Abstract
Failure of conventional treatments is often observed in cancer management and this requires the development of alternative therapeutic strategies. However, new drug development is known to be a high-failure process because of the possibility of a lower efficacy than expected for the drug or appearance of non-manageable side effects. Another way to find alternative therapeutic drugs consists in identifying new applications for drugs already approved for a particular disease: a concept named “drug repurposing”. In this context, several studies demonstrated the potential anti-tumour activity exerted by α1-adrenergic receptor antagonists and notably renewed interest for naftopidil as an anti-cancer drug. Naftopidil is used for benign prostatic hyperplasia management in Japan and a retrospective study brought out a reduced incidence of prostate cancer in patients that had been prescribed this drug. Further studies showed that naftopidil exerted anti-proliferative and cytotoxic effects on prostate cancer as well as several other cancer types in vitro, as well as ex vivo and in vivo. Moreover, naftopidil was demonstrated to modulate the expression of Bcl-2 family pro-apoptotic members which could be used to sensitise cancer cells to targeting therapies and to overcome resistance of cancer cells to apoptosis. For most of these anti-cancer effects, the molecular pathway is either not fully deciphered or shown to involve α1-adrenergic receptor-independent pathway, suggesting off target transduction signals. In order to improve its efficacy, naftopidil analogues were designed and shown to be effective in several studies. Thereby, naftopidil appears to display anti-cancer properties on different cancer types and could be considered as a candidate for drug repurposing although its anti-cancerous activities need to be studied more deeply in prospective randomized clinical trials.
1. Drug Repurposing: A Promising Tool for Cancer Management
Despite many improvements in prevention and therapy, failure of conventional treatments is often observed and cancer management requires the development of new therapeutic strategies. At this time, development of targeting therapies like PARP (Poly (ADP-ribose) polymerase) inhibitors, anti-angiogenic molecules or immunotherapy are booming [1,2,3]. Nevertheless, new drug development is time-consuming, expensive and is a high failure process because of the possibility of a lower efficacy than expected for the drug (for example if compensatory feedback loops take over) or appearance of non-manageable side effects. These hurdles mean that only one of every 5000–10,000 proposed anti-cancer drugs are approved by the Food and Drug Administration (FDA) [4].
In this context, drug repurposing (thank to computational and experimental approaches) aims at identifying new uses for drugs already approved or investigated for a particular disease and can be a mean to overcome those barriers. Actually, drug repurposing alleviates the pre-clinical steps of a classical drug development allowing significant time savings. Moreover, toxicity and safety profiles of the repurposed molecule are already known, as well as its pharmacokinetic and pharmacodynamic properties, reducing the risk of failure because of deleterious effects [5,6].
A bibliometric analysis conducted to appreciate the use of drug repurposing revealed that around 21,000 of the chemicals related to disease were associated with more than 1 disease suggesting a drug repurposing strategy. Among them, nearly 200 were connected for more than 300 diseases each [7]. This strategy has been successfully exploited many times and one of the most known examples is that of the phosphodiesterase inhibitor sildenafil, commercialized as Viagra by Pfizer and initially used for angina pectoris treatment. This drug has successfully been repurposed for erectile dysfunction management [8].
As for cancer management, recycling of drugs allows a rapid translation into clinical trial and appears as a promising strategy. In fact, some drugs used for non-cancerous diseases are also able to target the hallmarks of cancer cells including proliferation, resistance to cell death, angiogenesis, migration/invasion, tumour promoting inflammation and deregulated cellular metabolism and thereby represent promising candidates [9]. Thereupon, several non-cancer drugs exhibit anti-cancerous activities, such as the antidiabetic metformin which is efficient on various cancer types, notably lung, breast, prostate, colorectal and pancreatic cancers [10]. Actually, metformin seems to reduce cancer cells growth by activating the Adenosine Monophosphate-activated protein kinase (AMPK) and thereby modulating the activity of several metabolic pathways required for tumour growth [11]. Moreover, raloxifene used firstly for osteoporosis in postmenopausal women treatment was approved by the FDA for breast cancer prevention in high risk women [12] and the antiparasitic agent ivermectin or the anti-fungal agent itraconazole are already studied as potential repurposed drugs for ovarian cancer management [13].
In this context, several studies demonstrated the potential anti-tumour activity exerted by α1-adrenergic receptor (α1-AR) antagonists [14] which could make them to be considered as a potential strategy for cancer management and good candidates for drug repurposing concept.
2. Alpha1-Adrenergic Receptor Antagonists
The α1-AR are divided in three subtypes: α1A, α1B and α1D which are expressed in several human tissues. The distribution of these receptors has been performed largely by analysis of mRNA expression and revealed that α1B subtype is highly expressed by spleen, kidney, heart and brain tissues. Smooth muscles express the three α1-AR subtypes with a predominant α1A and α1D expression, which are also found in aorta and cerebral cortex tissues [15,16,17].
These receptors are classically coupled to Gq/11 proteins (but are also reported to be coupled to Gi proteins) and they are activated by catecholamines (adrenaline, noradrenaline, dopamine) [17,18]. The ligand binding on the receptor induces the dissociation of the Gq/11 protein which activates the phospholipase C (PLC). This enzyme cleaves its substrate: the membrane phospholipid phosphatydylinositol-4,5-bisphosphate (PIP2) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). The IP3 binds to its receptor, the IP3-receptor (IP3-R), located at the endoplasmic reticulum (ER) surface, allowing the exit of calcium from this organelle to the cytosol. As for DAG, it stimulates Protein Kinase C (PKC) that phosphorylates in its turn several proteins, leading to a cell specific response. DAG can also activate various calcium channels as voltage-gated calcium channel (VGCC) or transient receptor potential calcium channel (TRP). It could be noted that α1-AR are also known to be coupled to β-arrestin (that activates mitogen-activated protein kinase pathways and that allows receptor internalization), and to the small GTP binding protein Rho A that mediates calcium sensitization. Alpha1-adrenergic receptors have also been shown to induce arachidonic acid in a PLC-independent manner but probably through phospholipase A2 (PLA2) activation [16,17]. These different pathways converge to mitogenic response to catecholamines and to the release of calcium. The latter effect induces the contraction of smooth muscles in physiological conditions and participates in various physiological effects, such as mydriasis, increase of blood pressure and promotion of bladder continence, for example. The use of α1-AR antagonists disturbs intracellular calcium flux and induces a relaxation of smooth muscles; this is why these drugs are prescribed for benign prostatic hyperplasia (BPH) [19].
BPH is a non-cancerous proliferation of the prostate glandular epithelium, connective tissue and smooth muscle. BPH is a common disorder affecting 50% of men over 50 years old and reducing significantly their quality of life [20]. The widely held concept is that BPH leads to prostate enlargement that impinges upon the prostatic urethra and bladder outlet which is responsible of mechanical obstruction to urinary outflow and bladder detrusor muscle irritability. Moreover, the increase of smooth muscle tone at the prostate and the bladder neck accentuates the bladder outlet obstruction. The whole of these mechanisms results to lower urinary tract symptoms (LUTS), such as storage symptoms (urgency, frequency and nocturia) and voiding symptoms (weak stream, urinary retention and hesitancy) [19,20,21,22]. However, it is noteworthy that other studies questioned the causal relationship between prostatic enlargement, bladder outlet obstruction and LUTS in men with clinical BPH revealing that the mechanism by which BPH causes LUTS needs to be further investigated [23].
