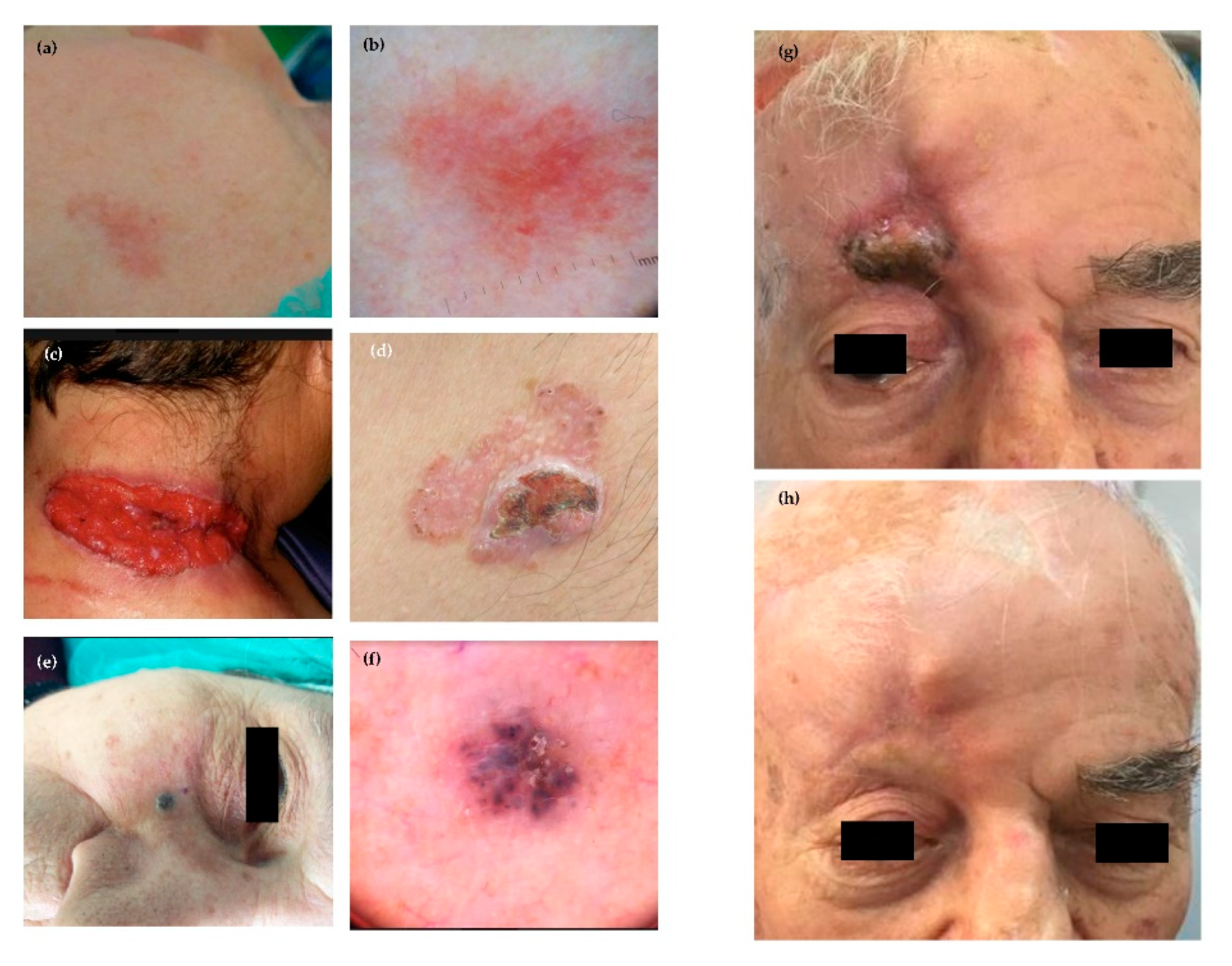
3.2. Epidemiology, Classification and Clinical Features
3.2.1. Nodular
3.2.2. Superficial
3.2.3. Fibroepithelial
3.2.4. Morpheaform
3.2.5. Infiltrative
3.2.6. Micronodular
3.2.7. Basosquamous
3.3. Therapeutic Options
3.3.1. Radiotherapy
3.3.2. Systemic Treatments
3.3.3. Innovative Therapeutic Strategies
4. Squamous Cell Carcinoma
4.1. The Molecular Features of SCC
4.2. The Clinical Characteristics of SCC
4.3. The Immune System and SCC
4.4. Systemic Treatments
5. Merkel Cell Carcinoma
5.1. Pathogenesis of MCC
5.2. The Clinical Presentation and Diagnosis of MCC
5.3. The Role of Sentinel Lymph Node Biopsy and of Systemic Treatments
6. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Veisani, Y.; Jenabi, E.; Khazaei, S.; Nematollahi, S. Global incidence and mortality rates in pancreatic cancer and the association with the Human Development Index: Decomposition approach. Public Health 2018, 156, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Zaar, O.; Gillstedt, M.; Lindelöf, B.; Wennberg-Larkö, A.-M.; Paoli, J. Merkel cell carcinoma incidence is increasing in Sweden. J. Eur. Acad. Derm. Venereol 2016, 30, 1708–1713. [Google Scholar] [CrossRef] [PubMed]
- Emerging trends in the treatment of advanced basal cell carcinoma. Public Health 2017, 64, 1–10.