The treatment of BPH and associated LUTS can be achieved using three major therapeutic strategies. Firstly, prostate development is controlled by the testosterone derivate dihydrotestosterone (DHT). Thereby, the use of the 5-α reductase inhibitors (5-ARI), such as Dutasteride or Finasteride, that impede the conversion of testosterone into DHT, reduces the serum and intraprostatic DHT concentration and allows the slowdown of BPH progression [24]. Secondly, the isoenzymes PDE5 (phosphodiesterase type 5), highly expressed in LUT tissues, degrade the cyclic Guanosine Monophosphate (cGMP) inducing vessels vasoconstriction. Hence, PDE5 inhibitors, such as Tadalafil, increase the cGMP level resulting in vasodilation through activation of the endothelial Nitric Oxide Synthase–Nitric Oxide–cGMP pathway, thereby allowing relaxation of LUT tissues [25,26]. At last, the use of α1-AR antagonists (also called α-blockers) turned out to be very effective. Three α1-AR subtypes are found in LUT tissues. The α1A-AR subtype is predominant and is located in urethra, bladder neck and in the prostate stroma and smooth muscle (although both mRNA of α1A- and α1D-AR were found in this tissue) and it mediates prostate contractility [27,28,29]. The α1B-AR subtype is expressed by vascular tissue and is less abundant than α1A- and α1D-AR in male ureters [30]. Finally, α1D-AR subtype is also importantly expressed. It is found in bladder and sacral region of the spinal cord [27,29] and it was demonstrated that bladder smooth muscle tissue obtained from surgical patients express predominantly the α1D-AR subtype at mRNA level, a result that was confirmed at a protein level by competition analysis assay [31]. Moreover α1D-AR subtype was described to be increased in expression and function in models of bladder hypertrophy [32]. This receptor subtype takes part in bladder contraction.
The selectivity to α1A- and α1D-AR antagonists presents several advantages. It allows these antagonists to better target prostate and bladder tissues without displaying side-effects, such as blood pressure disturbance driven by the α1B-AR subtype. Moreover, even if α1A-AR antagonists are very effective in relaxing prostate smooth muscle, their combination with α1D-AR antagonists is more effective to improve LUT symptom scores in men with BPH because α1D-AR antagonists also relieve bladder symptoms [33]. Finally, it was shown that the tissues of BPH display decreased expression of α1B-AR and increased expression of α1A- and α1D-AR subtypes mRNA, compared to normal prostatic tissue, leading to the suggestion that both α1A- and α1D-AR contribute to BPH development [34,35,36,37].
Combination disposals using an α-blocker and 5-ARI or PDE-5 inhibitor, anticholinergic agents or β-3-agonists can also be prescribed for the treatment of BPH [20,21,38].
Seven α1-AR antagonists are used for the treatment of BPH and associated LUTS. Silodosin is marketed worldwide and prazosin, alfuzosin, doxazosin, terazosin and tamsulosin are used in most western countries. Naftopidil is used for the same indication but only in Japan. Each α1-AR antagonist possesses a different selectivity for each α1-AR subtype. Alfuzosin, doxazosin, prazosin and terazosin block the three α1-AR subtypes, they are called non-selective α1-AR antagonists [16,17,27]. Conversely, silodosin is α1A-AR-selective antagonist and tamsulosin and naftopidil are more selective for α1A and α1D than for α1B [27]. Concerning naftopidil, the studied carried out by Michel et al., did not allow to observe any selectivity for naftopidil in cloned α1-AR subtypes transiently expressed in COS cells [39]. On the contrary, naftopidil was described to bind more specifically α1A- and α1D-AR [40]. In order to precise their results, experiments using membrane preparations from CHO cells stably expressing the cloned human α1-AR genes showed that naftopidil has 17- and 3-fold higher potency for α1D-AR than for the α1B- and α1A-AR, respectively [41]. Yuan’s team showed similar results through docking studies and on rat functional assay in vitro and highlighted that naftopidil used as a racemate, as well as its S- and R- enantiomers had similar blocking activity on α1-AR subtypes [42,43]. However, a recent work contradicted these previous observations and showed that naftopidil affinity is α1A > α1B > α1D [44]. These discrepancies are confusing but variations on affinities are frequently observed between laboratories and it could be suggested that global view of data of different laboratories is required to precise ligand binding affinity.
Naftopidil, named Flivas™, has been marketed in Japan for BPH and associated LUTS treatment by Asahi Kasei Corporation since 1999 and several clinical trials and prospective studies demonstrated that naftopidil appears efficient for the treatment of BPH and LUTS without major adverse effects [36,45]. Naftopidil which displays selectivity for α1D-AR expressed in the bladder, was reported to improve storage symptoms compared to tamsulosin [46,47,48]. Some studies did not find significant difference in IPPS (International Prostate Symptom Score) and quality of life between these two α1-AR antagonists [49], whereas other studies showed that naftopidil also increased quality of life parameter [50]. To have a global vision on naftopidil effect on LUTS associated with BPH, a meta-analysis was carried out by the Cochrane library [22]. This analysis included 22 studies with randomised 2223 participants and compared the efficacy of naftopidil to that of tamsulosin and silodosin on several outcomes. The conclusion revealed that compared to tamsulosin, naftopidil had similar effect on urological symptoms score, quality of life and adverse events. The same conclusion was obtained when naftopidil was compared with silodosin, but a substantial reduction of sexual adverse events was observed with naftopidil [22]. Thus, naftopidil seems to be as potent and tolerable as the other α1-AR antagonists. However, as studies evaluated naftopidil effects only on Asian men, it certainly led to bias and it would be required to test its effect on other populations.
In clinical practice, the optimal dose of naftopidil is ranging between 25 and 75 mg/day for Japanese men [45]. It has been demonstrated that, after oral administration of naftopidil, 80–95% of the dose is rapidly absorbed, widely distributed and its half-life not exceeds 3h [51]. After a single dose of 50mg, the maximal plasma concentration of naftopidil (plus its metabolites) is in the range of 300–600 nM [52]. Naftopidil is clinically used as a racemate. Its bioavailability in humans only reaches 18%, suggesting an important first-pass metabolism [52]. In this context, Zhu and colleagues showed that the hepatic metabolism associated isoenzymes CYP2C9 and CYP2C19 are involved in naftopidil metabolism, mainly by its demethylation and hydroxylation [53]. In addition, plasma levels and half-life times of naftopidil after oral administration are increased in patients with hepatic dysfunction [51], comforting the importance of hepatic metabolism in pharmacokinetic properties of naftopidil. A study carried out in rats confirmed this fist-bypass metabolism and showed that naftopidil S-enantiomere bioavailability is higher than racemate and twice higher than R-enantiomer after oral administration [54]. However, R-enantiomere was more widely distributed in peripheral tissues with high concentrations found in prostate, suggesting stereoselective pharmacokinetic [54]. Buccal films of naftopidil allowing its intra-oral administration are currently undergoing development and evaluation in order to overcome its hepatic by-pass and thereby to enhance its bioavailability [55]. Finally, this compound is well tolerated at up to 100 times the pharmacologically active dose and its therapeutic index is in the range of 4.4–6.7, supporting that naftopidil displays a broad therapeutic range [52].
In parallel of studies showing its activity for BPH treatment, several results demonstrated that naftopidil exhibited certain anti-cancer properties in vitro, in vivo as well as in clinic.
3. Anti-Cancerous Properties of Naftopidil
3.1. Cytostatic Effects of Naftopidil In Vitro
Several studies showed that naftopidil can exert anti-proliferative effects on cancer cell lines (Table 1).

Table 1.
Anti-cancerous properties of naftopidil.
In this context, Kanda et al., studied the effect of naftopidil on growth of human androgen sensitive, androgen receptor-positive LNCaP cell line and androgen insensitive, androgen receptor-negative PC-3 cell line [62]. They showed that naftopidil had an anti-proliferative effect on both cell lines with IC50 around 20 and 30 µM respectively and induced a cell cycle arrest with a blockade in G0/G1 phase. This anti-proliferative effect was also observed in androgen low-sensitive, androgen receptor-positive E9 cell line (that derives from LNCaP) [56] implying that the antiproliferative effect of naftopidil is not related with androgen sensitivity of the cells. Molecular mechanism analysis showed that in androgen sensitive cell line LNCaP, p27kip1 and p21cip1 were strongly up-regulated, whereas only the former was increased in E9 cell lines and only the latter was increased in PC-3 androgen insensitive ones. This result implies that the antiproliferative effect of naftopidil depends on cellular context. Moreover, whereas naftopidil had no effect on Akt activity in androgen-sensitive cell line (LNCaP), it inhibited Akt phosphorylation on ser473 in androgen-insensitive cell line, PC-3. Authors suggested that this result could account for p21cip1 inhibition in PC-3. Finally, naftopidil did not modulate p53 expression in both cell lines, ruling out p53 implication in naftopidil-induced p27kip1 and p21cip1 proteins [62]. Naftopidil was also demonstrated to have an anti-proliferative effect on the renal cancer cells lines ACHN an Caki-2 in the same range of concentrations. In fact, it reduced cell proliferation that was accompanied by an arrest in the G0/G1 phase of the cell cycle, a decrease of Cyclin dependent kinase-2 (Cdk-2) expression (which is required for the transition of cell cycle phases) and increase of p21cip1 [57]. As well, naftopidil was proven to reduce proliferation of colon adenocarcinoma cells HT29 [60] and that of ovarian cancer cell lines SKOV3 and IGROV1-R10 in a dose-dependent manner [61]. Even if this anti-proliferation effect was accompanied by an increase of p21cip1 and p27kip1 in SKOV3 cell line, the expression of these proteins was not disturbed in IGROV1-R10 cell lines supporting that naftopidil anti-proliferative effect was cell-context dependent (personal data). This antiproliferative effect was also observed in cells in the microenvironment of the cancer cells. For example, in Hori’s study, naftopidil also reduced the growth of the fibroblasts cells PrSC derived from the prostate stroma as well as their secretion of IL-6, a growth factor for most prostate cancer cells, suggesting that naftopidil could interfere with tumour-stroma interactions [56]. This drug also increased p21 expression in HUVEC cells preventing their proliferation in vitro and giving to naftopidil an anti-angiogenic property [57].
3.2. Cytotoxic Effects of Naftopidil In Vitro
Naftopidil was also described to exert cytotoxic effects in several cancer cell lines when used as a single agent (Table 1). Actually, naftopidil reduced cell viability in different models such as bladder and renal cancer cells [58,59]. Moreover, it was shown to induce apoptosis of mesothelioma cells NCI-H28, NCI-H2052, NCI-2452 and MSTO-211H by activating caspase 3 and 8 [63,64] and increasing TNF-α mRNA expression and Fas-Ligand secretion in NCI-H2052 however, this effect is only observed for high concentrations. Several results also demonstrated that this α1-AR antagonist also triggered prostate cancer, cervical and gastric cancer cells apoptosis [65,66].
In most models, naftopidil appeared cytotoxic in in vitro assays at concentrations higher than 50 µM [62,63,64,66]. Under this threshold, naftopidil exerted a cytotoxic activity when it was used in combination with other anti-neoplasic strategies. In fact, naftopidil displayed an additive cytotoxic effect with radiotherapy in PC-3 cells [67]. Moreover, by an indeterminate mechanism, naftopidil induced apoptosis of LNCaP and PC-3 prostate cancer cells in combination with docetaxel, but not that of prostate stromal cells PrSC [68]. As for ovarian cancer cells SKOV3 and IGROV1-R10, whereas naftopidil only exerted a cytostatic effect when used as a single agent, it transcriptionally up-regulated Bim, Puma and Noxa pro-apoptotic protein expression. The combination of naftopidil with the BH3-mimetic targeting Bcl-xL ABT-737 or the MEK inhibitor Trametinib increased the [pro]/[anti-apoptotic] ratio in favour of the pro-apoptotic proteins, leading these cancer cell lines and high-grade ovarian cancer Patient-Derived Organoids (PDO) to apoptosis [61]. It is noteworthy that in most studies, the anti-cancer effects of naftopidil were not assessed in non-malignant cell, a control condition that would permit to evaluate therapeutic margins. Its effect was however evaluated on T1074 cell (non-malignant ovarian epithelial cells). Naftopidil at 50 µM has an anti-proliferative effect; however, its combination with Trametinib did not trigger apoptosis as observed in malignant SKOV3 and IGROV1-R10 cell lines, suggesting that naftopidil only sensitises ovarian malignant cell to targeting therapies [61].
3.3. Anti-Cancerous Effects of Naftopidil In Vivo
To evaluate its efficacy in vivo, naftopidil anti-cancerous effects were analysed in xenografted mice models (Table 1). Studies highlighted that naftopidil is a well-tolerated molecule as it did not provoke weight reduction in the treated mice [59,63]. Moreover, naftopidil was proven to be an efficient cytotoxic drug in several cancer types as it reduced the volume of tumours [56,62]. This effect was accompanied by a decrease in Ki-67 index [56], and also an increase in p21 staining [62]. Naftopidil exhibited cytotoxic effects in xenografts of mesothelioma, renal carcinoma and bladder cancer cells [57,59,63]. Naftopidil also reduced PC-3 xenograft tumour growth alone and more drastically in combination with radiotherapy [67] or docetaxel [68], such as in in vitro experiments. Finally, naftopidil exerted anti-angiogenic properties through reduction of the microvessels density (MVD) in renal carcinoma cell line (ACHN) and prostate cancer cell (PC-3) xenograft models, but also, in-patient renal carcinoma xenografted into nude mice [57,62].
All these studies were carried out at the dose of 10 mg/kg/day naftopidil, or twice a week for Mikami’s study [63]. This suggests that this dose is tolerable and effective in the model of cancer tested. Moreover Kanda et al., showed that increasing the dose to 100 mg/kg/day did not improve naftopidil efficacy [62]. Taken together, these observations suggest that naftopidil exerts anti-cancerous properties in vitro and in vivo in several cancer types.
3.4. Clinical Evidence of Naftopidil Anti-Cancer Effects
The anti-cancer effect of naftopidil in clinic was observed thanks to a retrospective study that was carried out in Memorial Hospital in Tokyo [65]. In this study, prostate cancer incidence has been evaluated in patients who have received either naftopidil (n = 766) or tamsolusin (n = 1015) between 2003 and 2010. As expected, prostate cancer incidence was correlated with the level of PSA concentration. Surprisingly, this incidence was lower in the naftopidil group than in tamsulosin group from 3 months of treatment (1.8% versus 3.1% OR = 0.46 p = 0.035) and this difference is accentuated with the duration of treatment 0.46 (p = 0.081) for 12 months and 0.16 (p = 0.039) for 36 months. Moreover, these authors also showed that prostate cancer cells from men treated with naftopidil over-expressed p21 and under-expressed Bcl-2 compared with men exposed to tamsulosin or no treatment, suggesting that naftopidil acted through inhibition of cell cycle progression and perturbation of apoptosis member expression in clinic. These encouraging results allowed naftopidil to be considered as a promising candidate for prostate cancer management in chemoprevention although it should be kept in mind that retrospective cohort studies, even if results can be easily generalizable to real-world situation, are subjected to bias because of absence of randomization. Thus, randomized controlled trials are required to evaluate causal relationship between naftopidil treatment and reduction of prostate cancer incidence [69,70].