- García, J.B.; Suárez-Varela, M.M.; Vilata, J.J.; Marquina, A.; Pallardó, L.; Crespo, J. Risk factors for non-melanoma skin cancer in kidney transplant patients in a Spanish population in the Mediterranean region. Acta Derm. Venereol. 2013, 93, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.Y.; Popescu, N.A.; Su, W.P.; Chute, C.G. Squamous cell carcinoma. A population-based incidence study in Rochester, Minn. Arch. Derm. 1990, 126, 185–188. [Google Scholar] [CrossRef]
- Samarasinghe, V.; Madan, V. Nonmelanoma skin cancer. Oncotarget 2012, 5, 3–10. [Google Scholar] [CrossRef]
- Chuang, T.Y.; Popescu, A.; Su, W.P.; Chute, C.G. Basal cell carcinoma. A population-based incidence study in Rochester, Minnesota. J. Am. Acad. Derm. 1990, 22, 413–417. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer. J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Cribier, B.; Scrivener, Y.; Grosshans, E. Tumors arising in nevus sebaceus: A study of 596 cases. Cell 2000, 42, 263–268. [Google Scholar] [CrossRef]
- Euvrard, S.; Kanitakis, J.; Claudy, A. Skin cancers after organ transplantation. N Engl. J. Med. 2003, 348, 1681–1691. [Google Scholar] [CrossRef]
- Silverberg, M.J.; Leyden, W.; Warton, E.M.; Quesenberry, C.P.; Engels, E.A.; Asgari, M.M. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J. Natl. Cancer Inst. 2013, 105, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.S.; Bolger, P.M. Dietary arsenic intakes in the United States: FDA Total Diet Study, September 1991-December 1996. Mol. Nucleic. Acids 2001, 16, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Christenson, L.J.; Borrowman, T.A.; Vachon, C.M.; Tollefson, M.M.; Otley, C.C.; Weaver, A.L.; Roenigk, R.K. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA 2005, 294, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Rigel, D.S.; Friedman, R.J.; Kopf, A.W. Lifetime risk for development of skin cancer in the U.S. population: Current estimate is now 1 in 5. J. Am. Acad. Derm. 1996, 35, 1012–1013. [Google Scholar] [CrossRef]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef]
- Kripke, M.L. Skin cancer, photoimmunology, and urocanic acid. Photo-dermatology 1984, 1, 161–163. [Google Scholar]
- Carroll, R.P.; Ramsay, H.M.; Fryer, A.A.; Hawley, C.M.; Nicol, D.L.; Harden, P.N. Incidence and prediction of nonmelanoma skin cancer post-renal transplantation: A prospective study in Queensland, Australia. Oncotarget 2003, 41, 676–683. [Google Scholar] [CrossRef]
- Marzuka, A.G.; Book, S.E. Basal cell carcinoma: Pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J. Biol. Med. 2015, 88, 167–179. [Google Scholar]
- Kripke, M.L. Immunological unresponsiveness induced by ultraviolet radiation. Immunol. Rev. 1984, 80, 87–102. [Google Scholar] [CrossRef]
- Kripke, M.L. Effects of methoxsalen plus near-ultraviolet radiation or mid-ultraviolet radiation on immunologic mechanisms. Natl. Cancer Inst. Monogr. 1984, 66, 247–251. [Google Scholar]
- Moloney, F.J.; Comber, H.; Conlon, P.J.; Murphy, G.M. The role of immunosuppression in the pathogenesis of basal cell carcinoma. Br. J. Derm. 2006, 154, 790–791. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R.-M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Derm. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Kaskel, P.; Lange, U.; Sander, S.; Huber, M.A.; Utikal, J.; Leiter, U.; Krähn, G.; Meurer, M.; Kron, M. Ultraviolet exposure and risk of melanoma and basal cell carcinoma in Ulm and Dresden, Germany. J. Eur. Acad. Derm. Venereol. 2014, 29, 134–142. [Google Scholar] [CrossRef]
- Chang, N.-B.; Feng, R.; Gao, Z.; Gao, W. Skin cancer incidence is highly associated with ultraviolet-B radiation history. Int. J. Hyg. Environ. Health 2010, 213, 359–368. [Google Scholar] [CrossRef]
- Coelho, S.G.; Choi, W.; Brenner, M.; Miyamura, Y.; Yamaguchi, Y.; Wolber, R.; Smuda, C.; Batzer, J.; Kolbe, L.; Ito, S.; et al. Short- and long-term effects of UV radiation on the pigmentation of human skin. J. Investig. Derm. Symp. Proc. 2009, 14, 32–35. [Google Scholar] [CrossRef]
- Benjamin, C.L.; Melnikova, V.O.; Ananthaswamy, H.N. P53 protein and pathogenesis of melanoma and nonmelanoma skin cancer. Adv. Exp. Med. Biol. 2008, 624, 265–282. [Google Scholar]
- Marshall, S.E.; Bordea, C.; Haldar, N.A.; Mullighan, C.G.; Wojnarowska, F.; Morris, P.J.; Welsh, K.I. Glutathione S-transferase polymorphisms and skin cancer after renal transplantation. Kidney Int. 2000, 58, 2186–2193. [Google Scholar] [CrossRef]
- Cameron, M.C.; Lee, E.; Hibler, B.P.; Barker, C.A.; Mori, S.; Cordova, M.; Nehal, K.S.; Rossi, A.M. Basal cell carcinoma: Epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J. Am. Acad. Derm. 2018, 80, 303–317. [Google Scholar] [CrossRef]
- de Sá, T.R.C.; Silva, R.; Lopes, J.M. Basal cell carcinoma of the skin (part 1): Epidemiology, pathology and genetic syndromes. Future Oncol. 2015, 11, 3011–3021. [Google Scholar] [CrossRef]
- Bresler, S.C.; Padwa, B.L.; Granter, S.R. Nevoid Basal Cell Carcinoma Syndrome (Gorlin Syndrome). Head Neck Pathol. 2016, 10, 119–124. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharm. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.; Reiter, J.F. The primary cilium at the crossroads of mammalian hedgehog signaling. Curr. Top. Dev. Biol. 2009, 85, 225–260. [Google Scholar]
- di Magliano, M.P.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2004, 3, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Levesque, M.P.; Dummer, R.; Kabashima, K. Hedgehog signaling in basal cell carcinoma. Trends Immunol. 2015, 78, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.R.; Choksi, P.K.; Krup, A.L.; Mayer, W.; Santos, N.; Reiter, J.F. Hedgehog signaling drives medulloblastoma growth via CDK6. Nat. Med. 2017, 128, 120–124. [Google Scholar] [CrossRef]
- Monkkonen, T.; Lewis, M.T. New paradigms for the Hedgehog signaling network in mammary gland development and breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef]
- Bushman, W. Hedgehog Signaling in Prostate Development, Regeneration and Cancer. J. Dev. Biol 2016, 4, 30. [Google Scholar] [CrossRef]
- Song, L.; Li, Z.-Y.; Liu, W.-P.; Zhao, M.-R. Crosstalk between Wnt/β-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Bai, Y.; Bai, Y.; Dong, J.; Li, Q.; Jin, Y.; Chen, B.; Zhou, M. Hedgehog Signaling in Pancreatic Fibrosis and Cancer. Medicine (Baltimore) 2016, 95, e2996. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Gutzmer, R.; Kieran, M.W.; Solomon, J.A. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat. Rev. 2019, 76, 41–50. [Google Scholar] [CrossRef]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.-S.; Chang, C.-F.; Lin, S.-S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Adv. Med. Oncol. 2011, 2, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chi, S.; Xie, J. Hedgehog signaling in skin cancers. J. Control. Release 2011, 23, 1235–1243. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; García-Rodrigo, C.G.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef]
- McCusker, M.; Basset-Seguin, N.; Dummer, R.; Lewis, K.; Schadendorf, D.; Sekulic, A.; Hou, J.; Wang, L.; Yue, H.; Hauschild, A. Metastatic basal cell carcinoma: Prognosis dependent on anatomic site and spread of disease. Trends Pharm. Sci. 2014, 50, 774–783. [Google Scholar] [CrossRef]
- Wong, S.L.; Kattan, M.W.; McMasters, K.M.; Coit, D.G. A nomogram that predicts the presence of sentinel node metastasis in melanoma with better discrimination than the American Joint Committee on Cancer staging system. Ann. Surg. Oncol. 2005, 12, 282–288. [Google Scholar] [CrossRef]
- Staples, M.P.; Elwood, M.; Burton, R.C.; Williams, J.L.; Marks, R.; Giles, G.G. Non-melanoma skin cancer in Australia: The 2002 national survey and trends since 1985. Med. J. Aust. 2006, 184, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Richmond-Sinclair, N.M.; Pandeya, N.; Ware, R.S.; Neale, R.E.; Williams, G.M.; van der Pols, J.C.; Green, A.C. Incidence of basal cell carcinoma multiplicity and detailed anatomic distribution: Longitudinal study of an Australian population. J. Investig. Derm. Symp. Proc. 2008, 129, 323–328. [Google Scholar] [CrossRef]
- Zoledronic acid prevents vertebral fractures in men with osteoporosis. Bonekey Rep. 2013, 2, 294.
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Derm. 2012, 166, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Trakatelli, M.; Morton, C.; Nagore, E.; Ulrich, C.; Del Marmol, V.; Peris, K.; Basset-Seguin, N. Update of the European guidelines for basal cell carcinoma management. Eur. J. Derm. 2014, 24, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Kauvar, A.N.B.; Cronin, T.; Roenigk, R.; Hruza, G.; Bennett, R. Consensus for nonmelanoma skin cancer treatment: Basal cell carcinoma, including a cost analysis of treatment methods. Derm. Surg. 2015, 41, 550–571. [Google Scholar] [CrossRef]
- Di Stefani, A.; Chimenti, S. Basal cell carcinoma: Clinical and pathological features. G Ital. Derm. Venereol. 2015, 150, 385–391. [Google Scholar]
- Cullen, R.; Hasbún, P.; Campos-Villenas, M. Superficial basal cell carcinoma. Exp. Hematol. 2016, 149, 140. [Google Scholar] [CrossRef]
- Abbas, O.; Richards, J.E.; Mahalingam, M. Fibroblast-activation protein: A single marker that confidently differentiates morpheaform/infiltrative basal cell carcinoma from desmoplastic trichoepithelioma. Mod. Pathol. 2010, 23, 1535–1543. [Google Scholar] [CrossRef]
- Betti, R.; Menni, S.; Radaelli, G.; Bombonato, C.; Crosti, C. Micronodular basal cell carcinoma: A distinct subtype? Relationship with nodular and infiltrative basal cell carcinomas. J. Derm. 2010, 37, 611–616. [Google Scholar] [CrossRef]
- Tan, C.Z.; Rieger, K.E.; Sarin, K.Y. Basosquamous Carcinoma: Controversy, Advances, and Future Directions. Derm. Surg. 2016, 43, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Drucker, A.M.; Adam, G.P.; Rofeberg, V.; Gazula, A.; Smith, B.; Moustafa, F.; Weinstock, M.A.; Trikalinos, T.A. Treatments of Primary Basal Cell Carcinoma of the Skin: A Systematic Review and Network Meta-analysis. Ann. Intern. Med. 2018, 169, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.M.; Baker, D.R.; Coldiron, B.M.; Fazio, M.J.; Storrs, P.A.; Vidimos, A.T.; Zalla, M.J.; Brewer, J.D.; Begolka, W.S.; Berger, T.G.; et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: A report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J. Control. Release 2012, 67, 531–550. [Google Scholar]