4. Molecular Mechanisms Involved in the Anti-Cancerous Properties of Naftopidil
4.1. Involvement of the Chemical Structure
The chemical structure of α1-AR antagonists seems to play a major role in the capacity of these molecules to display anti-cancer properties and Kyprianou and colleagues suggested that their anti-cancer activity is quinazoline/piperazine-dependent. Actually, it has been demonstrated that exposure to the quinazoline-based α1-AR antagonists’ doxazosin, prazosin and terazosin induced apoptosis, anoikis, decreased cell growth in bladder and in several prostate cancer cell lines in vitro as well as in vivo, exerted anti-angiogenic properties and sensitised human cervical carcinoma cell lines to chemotherapy through inhibition of MDR-1 mediated drug efflux. These results have been comprehensively collected by Batty et al., [71]. On the contrary, tamsulosin, a sulphonamide-based α1-AR antagonist that does not possess a quinazoline or piperazine group displayed no anti-cancer property [72,73]. Interestingly, a significant decrease in the incidence of prostate cancer was also observed in clinic for the quinazoline-based α1-AR antagonists. Indeed, the result of a retrospective cohort study showed that men treated with doxazosin and terazosin have a 1.46 times lower relative risk to develop prostate cancer compared with non-treated men [74]. Although prospective clinical studies are lacking to support these arguments, these results encourage further studies.
Naftopidil is an aryl-piperazine based α1-AR antagonist possessing a naphthalene group (Figure 1) and its anti-cancerous properties could be dependent of these chemical groups.
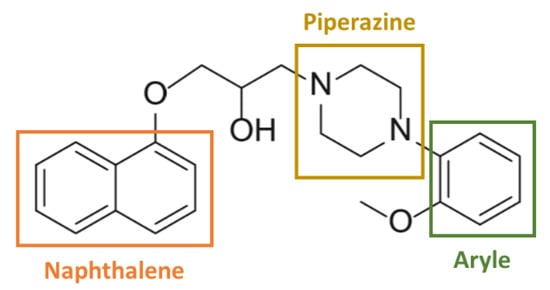
Figure 1.
Chemical structure of naftopidil (according to [65,71,75]). Naftopidil is an aryl-piperazine based α1-AR antagonist possessing a naphthalene group.
Piperazine is a core scaffold for synthesis of a plethora of bioactive molecules and piperazine-based drugs possess various pharmacological activities such as anti-fungal, anti-viral, anti-depressant properties [76,77,78] and also exert anti-cancerous properties. For example, an 1-(2-aryl-2-adamantyl)piperazine derivate reduced viability of cervical, breast, pancreatic and lung cancer cells in a dose-dependent manner without exerting any toxicity on normal cell lines [79] while others seem to have HDAC (Histone deacetylase) inhibitor properties [80]. Moreover, several aryl-piperazine derivatives containing the saccharin moiety were shown to reduce cell viability of cancer prostate models in a dose-dependent manner [81]. These structure-activity relationships open the perspective of their use as anti-cancer drugs as illustrated by the anti-cancer effects of naftopidil in vitro and in vivo but also led to synthesis of naftopidil derivatives as HUHS1015 and compound 12 to improve its efficacy as it is discussed thereafter.
4.2. Alpha1-AR Independent Anti-Cancerous Action
As α1-AR antagonists, naftopidil and its derivatives anti-cancer actions were supposed to involve α1-AR pathway. In this context, it has been shown that the endogenous α1-AR agonist noradrenaline protected prostate cancer cells against the anti-proliferative effect of the naftopidil derivate, compound 12, suggesting that this naftopidil analogue acted through α1-AR binding to exert its anti-cancerous activity [82]. It should be remarked that even noradrenaline has a high affinity for α1-AR, this catecholamine can also bind other adrenoreceptors as α2- or β-AR to exert its proliferative activity. So, the noradrenaline protective effect observed does not necessarily attest that compound 12 acts through α1-AR and other pathways could not be excluded. As for naftopidil, several studies described that its anti-proliferative and cytotoxic effects involved α1-AR independent mechanisms. In fact, naftopidil reduced mesothelioma cell viability, while the α1D-AR knock-down enhanced it [64]. Moreover, the α1D-AR stimulation led to Protein Kinase A (PKA) and PKC activation, thereby their inhibition should enhance naftopidil effects. However, the PKC inhibitor GF109203X attenuated naftopidil-induced apoptosis of mesothelioma cells [64]. Moreover, neither the PKA inhibitor H89 nor the GF109203X increased naftopidil cytoxic effects on bladder cancer cells. Furthermore, the α1-AR agonists’ methoxamine and phenylephrine should counteract the effects of naftopidil, which was not observed in bladder cancer models [59]. Similarly, neither the cytostatic effect of naftopidil nor its capacity to induce BH3-only protein expression in ovarian cancer cell lines were counteracted by methoxamine. Moreover, these effects were not mimicked by the other α1A/α1D-AR-selective drug BMY-7378 that also displays a phenylpiperazine moiety [61]. Lastly, naftopidil reduced proliferation of the prostate cancer cells AIDL (Androgen-Independent LNCaP), although this cell line does not express the α1D-AR subtype [56].
Interestingly, it has also been described that the α1-AR antagonists’ doxazosin and terazosin induced apoptosis of prostate cancer cells independently of α1-AR. In fact, these drugs induced apoptosis of PC-3 cells that was not abrogated by phenoxybenzamine, an irreversible α1-adrenoreceptor antagonist [72]. Moreover, these compounds induced apoptosis in DU-145 cells line that lacks α1-AR [73]. This suggests that naftopidil is not the only α1-AR antagonist exerting anti-cancerous properties independently of α1-AR. Finally, it should be remarked that, in all of these studies, concentrations required for anti-cancerous activities are higher than those required for α1-AR inhibition [34,35,36]. This finding is also an argument to support that the anti-cancerous properties of α1-AR antagonists probably do not involve α1-AR pathway.
4.3. Other Pathways Involved
Naftopidil was found to modulate the activity of different signaling pathways. Firstly, naftopidil reduced Akt phosphorylation in prostate and gastric cancer cells [62,66,68]. However, this effect was not observed in ovarian cancer cell lines [61].
Moreover, this α1-AR antagonist was described to reduce activity of the TGF-β pathway by decreasing Smad2 phosphorylation in HeLa cells but the entire molecular pathways is not described [65].
Depending on cellular context in ovarian cancer cell lines, naftopidil induced either ER stress-activated ATF4 transcription factor or JNK/c-Jun phosphorylation; both pathways leading to BH3-only protein up-regulation. Interestingly, these molecular transduction pathways are known to be activated by destabilization of microtubules [83,84] and naftopidil was described to disturb microtubules polymerization [60]. In fact, in this study, Ishii and colleagues showed that naftopidil, as well as other phenylpiperazine derivates RS100329, BMY-7378, and KN-62, were able to bind tubulin and to inhibit its polymerization. In contrast, the quinazoline-based α1-AR antagonist doxazosin increased the tubulin polymerization and tamsulosin or silodosin, which are respectively carboxamide- or sulphonamide-based α1-AR blockers, did not show any effect. Thereby, the ability to bind tubulin appears to be a specific feature of piperazine-based drugs [60]. This property was supported by the fact that AK301, a piperazine-based compound, was also known to inhibit tubulin polymerization and to induce a blockade of colon cancer cells in the G2/M phase of the cell cycle which restored their apoptosis in presence of TNF-α [85]. Microtubule-targeting agents are known to modulate the activity of several molecular pathways by disturbing microtubule dynamic and thereby exert anti-cancerous properties [83]. It could then be hypothesized that through interaction with tubulin, naftopidil could activate ER stress and JNK/c-Jun pathways, increasing pro-apoptotic protein expression and allowing naftopidil to sensitise ovarian cancer cells to pro-apoptotic strategies [61]. However, the ability of the naftopidil analogues sharing aryl-piperazine moiety to bind tubulin has not been demonstrated yet and further studies are needed.
Moreover, an in-silico drug repositioning-approach carried out to predict drugs that can modulate transcription factor activity identified naftopidil among more than 6700 drugs as a potential activator of p53 [86]. This pathway could besides be involved in cell cycle arrest and apoptosis [87] which could explain the anti-cancerous properties of naftopidil on several models, even if p53 implication was ruled out in naftopidil-induced BH3- only increase in ovarian cancer cells [61].
Taken together, these observations strongly suggest that naftopidil acts as an anti-cancer agent independently of its α1-AR antagonist role. However, the molecular targets of AR-independent effects remain unknown and certainly depend on cellular context.