- Lazarevic, D.; Ramelyte, E.; Dummer, R.; Imhof, L. Radiotherapy in Periocular Cutaneous Malignancies: A Retrospective Study. Dermatology (Basel) 2019, 235, 234–239. [Google Scholar] [CrossRef]
- Guinot, J.L.; Rembielak, A.; Perez-Calatayud, J.; Rodríguez-Villalba, S.; Skowronek, J.; Tagliaferri, L.; Guix, B.; Gonzalez-Perez, V.; Valentini, V.; Kovacs, G. GEC-ESTRO ACROP recommendations in skin brachytherapy. Radiother. Oncol. 2018, 126, 377–385. [Google Scholar] [CrossRef]
- Peris, K.; Fargnoli, M.C.; Garbe, C.; Kaufmann, R.; Bastholt, L.; Seguin, N.B.; Bataille, V.; Del Marmol, V.; Dummer, R.; Harwood, C.A.; et al. Diagnosis and treatment of basal cell carcinoma: European consensus-based interdisciplinary guidelines. Eur. J. Cancer 2019, 118, 10–34. [Google Scholar] [CrossRef]
- Dummer, R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kaatz, M.; Loquai, C.; Stratigos, A.J. The 12-month analysis from Basal Cell Carcinoma Outcomes with LDE225 Treatment (BOLT): A phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J. Am. Acad. Derm. 2016, 75, 113–125. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Dummer, R.; Guminksi, A.; Gutzmer, R.; Lear, J.T.; Lewis, K.D.; Chang, A.L.S.; Combemale, P.; Dirix, L.; Kaatz, M.; Kudchadkar, R.; et al. Long-term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42-month analysis of the phase II randomized, double-blind BOLT study. Br. J. Derm. 2019, 182, 1369–1378. [Google Scholar] [CrossRef]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L.S. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2015, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Smith, B.D.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Oncotarget 2018, 93, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Basset-Séguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. J. Control. Release 2017, 86, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Trends Immunol. 2015, 27, 327–341. [Google Scholar] [CrossRef]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef]
- Dréno, B.; Kunstfeld, R.; Hauschild, A.; Fosko, S.; Zloty, D.; Labeille, B.; Grob, J.-J.; Puig, S.; Gilberg, F.; Bergström, D.; et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): A randomised, regimen-controlled, double-blind, phase 2 trial. Oncotarget 2017, 18, 404–412. [Google Scholar] [CrossRef]
- Lear, J.T.; Migden, M.R.; Lewis, K.D.; Chang, A.L.S.; Guminski, A.; Gutzmer, R.; Dirix, L.; Combemale, P.; Stratigos, A.; Plummer, R.; et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. J. Eur. Acad. Derm. Venereol. 2017, 32, 372–381. [Google Scholar] [CrossRef]
- Bridge, J.A.; Lee, J.C.; Daud, A.; Wells, J.W.; Bluestone, J.A. Cytokines, Chemokines, and Other Biomarkers of Response for Checkpoint Inhibitor Therapy in Skin Cancer. Front. Immunol. 2018, 5, 351. [Google Scholar] [CrossRef]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef]
- Zhao, X.; Pak, E.; Ornell, K.J.; Pazyra-Murphy, M.F.; MacKenzie, E.L.; Chadwick, E.J.; Ponomaryov, T.; Kelleher, J.F.; Segal, R.A. A Transposon Screen Identifies Loss of Primary Cilia as a Mechanism of Resistance to SMO Inhibitors. Cancer Discov. 2017, 7, 1436–1449. [Google Scholar] [CrossRef]
- Gruber, W.; Hutzinger, M.; Elmer, D.P.; Parigger, T.; Sternberg, C.; Cegielkowski, L.; Zaja, M.; Leban, J.; Michel, S.; Hamm, S.; et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget 2016, 7, 7134–7148. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Leiter, U.; Keim, U.; Eigentler, T.; Katalinic, A.; Holleczek, B.; Martus, P.; Garbe, C. Incidence, Mortality, and Trends of Nonmelanoma Skin Cancer in Germany. Eur. J. Cancer 2017, 137, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.J.; Garbe, C.; Dessinioti, C.; Bataille, V.; Bastholt, L.; Fargnoli, M.C.; Forsea, A.M.; Frenard, C.; Harwood, C.A.; Hauschild, A.; et al. European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part 2. Treatment. J. Control. Release 2020, 128, 60–82. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic. Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Ventura, A.; Pellegrini, C.; Cardelli, L.; Rocco, T.; Ciciarelli, V.; Peris, K.; Fargnoli, M.C. Telomeres and Telomerase in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 1333. [Google Scholar] [CrossRef]
- García-Sancha, N.; Corchado-Cobos, R.; Pérez-Losada, J.; Cañueto, J. MicroRNA Dysregulation in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar] [CrossRef]
- Yilmaz, A.S.; Ozer, H.G.; Gillespie, J.L.; Allain, D.C.; Bernhardt, M.N.; Furlan, K.C.; Castro, L.T.F.; Peters, S.B.; Nagarajan, P.; Kang, S.Y.; et al. Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors. Cancer 2016, 123, 1184–1193. [Google Scholar] [CrossRef]