4.4. Anti-Cancerous Properties of Naftopidil Analogues
Interestingly, the naftopidil major metabolite HUHS190, also reduced cell viability of prostate, bladder and renal cancer cell lines in dose-dependent manner [88]. In this context, and in order to obtain more effective anti-cancer drugs, different naftopidil analogues were synthesized. Thus, naftopidil derivates containing methyl phenylacetate moiety exhibited α1-AR antagonist properties but their potential anti-cancerous activity has not been studied yet [89]. Other naftopidil-based aryl-piperazine analogues were developed and among them, some exert anti-cancerous properties against prostate cancer cells, such as derivates containing a bromophenol moiety [75,90].
Among all naftopidil analogues, the lead molecule HUHS1015 was widely studied such as another drug called “compound 12” (Figure 2) (Table 2).
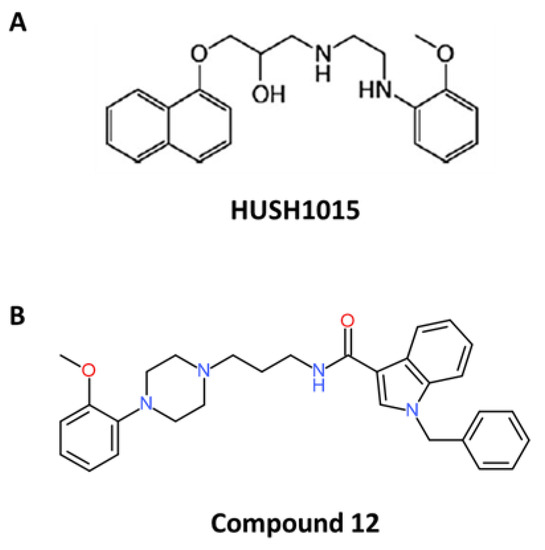
Figure 2.
Chemical structures of naftopidil analogues (according to [82,91]). (A) Chemical structure of HUHS1015 and (B) chemical structure of compound 12.
Table 2.
Anti-cancerous properties of naftopidil analogues HUHS1015 and compound 12.
4.5. HUHS1015
Nishizaki and colleagues synthesized 21 naftopidil analogues and among them HUHS1015 seemed to present interesting anti-cancerous properties [92].
In vitro, HUHS1015 reduced mesothelioma cell lines proliferation by inducing an arrest in G0/G1 phase of the cell cycle at 10 µM and induced their necrosis and apoptosis at 15 µM after a 24 h treatment [91]. Moreover, HUHS1015 was able to reduce cell viability in other several models, notably it induced cell death in lung, liver, gastric, bladder and renal cancer cells [93]. Concerning mesothelioma cell lines, it is interesting to note that HUHS1015 is less cytotoxic for the non-malignant MeT-5A cell line than for malignant ones.
Molecular pathways involved in mesothelioma cell lines showed that caspase 3 and 4 are activated, but not caspase 8 and 9, suggesting that HUHS1015 induced apoptosis not through canonical extrinsic and intrinsic pathways but perhaps through reticulum stress (known to activate apoptosis through caspase 4) or through independent-caspase pathway [93].
In addition, a 24h-treament with 15 µM HUHS1015 induced both necrosis and apoptosis in cisplatin-resistant MKN28 and MKN45 gastric cancer cells [94]. Interestingly, HUHS1015 triggered MKN45 cells apoptosis by activating caspase 3, 4 and 8 while it acted by a caspase-independent way, probably through nuclear accumulation of AMID (Apoptosis-inducing factor-homologous Mitochondrion-associated Inducer of Death) in MKN28 cell line [95].
HUHS1015 was also able to bring colorectal cancer cell lines Caco-2 and CW2 in apoptosis by inducing mitochondria damages [96].
Moreover, HUHS1015 modulated the expression of proteins implicated in apoptosis regulation, which could explain its ability to induce this type of cell death. Indeed, in mesothelioma cells, 15 µM HUHS1015 increased the mRNA expression of Puma, Hrk and Noxa, implicated in the intrinsic apoptosis pathway control [91]. It also increased the mRNA expression of Bax and Bad in colorectal cancer cells [96], such as naftopidil was able to transcriptionally induce BH3-only proteins expression in ovarian cancer cells [61]. Like naftopidil in mesothelioma cells, HUHS1015 also increased expression of factors implicated in the extrinsic apoptosis pathway, such as the cytokine TNF-α in MKN45 gastric cells; however, this effect has been called into question due to the very high concentrations of HUHS1015 used [63,94].
In vivo, HUHS1015 was described to be well tolerated by mice, as it did not induce a loss of weight and it reduced the tumour volume in mesothelioma, gastric and colorectal cancer cells xenografted mice [91,94,96]. Moreover, HUHS1015 seemed to be more efficient than naftopidil on gastric and colorectal cancer models as it induced a stronger decrease of tumour for the same dose used [94,96].
4.6. Compound 12
Huang and colleagues designed eleven compounds, compound 2 to compound 12, derived from naftopidil [82]. For all these derivates, the 2-hydroxypropane of naftopidil was replaced by an amide structure in order to improve their α1A/1D binding affinity and an indole substituent was introduced because it was suggested to enhance their anti-cancerous properties. Among these novel synthesised molecules, compound 12 possessed a large benzyl group at the indole-N-position that increased its flexibility and was suggested to strengthen its affinity to α1A/1D-AR, lower its binding to α1B-AR through its hydrophobic properties and favour anti-cancerous properties of α1-AR antagonists [82,97]. Compound 12 showed the highest cytotoxic activity against the PC-3, DU145, and LNCaP prostate cancer cell lines in vitro and induced an arrest in the G0/G1 phase of the cell cycle, such as naftopidil and HUHS1015 [82]. Beyond 20 µM, compound 12 induced apoptosis of prostate cancer cells, as observed by activated caspase 3 and phosphatidylserine exposure. As HUHS1015, compound 12 was able to modulate Bcl-2 family member expression by inducing Bax and Bcl-2 mRNA expression [82]. To our knowledge, this compound was not used in in vivo experiments.
Taken together, naftopidil anti-cancerous properties allowed the development of several analogues. Among them, HUHS1015 and compound 12 display anti-cancerous activities in several cancer types at lower concentrations than naftopidil, suggesting their better efficacy. However, other studies are required to decipher molecular pathways involved and to evaluate the clinical safety of these two analogues.
5. Discussion
Collectively, naftopidil shows anti-cancer properties on several cancer models. In vitro, naftopidil has an anti-proliferative effect, presumably by modifying the expression of proteins regulating cell cycle progression [56,57,60,61,62]. As it can inhibit cancer cell growth when used as a single agent, naftopidil could be regarded as a cytostatic drug which could slow proliferation and generate a lap of disease stabilisation that will delay the introduction of cytotoxic drugs and improve time of progression, quality of life and survival. Yamada’s study suggested that naftopidil reduced prostate cancer incidence and that this compound could be seen as a potential chemopreventive treatment. However, its antiproliferative action could also allow this compound to be suggested as a possible maintenance treatment for spacing out recurrence episodes. Other clinical studies are required to evaluate this point.
Naftopidil can also exert cytotoxic effects when used as a single agent [58,59,62,63,64,65,66], but in other models it was also able to induce apoptosis only when combined with other antineoplasic drugs in vitro, in vivo as well as ex vivo in high grade serous ovarian cancer PDO models [61,67,68]. This result suggests that naftopidil could be regarded as a possible chemotherapeutic adjuvant due to its capacity to sensitise cancer cells to other therapies. A combination with other molecules seems to be an interesting strategy as it could target different pathways that act synergistically. This could allow reduction of dose and thereby limitation of possible side effects. Studies deciphering molecular pathways activated by naftopidil would also help to find new relevant therapeutic combinations to empower naftopidil efficacy. However, these studies are lacking and efforts have to be made to fill the gap.