- Cho, R.J.; Alexandrov, L.B.; Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, 1126. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.A.; Bener, A.; Teebi, A.S. The incidence patterns of Down syndrome in Qatar. Clin. Genet. 2006, 69, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Chantrain, C.F.; Henriet, P.; Jodele, S.; Emonard, H.; Feron, O.; Courtoy, P.J.; DeClerck, Y.A.; Marbaix, E. Mechanisms of pericyte recruitment in tumour angiogenesis: A new role for metalloproteinases. Nat. Med. 2006, 42, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Dirschka, T.; Gupta, G.; Micali, G.; Stockfleth, E.; Dummer, R.; Jemec, G.B.E.; Malvehy, J.; Peris, K.; Puig, S.; Stratigos, A.J.; et al. Real-world approach to actinic keratosis management: Practical treatment algorithm for office-based dermatology. J. Dermatol. Treat. 2016, 28, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.; Garbe, C.; Malvehy, J.; Del Marmol, V.; Pehamberger, H.; Peris, K.; Becker, J.C.; Zalaudek, I.; Saiag, P.; Middleton, M.R.; et al. Diagnosis and treatment of invasive squamous cell carcinoma of the skin: European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 1989–2007. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.J.; Garbe, C.; Dessinioti, C.; Bataille, V.; Bastholt, L.; Fargnoli, M.C.; Forsea, A.M.; Frenard, C.; Harwood, C.A.; Hauschild, A.; et al. European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part 1. epidemiology, diagnostics and prevention. J. Control. Release 2020, 128, 60–82. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Nissinen, L.; Knuutila, J.; Nezhad, P.R.; Viiklepp, K.; Kähäri, V.-M. Complement System in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 3550. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019, 19, 688–701. [Google Scholar] [CrossRef]
- Motwani, M.P.; Gilroy, D.W. Macrophage development and polarization in chronic inflammation. J. Control. Release 2015, 27, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514. [Google Scholar] [CrossRef]
- Tucci, M.; Ciavarella, S.; Strippoli, S.; Brunetti, O.; Dammacco, F.; Silvestris, F. Immature dendritic cells from patients with multiple myeloma are prone to osteoclast differentiation in vitro. Exp. Hematol. 2011, 39, 773–783. [Google Scholar] [CrossRef]
- Fernandez, T.L.; Van Lonkhuyzen, D.R.; Dawson, R.A.; Kimlin, M.G.; Upton, Z. Characterization of a human skin equivalent model to study the effects of ultraviolet B radiation on keratinocytes. Tissue Eng. Part. C Methods 2014, 20, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef]
- Norval, M.; Simpson, T.J.; Ross, J.A. Urocanic acid and immunosuppression. Photochem. Photobiol. 1989, 50, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.R.; Woodward, S.; Sabharwal, S.C. Alpha 2-adrenergic mechanism in menopausal hot flushes. Obs. Gynecol. 1990, 76, 573–578. [Google Scholar]
- Szélig, L.; Kun, S.; Woth, G.; Molnár, G.A.; Zrínyi, Z.; Kátai, E.; Lantos, J.; Wittmann, I.; Bogár, L.; Miseta, A.; et al. Time courses of changes of para-, meta-, and ortho-tyrosine in septic patients: A pilot study. Redox Rep. 2016, 21, 180–189. [Google Scholar] [CrossRef]
- Tucci, M.; Mannavola, F.; Passarelli, A.; Stucci, L.S.; Cives, M.; Silvestris, F. Exosomes in melanoma: A role in tumor progression, metastasis and impaired immune system activity. Oncotarget 2018, 9, 20826–20837. [Google Scholar] [CrossRef]
- Mannavola, F.; Tucci, M.; Felici, C.; Passarelli, A.; D’Oronzo, S.; Silvestris, F. Tumor-derived exosomes promote the in vitro osteotropism of melanoma cells by activating the SDF-1/CXCR4/CXCR7 axis. J. Transl. Med. 2019, 17, 230. [Google Scholar] [CrossRef]
- Mannavola, F.; Pezzicoli, G.; Tucci, M. DLC-1 down-regulation via exosomal miR-106b-3p exchange promotes CRC metastasis by the epithelial-to-mesenchymal transition. Mol. Nucleic. Acids 2020, 134, 955–959. [Google Scholar]
- Mannavola, F.; D’Oronzo, S.; Cives, M.; Stucci, L.S.; Ranieri, G.; Silvestris, F.; Tucci, M. Extracellular Vesicles and Epigenetic Modifications Are Hallmarks of Melanoma Progression. Int. J. Mol. Sci. 2019, 21, 52. [Google Scholar] [CrossRef]
- Mannavola, F.; Salerno, T.; Passarelli, A.; Tucci, M.; Internò, V.; Silvestris, F. Revisiting the Role of Exosomes in Colorectal Cancer: Where Are We Now? Front. Oncol. 2019, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Stucci, L.S.; Mannavola, F.; Passarelli, A.; D’Oronzo, S.; Lospalluti, L.; Giudice, G.; Silvestris, F. Defective levels of both circulating dendritic cells and T-regulatory cells correlate with risk of recurrence in cutaneous melanoma. Clin. Transl. Oncol. 2019, 21, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Stucci, L.S.; Ascierto, P.A.; Capone, M.; Madonna, G.; Lopalco, P.; Silvestris, F. Serum exosomes as predictors of clinical response to ipilimumab in metastatic melanoma. Oncoimmunology 2017, 7, e1387706. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Mueller, S.A.; Gauthier, M.-E.A.; Ashford, B.; Gupta, R.; Gayevskiy, V.; Ch’ng, S.; Palme, C.E.; Shannon, K.; Clark, J.R.; Ranson, M.; et al. Mutational Patterns in Metastatic Cutaneous Squamous Cell Carcinoma. J. Investig. Derm. 2019, 139, 1449–1458.e1. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Hernández-Guerrero, T.; Doger, B.; Moreno, V. Cemiplimab for the treatment of advanced cutaneous squamous cell carcinoma. Drugs Today 2019, 55, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Bertino, G.; Sersa, G.; De Terlizzi, F.; Occhini, A.; Plaschke, C.C.; Groselj, A.; Langdon, C.; Grau, J.J.; McCaul, J.A.; Heuveling, D.; et al. European Research on Electrochemotherapy in Head and Neck Cancer (EURECA) project: Results of the treatment of skin cancer. Eur. J. Cancer 2016, 63, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef]