Interestingly, anti-cancerous effects of naftopidil seem to be independent of its α1-AR antagonist property [56,59,61,64] and, depending on cellular context, naftopidil is able to modulate activity of various signaling pathways [60,61,62,65,86].
The anti-cancerous properties of naftopidil has promoted the development of numerous derivatives to improve α1-AR affinity for prostate targeting or cytostatic/cytotoxic activity [75,82,90,91,92,93,94,95,96]. Indeed, naftopidil cytostatic and cytotoxic properties are reached at high concentrations in vitro and reducing effective concentrations through more effective compounds would facilitate is use in clinic. It is interesting to note that Colciago et al., synthesised a compound derived from WB4101: A175, that binds with a strong affinity α1D-AR [98]. They demonstrated that this compound had an anti-proliferative effect in androgen-insensitive prostate cancer cells PC3 that strongly expressed α1D-AR at the mRNA level but had no effect on DU145 cells that did not express this receptor. This study showed that mRNA expression of α1A-AR was highly expressed in less aggressive and androgen-sensitive prostate cancer cells suggesting that α1A-AR antagonists might be more useful to counteract cell proliferation in the first steps of cancer. This is in agreement with Thebault’s work showing that LNCaP cells expressed α1A-AR and that the inhibition of these receptors prevented cancer epithelial cell proliferation [99]. Conversely, α1D-AR was expressed in androgen-independent prostate cancer cells and α1D-AR antagonists could be more useful in this type of cancer. So, the improvement of α1-AR selectivity (as made for compound 12) could be important in prostate cancer management, especially because it was suggested that α1D-AR is overexpressed in many untreated patients with advanced prostate cancer and thus could be regarded as an interesting target [100]. For other types of cancer, it would be interesting to improve the anti-proliferative effects of the analogues by disrupting the dynamics of tubulin (as arylpiperazine compounds do) or by blocking the cell cycle in the G0/G1 phase. These types of compounds have been shown to be good adjuvants for sensitizing to conventional chemotherapies and allowing for the spacing of treatment cycles. In addition, it would prevent the acquisition of mutations and the development of chemoresistance.
Although there is evidence suggesting that naftopidil may be considered a potential candidate for drug repurposing, some questions arise. Naftopidil is approved for BPH/LUTS treatment in Japan, but not in other countries, because of the lack of non-Asian randomized clinical trials and placebo-controlled trials [22]. As the Asian population displays a metabolic phenotype different to that of the Caucasian or African population, one could then argue that naftopidil would not be metabolized in the same way, which could lead to side-effects or lack of efficacy. Moreover, naftopidil is used for BPH/LUTS treatment in men and it has never been used on women; clinical trials on women are then needed to prove is safety without major adverse effects. Finally, naftopidil safety was not evaluated beyond 18 weeks of treatment in clinical trials [101]. As its possible use in cancer management would need longer courses of treatment, clinical trials are required to ensure its safety.
Taken together, naftopidil displays cytostatic and cytotoxic properties in several in vitro and in vivo models. Its capacity to slow cancer cell proliferation, the result from Yamada’s study and its good tolerability could enable naftopidil to be considered as a potential candidate for cancer prevention or maintenance treatment. Moreover, it could also be considered as a good adjuvant due to its capacity to potentialize anti-cancer therapies. However, its molecular targets need to be more deeply investigated to relevantly sensitise cancer cells to other anti-tumoural drugs. Finally, its anti-tumoural efficacy has to be confirmed and evaluated in large cohort prospective clinical studies. Naftopidil still has a long way to clinically prove its efficacy and tolerability in cancer management, but the encouraging results make naftopidil an interesting candidate to drug repurposing and incite to pursuit investigations on its anti-cancerous properties.
Author Contributions
Conceptualization, R.F. and M.N.; methodology, R.F. and M.N.; validation, R.F., M.N. and L.P.; formal analysis, R.F. and M.N.; investigation, R.F.; resources, R.F. and M.N.; writing—original draft preparation, R.F. and M.N.; writing—review and editing, M.N.; visualization, M.N.; supervision, M.N.; project administration, M.N.; funding acquisition, M.N. All authors have read and agreed to the published version of the manuscript.
Funding
R.F. was funded by a grant from Normandy Regional Council.
Acknowledgments
The authors thank Jana Sopkova-de Oliveira Santos for her careful reading of the article and for her help in the part of the review dealing with chemistry.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| α1-AR | α1-Adrenergic Receptors |
| 5-ARI | 5-α Reductase Inhibitors |
| Akt | protein kinase B |
| AMID | Apoptosis-inducing factor-homologous Mitochondrion-associated Inducer of Death |
| ATF4 | Activating Transcription Factor 4 |
| AMPK | Adenosine Monophosphate-actived Protein Kinase |
| Bad | Bcl2-Associated agonist of cell death |
| Bax | Bcl-2–associated X |
| Bcl-2 | B-cell lymphoma 2 |
| Bim | Bcl-2 like 11 |
| Bcl-xL | B-cell lymphoma X long isoform |
| BPH | Benign Prostatic Hyperplasia |
| Cdk-2 | Cyclin dependent kinase-2 |
| cGMP | cyclic Guanosine Monophosphate |
| CYP2C9 | Cytochrome P450 2C9 |
| CYP2C19 | Cytochrome P450 2C19 |
| DAG | Diacylglycerol |
| DHT | Dihydrotestosterone |
| ER | Endoplasmic Reticulum |
| FDA | Food and Drug Administration |
| HDAC | Histone Deacetylase |
| Hrk | Harakiri |
| IP3 | Inositol 1,4,5-triphosphate |
| IP3-R | IP3-Receptor |
| JNK | c-Jun NH2-terminal Kinase |
| LUTS | Lower Urinary Tract Symptoms |
| Mcl-1 | Myeloid cell leukemia-1 |
| MVD | Microvessels Density |
| Noxa | Phorbol-12-myristate-13-acetate-induced protein 1 |
| PARP | Poly (ADP-Ribose) Polymerase |
| PDE5 | Phosphodiesterase type 5 |
| PDO | Patient-Derived Organoids |
| PLC | phospholipase C |
| PI3K | Phosphoinositide 3-Kinase |
| PIP2 | Phospholipid phosphatydylinositol-4,5-bisphosphate |
| PKA | Protein Kinase A |
| PKC | Protein Kinase C |
| Puma | p53 Up-regulated Modulator of Apoptosis |
| TGF-β | Tumour Growth Factor-β |
| TNF-α | Tumour Necrosis Factor-α |
| TRP | Transient Receptor Potential calcium channel |
| VGCC | Voltage-Gated Calcium Channel |
References