- Simon, B.; Uslu, U. CAR-T cell therapy in melanoma: A future success story? Exp. Derm. 2018, 27, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schaft, N.; Schuler, G.; Dörrie, J.; Uslu, U. The siRNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in vitro CAR-T-cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp. Derm. 2018, 27, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Derm. 2017, 78, 457–463.e2. [Google Scholar] [CrossRef]
- Fondain, M.; Dereure, O.; Uhry, Z.; Guizard, A.V.; Woronoff, A.S.; Colonna, M.; Molinie, F.; Bara, S.; Velten, M.; Marrer, E.; et al. Merkel cell carcinoma in France: A registries-based, comprehensive epidemiological survey. J. Eur. Acad. Derm. Venereol. 2018, 32, 1292–1296. [Google Scholar] [CrossRef]
- Youlden, D.R.; Soyer, H.P.; Youl, P.H.; Fritschi, L.; Baade, P.D. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993–2010. JAMA Derm. 2014, 150, 864–872. [Google Scholar] [CrossRef]
- Reichgelt, B.A.; Visser, O. Epidemiology and survival of Merkel cell carcinoma in the Netherlands. A population-based study of 808 cases in 1993–2007. Mol. Nucleic. Acids 2010, 47, 579–585. [Google Scholar] [CrossRef]
- Albores-Saavedra, J.; Batich, K.; Chable-Montero, F.; Sagy, N.; Schwartz, A.M.; Henson, D.E. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: A population based study. J. Cutan. Pathol. 2009, 37, 20–27. [Google Scholar] [CrossRef]
- Stang, A.; Becker, J.C.; Nghiem, P.; Ferlay, J. The association between geographic location and incidence of Merkel cell carcinoma in comparison to melanoma: An international assessment. J. Control. Release 2018, 94, 47–60. [Google Scholar] [CrossRef]
- Koljonen, V.; Kukko, H.; Pukkala, E.; Sankila, R.; Böhling, T.; Tukiainen, E.; Sihto, H.; Joensuu, H. Chronic lymphocytic leukaemia patients have a high risk of Merkel-cell polyomavirus DNA-positive Merkel-cell carcinoma. Br. J. Cancer 2009, 101, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, F.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Longino, N.V.; Yang, J.; Iyer, J.G.; Ibrani, D.; Chow, I.-T.; Laing, K.J.; Campbell, V.L.; Paulson, K.G.; Kulikauskas, R.M.; Church, C.D.; et al. Human CD4+ T Cells Specific for Merkel Cell Polyomavirus Localize to Merkel Cell Carcinomas and Target a Required Oncogenic Domain. Cancer Immunol. Res. 2019, 7, 1727–1739. [Google Scholar] [CrossRef]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.; Lai, I.; et al. Merkel cell polyomavirus-specific CD8⁺ and CD4⁺ T-cell responses identified in Merkel cell carcinomas and blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar] [CrossRef]
- Afanasiev, O.K.; Yelistratova, L.; Miller, N.; Nagase, K.; Paulson, K.; Iyer, J.G.; Ibrani, D.; Koelle, D.M.; Nghiem, P. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin. Cancer Res. 2013, 19, 5351–5360. [Google Scholar] [CrossRef]
- Jing, L.; Ott, M.; Church, C.D.; Kulikauskas, R.M.; Ibrani, D.; Iyer, J.G.; Afanasiev, O.K.; Colunga, A.; Cook, M.M.; Xie, H.; et al. Prevalent and Diverse Intratumoral Oncoprotein-Specific CD8+ T Cells within Polyomavirus-Driven Merkel Cell Carcinomas. Cancer Immunol. Res. 2020, 8, 648–659. [Google Scholar] [CrossRef]
- Colunga, A.; Pulliam, T.; Nghiem, P. Merkel Cell Carcinoma in the Age of Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2017, 24, 2035–2043. [Google Scholar] [CrossRef]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef]
- Dabner, M.; McClure, R.J.; Harvey, N.T.; Budgeon, C.A.; Beer, T.W.; Amanuel, B.; Wood, B.A. Merkel cell polyomavirus and p63 status in Merkel cell carcinoma by immunohistochemistry: Merkel cell polyomavirus positivity is inversely correlated with sun damage, but neither is correlated with outcome. Pathology 2014, 46, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Garneski, K.M.; Warcola, A.H.; Feng, Q.; Kiviat, N.B.; Leonard, J.H.; Nghiem, P. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J. Investig. Derm. Symp. Proc. 2008, 129, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Mangana, J.; Dziunycz, P.; Kerl, K.; Dummer, R.; Cozzio, A. Prevalence of Merkel cell polyomavirus among Swiss Merkel cell carcinoma patients. Dermatology (Basel) 2010, 221, 184–188. [Google Scholar] [CrossRef]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2015, 7, 3403–3415. [Google Scholar] [CrossRef]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef]
- Harms, K.L.; de la Vega, L.L.; Hovelson, D.H.; Rahrig, S.; Cani, A.K.; Liu, C.-J.; Fullen, D.R.; Wang, M.; Andea, A.A.; Bichakjian, C.K.; et al. Molecular Profiling of Multiple Primary Merkel Cell Carcinoma to Distinguish Genetically Distinct Tumors from Clonally Related Metastases. JAMA Derm. 2017, 153, 505–512. [Google Scholar] [CrossRef]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.-M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef]