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Mander, K.; Finnie, J. Tumour angiogenesis, anti-angiogenic therapy and chemotherapeutic resistance. Aust. Vet. J. 2018, 96, 371–378. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharm. 2013, 34, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.C.; Ekins, S.; Williams, A.J.; Tropsha, A. A bibliometric review of drug repurposing. Drug Discov. Today 2018, 23, 661–672. [Google Scholar] [CrossRef]
- Goldstein, I.; Tseng, L.-J.; Creanga, D.; Stecher, V.; Kaminetsky, J.C. Efficacy and safety of sildenafil by age in men with erectile dysfunction. J. Sex. Med. 2016, 13, 852–859. [Google Scholar] [CrossRef]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharm. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef]
- Sauter, E.R. Breast cancer prevention: Current approaches and future directions. Eur. J. Breast Health 2018. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Banno, K.; Kunitomi, H.; Tominaga, E.; Aoki, D. Current state and outlook for drug repositioning anticipated in the field of ovarian cancer. J. Gynecol. Oncol. 2019, 30, e10. [Google Scholar] [CrossRef] [PubMed]
- Hanusova, V.; Skalova, L.; Kralova, V.; Matouskova, P. Potential anti-cancer drugs commonly used for other indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Price, D.T.; Lefkowitz, R.J.; Caron, M.G.; Berkowitz, D.; Schwinn, D.A. Localization of mRNA for three distinct alpha 1-adrenergic receptor subtypes in human tissues: Implications for human alpha-adrenergic physiology. Mol. Pharm. 1994, 45, 171–175. [Google Scholar]
- Zhong, H.; Minneman, K.P. Alpha1-adrenoceptor subtypes. Eur. J. Pharm. 1999, 375, 261–276. [Google Scholar] [CrossRef]
- Docherty, J.R. The pharmacology of α1-adrenoceptor subtypes. Eur. J. Pharm. 2019, 855, 305–320. [Google Scholar] [CrossRef]
- Petitcolin, M.A.; Spitzbarth-Régrigny, E.; Bueb, J.L.; Capdeville-Atkinson, C.; Tschirhart, E. Role of G(i)-proteins in norepinephrine-mediated vasoconstriction in rat tail artery smooth muscle. Biochem. Pharm. 2001, 61, 1169–1175. [Google Scholar] [CrossRef]
- Kaplan, S.A. Current role of alpha-blockers in the treatment of benign prostatic hyperplasia. BJU Int. 2008, 102 (Suppl. S2), S3–S7. [Google Scholar] [CrossRef]
- Kim, E.H.; Larson, J.A.; Andriole, G.L. Management of benign prostatic hyperplasia. Annu. Rev. Med. 2016, 67, 137–151. [Google Scholar] [CrossRef]
- Asseldonk, B.V.; Barkin, J.; Elterman, D.S. Medical therapy for benign prostatic hyperplasia: A review. Can. J. Urol. 2015, 22 (Suppl. S1), S7–S17. [Google Scholar]
- Hwang, E.C.; Gandhi, S.; Jung, J.H.; Imamura, M.; Kim, M.H.; Pang, R.; Dahm, P. Naftopidil for the treatment of lower urinary tract symptoms compatible with benign prostatic hyperplasia. Cochrane Database Syst. Rev. 2018, 10, CD007360. [Google Scholar] [CrossRef] [PubMed]
- Lepor, H. Pathophysiology of lower urinary tract symptoms in the aging male population. Rev. Urol. 2005, 7, 3–11. [Google Scholar] [PubMed]
- Kim, E.H.; Brockman, J.A.; Andriole, G.L. The use of 5-alpha reductase inhibitors in the treatment of benign prostatic hyperplasia. Asian J. Urol. 2018, 5, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y. Lower urinary tract symptoms/benign prostatic hypertrophy and vascular function: Role of the nitric oxide-phosphodiesterase type 5-cyclic guanosine 3’, 5’-monophosphate pathway. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 2017, 24, 412–424. [Google Scholar] [CrossRef]
- Tinel, H.; Stelte-Ludwig, B.; Hütter, J.; Sandner, P. Pre-clinical evidence for the use of phosphodiesterase-5 inhibitors for treating benign prostatic hyperplasia and lower urinary tract symptoms. BJU Int. 2006, 98, 1259–1263. [Google Scholar] [CrossRef]
- Schwinn, D.A.; Roehrborn, C.G. α1-Adrenoceptor subtypes and lower urinary tract symptoms: α1-Adrenoceptor subtypes & LUTS. Int. J. Urol. 2008, 15, 193–199. [Google Scholar] [CrossRef]
- Kojima, Y.; Sasaki, S.; Shinoura, H.; Hayase, M.; Kubota, Y.; Hayashi, Y.; Tsujimoto, G.; Kohri, K. Change of expression levels of alpha1-adrenoceptor subtypes by administration of alpha1d-adrenoceptor-subtype-selective antagonist naftopidil in benign prostate hyperplasia patients. Prostate 2007, 67, 1285–1292. [Google Scholar] [CrossRef]
- Lepor, H. Medical treatment of benign prostatic hyperplasia. Rev. Urol. 2011, 13, 20–33. [Google Scholar] [PubMed]
- Sigala, S.; Dellabella, M.; Milanese, G.; Fornari, S.; Faccoli, S.; Palazzolo, F.; Peroni, A.; Mirabella, G.; Cunico, S.C.; Spano, P.; et al. Evidence for the presence of alpha1 adrenoceptor subtypes in the human ureter. Neurourol. Urodyn. 2005, 24, 142–148. [Google Scholar] [CrossRef]
- Malloy, B.J.; Price, D.T.; Price, R.R.; Bienstock, A.M.; Dole, M.K.; Funk, B.L.; Rudner, X.L.; Richardson, C.D.; Donatucci, C.F.; Schwinn, D.A. Alpha1-adrenergic receptor subtypes in human detrusor. J. Urol. 1998, 160, 937–943. [Google Scholar] [CrossRef]
- Hampel, C.; Dolber, P.C.; Smith, M.P.; Savic, S.L.; Th roff, J.W.; Thor, K.B.; Schwinn, D.A. Modulation of bladder alpha1-adrenergic receptor subtype expression by bladder outlet obstruction. J. Urol. 2002, 167, 1513–1521. [Google Scholar] [CrossRef]
- Michel, M.C. Potential role of a1-adrenoceptors in the aetiology of LUTS. Eur. Urol. Suppl. 2002, 5–13. [Google Scholar] [CrossRef]
- Nasu, K.; Moriyama, N.; Kawabe, K.; Tsujimoto, G.; Murai, M.; Tanaka, T.; Yano, J. Quantification and distribution of alpha 1-adrenoceptor subtype mRNAs in human prostate: Comparison of benign hypertrophied tissue and non-hypertrophied tissue. Br. J. Pharm. 1996, 119, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Sasaki, S.; Kubota, Y.; Hayase, M.; Hayashi, Y.; Shinoura, H.; Tsujimoto, G.; Kohri, K. Expression of alpha1-adrenoceptor subtype mRNA as a predictor of the efficacy of subtype selective alpha1-adrenoceptor antagonists in the management of benign prostatic hyperplasia. J. Urol. 2008, 179, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Masumori, N. Naftopidil for the treatment of urinary symptoms in patients with benign prostatic hyperplasia. Ther. Clin. Risk Manag. 2011, 7, 227–238. [Google Scholar] [CrossRef]
- Walden, P.D.; Gerardi, C.; Lepor, H. Localization and expression of the alpha1A-1, alpha1B and alpha1D-adrenoceptors in hyperplastic and non-hyperplastic human prostate. J. Urol. 1999, 161, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.; Feibus, A.; Baum, N. Benign prostatic hyperplasia and urinary symptoms: Evaluation and treatment. Postgrad. Med. 2015, 127, 301–307. [Google Scholar] [CrossRef]
- Michel, M.C.; Grübbel, B.; Taguchi, K.; Verfürth, F.; Otto, T.; Kröpfl, D. Drugs for treatment of benign prostatic hyperplasia: Affinity comparison at cloned alpha 1-adrenoceptor subtypes and in human prostate. J. Auton. Pharm. 1996, 16, 21–28. [Google Scholar] [CrossRef]
- Take, H.; Shibata, K.; Awaji, T.; Hirasawa, A.; Ikegaki, I.; Asano, T.; Takada, T.; Tsujimoto, G. Vascular alpha1-adrenoceptor subtype selectivity and alpha1-blocker-induced orthostatic hypotension. Jpn. J. Pharm. 1998, 77, 61–70. [Google Scholar] [CrossRef][Green Version]