- Harms, K.L.; Healy, M.A.; Nghiem, P.; Sober, A.J.; Johnson, T.M.; Bichakjian, C.K.; Wong, S.L. Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann. Surg. Oncol. 2016, 23, 3564–3571. [Google Scholar] [CrossRef]
- Vandeven, N.; Lewis, C.W.; Makarov, V.; Riaz, N.; Paulson, K.G.; Hippe, D.; Bestick, A.; Doumani, R.; Marx, T.; Takagishi, S.; et al. Merkel Cell Carcinoma Patients Presenting Without a Primary Lesion Have Elevated Markers of Immunity, Higher Tumor Mutation Burden, and Improved Survival. Clin. Cancer Res. 2017, 24, 963–971. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers. 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.T.; Papavasiliou, P.; Edwards, K.; Zhu, F.; Perlis, C.; Wu, H.; Turaka, A.; Berger, A.; Farma, J.M. A better prognosis for Merkel cell carcinoma of unknown primary origin. J. Control. Release 2013, 206, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.L.; Dedivitis, R.A.; Kulcsar, M.A.V.; de Mello, E.S.; Alves, V.A.F.; Cernea, C.R. External validation of the AJCC Cancer Staging Manual, 8th edition, in an independent cohort of oral cancer patients. J. Control. Release 2017, 71, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Concannon, R.; Larcos, G.S.; Veness, M. The impact of (18)F-FDG PET-CT scanning for staging and management of Merkel cell carcinoma: Results from Westmead Hospital, Sydney, Australia. Eur. J. Cancer 2010, 62, 76–84. [Google Scholar]
- Samimi, M.; Molet, L.; Fleury, M.; Laude, H.; Carlotti, A.; Gardair, C.; Baudin, M.; Gouguet, L.; Maubec, E.; Avenel-Audran, M.; et al. Prognostic value of antibodies to Merkel cell polyomavirus T antigens and VP1 protein in patients with Merkel cell carcinoma. Br. J. Derm. 2016, 174, 813–822. [Google Scholar] [CrossRef]
- Paulson, K.G.; Carter, J.J.; Johnson, L.G.; Cahill, K.W.; Iyer, J.G.; Schrama, D.; Becker, J.C.; Madeleine, M.M.; Nghiem, P.; Galloway, D.A. Antibodies to merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in merkel cell carcinoma patients. Cancer Res. 2010, 70, 8388–8397. [Google Scholar] [CrossRef]
- Harrington, C.; Kwan, W. Outcomes of Merkel cell carcinoma treated with radiotherapy without radical surgical excision. Ann. Surg. Oncol. 2014, 21, 3401–3405. [Google Scholar] [CrossRef]
- Strom, T.; Carr, M.; Zager, J.S.; Naghavi, A.; Smith, F.O.; Cruse, C.W.; Messina, J.L.; Russell, J.; Rao, N.G.; Fulp, W.; et al. Radiation Therapy is Associated with Improved Outcomes in Merkel Cell Carcinoma. Ann. Surg. Oncol. 2016, 23, 3572–3578. [Google Scholar] [CrossRef]
- Bhatia, S.; Storer, B.E.; Iyer, J.G.; Moshiri, A.; Parvathaneni, U.; Byrd, D.; Sober, A.J.; Sondak, V.K.; Gershenwald, J.E.; Nghiem, P. Adjuvant Radiation Therapy and Chemotherapy in Merkel Cell Carcinoma: Survival Analyses of 6908 Cases from the National Cancer Data Base. J. Natl. Cancer Inst. 2016, 108, djw042. [Google Scholar] [CrossRef]
- Topalian, S.L.; Bhatia, S.; Amin, A.; Kudchadkar, R.R.; Sharfman, W.H.; Delord, J.-P.; Dunn, L.A.; Shinohara, M.M.; Kulikauskas, R.; Chung, C.H.; et al. Neoadjuvant Nivolumab for Patients with Resectable Merkel Cell Carcinoma in the CheckMate 358 Trial. J. Clin. Oncol. 2020, JCO2000201. [Google Scholar] [CrossRef]
- Tai, P.T.; Yu, E.; Winquist, E.; Hammond, A.; Stitt, L.; Tonita, J.; Gilchrist, J. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: Case series and review of 204 cases. J. Clin. Oncol. 2000, 18, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Voog, E.; Biron, P.; Martin, J.P.; Blay, J.Y. Chemotherapy for patients with locally advanced or metastatic Merkel cell carcinoma. Cancer 1999, 85, 2589–2595. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Marquez-Rodas, I.; de la Cruz-Merino, L.; Martinez-Trufero, J.; Cabrera, M.A.; Piulats, J.M.; Capdevila, J.; Grande, E.; Berrocal, A. Recent Therapeutic Advances and Change in Treatment Paradigm of Patients with Merkel Cell Carcinoma. Oncologist 2019, 24, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Cowey, C.L.; Mahnke, L.; Espirito, J.; Helwig, C.; Oksen, D.; Bharmal, M. Real-world treatment outcomes in patients with metastatic Merkel cell carcinoma treated with chemotherapy in the USA. Future Oncol. 2017, 13, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Lorenz, E.; Ugurel, S.; Eigentler, T.K.; Kiecker, F.; Pföhler, C.; Kellner, I.; Meier, F.; Kähler, K.; Mohr, P.; et al. Evaluation of real-world treatment outcomes in patients with distant metastatic Merkel cell carcinoma following second-line chemotherapy in Europe. Oncotarget 2017, 8, 79731–79741. [Google Scholar] [CrossRef]
- Pommier, Y.; Sordet, O.; Antony, S.; Hayward, R.L.; Kohn, K.W. Apoptosis defects and chemotherapy resistance: Molecular interaction maps and networks. Oncogene 2004, 23, 2934–2949. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Milella, M.; Brownell, I.; et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Russell, J.; Lebbé, C.; Chmielowski, B.; Gambichler, T.; Grob, J.-J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I.; et al. Efficacy and Safety of First-line Avelumab Treatment in Patients with Stage IV Metastatic Merkel Cell Carcinoma: A Preplanned Interim Analysis of a Clinical Trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef]
- Walker, J.W.; Grignani, G.; Nathan, P.; Dirix, L.; Fenig, E.; Ascierto, P.A.; Sandhu, S.; Munhoz, R.; Benincasa, E.; Flaskett, S.; et al. Efficacy and safety of avelumab treatment in patients with metastatic Merkel cell carcinoma: Experience from a global expanded access program. J. Immunother. Cancer 2020, 8, e000313. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients with Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Lara, K.M.; In, G.K.; Matcuk, G.R.; Mehta, A.; Hu, J.S. Talimogene laherparepvec in combination with pembrolizumab leads to a complete response in a patient with refractory Merkel cell carcinoma. JAAD Case Rep. 2018, 4, 1004–1006. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Syndrome | Type of NMSC | Clinical Features |
|---|---|---|
| Xeroderma Pigmentosus | BCC SCC | Autosomal recessive disorder characterized by defects in the mechanisms of DNA repair. NMSCs are frequently developed by younger (≤20 years). The risk is directly associated to UVR exposition. |
| Oculocutaneous Albinism | SCC | Autosomal recessive disease showing a pigmentary dilution of the skin, eyes, and hair. SCCs are frequently developed by young subjects exposed to UVR. A high incidence of metastatic SCC has been described in black population. |
| Epidermodysplasia Verruciformis | SCC | Rare and usually recessively inherited disorder characterized by a colonization of the skin by HPV. The majority of patients develop NMSC as adults, usually in sun-exposed regions, but earlier with respect to the general population. Aggressive biological behavior includes perineural spread, metastases and death. |
| Dystrophic epidermolysis bullosa | SCC | It is characterized by both dominant and recessive mutations of the type VII collagen gene. The majority of recessive patients develop SCCs, that occur during the third-fifth decade of life, frequently multiple and with high attitude to local recurrence and spreading to distant metastatic sites. |
| Basal Cell Nevous Syndrome (Gorlin) | BCC | It is an autosomal dominant disorder caused by inactivating mutations of PTCH1 or rarely PTCH2. Clinical features include early NMSC, involvement of multiple sites on course of life, odontogenic keratocysts of the jaw, palmo-plantar pits calcifications of the falx cerebri and abnormalities of the skeleton. Patients also develop multiple cancers of the SNC, ovary and heart, as well as other skin defects such as epidermoid cysts and facial milia. |
| Bazex-Dupré-Christol Syndrome | BCC | It is a rare condition characterized by follicular atrophoderma of the hands and feet, hypotrichosis, localized hypohidrosis, epidermoid cysts and multiple BCCs developed during the second decade of lifer showing a trichoepithelioma-like histology. The inheritance pattern is often X-linked. |
| Rombo Syndrome | BCC | Patients have an atrophoderma vermiculatum-like appearance on the cheeks with evidence of sweat duct proliferation. They often suffer of hypotrichosis, blepharitis, peripheral erythema, trichoepithelioma and skin cancer. |
| Clinical Features | LOW RISK | HIGH RISK |
| Site and Size | Area L; <20 mm Area M; <10 mm | Area L; ≥20 mm Area M; ≥10 mm Area H; |
| Margins | Well defined; R0 | Undefined or R1 |
| Immune Suppression | Absence | Presence |
| Exposition to radiotherapy or chronic inflammatory process | Absence | Presence |
| Rapid growth | Absence | Presence |
| Neurological symptoms | Absence | Presence |
| Histology | LOW RISK | HIGH RISK |
| Grading | G1 or G2 | G3 |
| Adenoid-squamous, Adenoid-cystic, Desmoplastic, Metaplastic, Carcinosarcoma | Absence | Any Variant |
| Thickness or level of invasion | ≤6 mm and no subcutaneous invasion | >6 mm or subcutaneous invasion |
| Perineural, lymphatic or vascular invasion | Absence | Presence |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cives, M.; Mannavola, F.; Lospalluti, L.; Sergi, M.C.; Cazzato, G.; Filoni, E.; Cavallo, F.; Giudice, G.; Stucci, L.S.; Porta, C.; et al. Non-Melanoma Skin Cancers: Biological and Clinical Features. Int. J. Mol. Sci. 2020, 21, 5394. https://doi.org/10.3390/ijms21155394
Cives M, Mannavola F, Lospalluti L, Sergi MC, Cazzato G, Filoni E, Cavallo F, Giudice G, Stucci LS, Porta C, et al. Non-Melanoma Skin Cancers: Biological and Clinical Features. International Journal of Molecular Sciences. 2020; 21(15):5394. https://doi.org/10.3390/ijms21155394
Chicago/Turabian StyleCives, Mauro, Francesco Mannavola, Lucia Lospalluti, Maria Chiara Sergi, Gerardo Cazzato, Elisabetta Filoni, Federica Cavallo, Giuseppe Giudice, Luigia Stefania Stucci, Camillo Porta, and et al. 2020. "Non-Melanoma Skin Cancers: Biological and Clinical Features" International Journal of Molecular Sciences 21, no. 15: 5394. https://doi.org/10.3390/ijms21155394
APA StyleCives, M., Mannavola, F., Lospalluti, L., Sergi, M. C., Cazzato, G., Filoni, E., Cavallo, F., Giudice, G., Stucci, L. S., Porta, C., & Tucci, M. (2020). Non-Melanoma Skin Cancers: Biological and Clinical Features. International Journal of Molecular Sciences, 21(15), 5394. https://doi.org/10.3390/ijms21155394