- Takei, R.; Ikegaki, I.; Shibata, K.; Tsujimoto, G.; Asano, T. Naftopidil, a novel alpha1-adrenoceptor antagonist, displays selective inhibition of canine prostatic pressure and high affinity binding to cloned human alpha1-adrenoceptors. Jpn. J. Pharm. 1999, 79, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-J.; Cai, Y.; Yi, Y.-Z.; Huang, M.-Y.; Zhu, L.; He, F.; Liu, X.-W.; Huang, B.-Y.; Yuan, M. Pharmaceutical evaluation of naftopidil enantiomers: Rat functional assays in vitro and estrogen/androgen induced rat benign prostatic hyperplasia model in vivo. Eur. J. Pharm. 2016, 791, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Huang, J.; Jiang, R.; Yuan, M. Crystal structures, absolute configurations and molecular docking studies of naftopidil enantiomers as α1D-adrenoceptor antagonists. Acta Pharm. Sin. B 2017, 7, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Proudman, R.G.W.; Pupo, A.S.; Baker, J.G. The affinity and selectivity of α-adrenoceptor antagonists, antidepressants, and antipsychotics for the human α1A, α1B, and α1D-adrenoceptors. Pharm. Res. Perspect. 2020, 8, e00602. [Google Scholar] [CrossRef]
- Hara, N.; Mizusawa, T.; Obara, K.; Takahashi, K. The role of naftopidil in the management of benign prostatic hyperplasia. Ther. Adv. Urol. 2013, 5, 111–119. [Google Scholar] [CrossRef]
- Nishino, Y.; Masue, T.; Miwa, K.; Takahashi, Y.; Ishihara, S.; Deguchi, T. Comparison of two alpha1-adrenoceptor antagonists, naftopidil and tamsulosin hydrochloride, in the treatment of lower urinary tract symptoms with benign prostatic hyperplasia: A randomized crossover study. BJU Int. 2006, 97, 747–751. [Google Scholar] [CrossRef]
- Ukimura, O.; Kanazawa, M.; Fujihara, A.; Kamoi, K.; Okihara, K.; Miki, T. Kyoto Prefectural University of Medicine Benign Prostatic Hypertrophy Research Group Naftopidil versus tamsulosin hydrochloride for lower urinary tract symptoms associated with benign prostatic hyperplasia with special reference to the storage symptom: A prospective randomized controlled study. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 2008, 15, 1049–1054. [Google Scholar] [CrossRef]
- Perumal, C.; Chowdhury, P.S.; Ananthakrishnan, N.; Nayak, P.; Gurumurthy, S. A comparison of the efficacy of naftopidil and tamsulosin hydrochloride in medical treatment of benign prostatic enlargement. Urol. Ann. 2015, 7, 74–78. [Google Scholar] [CrossRef]
- Ikemoto, I.; Kiyota, H.; Ohishi, Y.; Abe, K.; Goto, H.; Kishimoto, K.; Miki, K. Usefulness of tamsulosin hydrochloride and naftopidil in patients with urinary disturbances caused by benign prostatic hyperplasia: A comparative, randomized, two-drug crossover study. Int. J. Urol. 2003, 10, 587–594. [Google Scholar] [CrossRef]
- Griwan, M.S.; Karthikeyan, Y.R.; Kumar, M.; Singh, B.J.; Singh, S.K. Comparative evaluation of naftopidil and tamsulosin in the treatment of patients with lower urinary tract symptoms with benign prostatic hyperplasia. Urol. Ann. 2014, 6, 181–186. [Google Scholar] [CrossRef]
- Farthing, M.J.; Alstead, E.M.; Abrams, S.M.; Haug, G.; Johnston, A.; Hermann, R.; Niebch, G.; Ruus, P.; Molz, K.H.; Turner, P. Pharmacokinetics of naftopidil, a novel anti-hypertensive drug, in patients with hepatic dysfunction. Postgrad. Med. J. 1994, 70, 363–366. [Google Scholar] [CrossRef][Green Version]
- Himmel, H.M. Naftopidil, a novel antihypertensive drug. Cardiovasc. Drug Rev. 1994, 12, 232–247. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, X.; Zhu, L.; Zhang, X.; Fu, X.; Huang, J.; Yuan, M. Identification of human cytochrome P450 isozymes involved in the metabolism of naftopidil enantiomers in vitro: The metabolism of naftopidil enantiomers. J. Pharm. Pharm. 2014, 66, 1534–1551. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Huang, J.; Rong, Y.; Luo, C.; Guo, J.; Zhu, L.; Huang, B.; Yuan, M. Enantiospecific determination of naftopidil by RRLC-MS/MS reveals stereoselective pharmacokinetics and tissue distributions in rats. J. Pharm. Biomed. Anal. 2015, 112, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Elagamy, H.I.; Essa, E.A.; Nouh, A.; El Maghraby, G.M. Development and evaluation of rapidly dissolving buccal films of naftopidil: In vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2019, 45, 1695–1706. [Google Scholar] [CrossRef]
- Hori, Y.; Ishii, K.; Kanda, H.; Iwamoto, Y.; Nishikawa, K.; Soga, N.; Kise, H.; Arima, K.; Sugimura, Y. Naftopidil, a selective {alpha}1-adrenoceptor antagonist, suppresses human prostate tumor growth by altering interactions between tumor cells and stroma. Cancer Prev. Res. 2011, 4, 87–96. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Ishii, K.; Sasaki, T.; Kato, M.; Kanda, H.; Yamada, Y.; Arima, K.; Shiraishi, T.; Sugimura, Y. Oral naftopidil suppresses human renal-cell carcinoma by inducing G(1) cell-cycle arrest in tumor and vascular endothelial cells. Cancer Prev. Res. 2013, 6, 1000–1006. [Google Scholar] [CrossRef]
- Gotoh, A.; Nagaya, H.; Kanno, T.; Nishizaki, T. Antitumor action of alpha(1)-adrenoceptor blockers on human bladder, prostate and renal cancer cells. Pharmacology 2012, 90, 242–246. [Google Scholar] [CrossRef]
- Nakagawa, Y.U.; Nagaya, H.; Miyata, T.; Wada, Y.; Oyama, T.; Gotoh, A. Piperazine-based Alpha-1 AR blocker, Naftopidil, selectively suppresses malignant human bladder cells via induction of apoptosis. Anticancer Res. 2016, 36, 1563–1570. [Google Scholar] [PubMed]
- Ishii, K.; Sugimura, Y. Identification of a new pharmacological activity of the phenylpiperazine derivative naftopidil: Tubulin-binding drug. J. Chem. Biol. 2015, 8, 5–9. [Google Scholar] [CrossRef][Green Version]
- Florent, R.; Weiswald, L.-B.; Lambert, B.; Brotin, E.; Abeilard, E.; Louis, M.-H.; Babin, G.; Poulain, L.; N’Diaye, M. Bim, Puma and Noxa upregulation by Naftopidil sensitizes ovarian cancer to the BH3-mimetic ABT-737 and the MEK inhibitor Trametinib. Cell Death Dis. 2020, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Ishii, K.; Ogura, Y.; Imamura, T.; Kanai, M.; Arima, K.; Sugimura, Y. Naftopidil, a selective alpha-1 adrenoceptor antagonist, inhibits growth of human prostate cancer cells by G1 cell cycle arrest. Int. J. Cancer 2008, 122, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Mikami, K.; Nagaya, H.; Gotoh, A.; Kanno, T.; Tsuchiya, A.; Nakano, T.; Nishizaki, T. Naftopidil is useful for the treatment of malignant pleural mesothelioma. Pharmacology 2014, 94, 163–169. [Google Scholar] [CrossRef]
- Masachika, E.; Kanno, T.; Nakano, T.; Gotoh, A.; Nishizaki, T. Naftopidil induces apoptosis in malignant mesothelioma cell lines independently of alpha1-adrenoceptor blocking. Anticancer Res. 2013, 33, 887–894. [Google Scholar] [PubMed]
- Yamada, D.; Nishimatsu, H.; Kumano, S.; Hirano, Y.; Suzuki, M.; Fujimura, T.; Fukuhara, H.; Enomoto, Y.; Kume, H.; Homma, Y. Reduction of prostate cancer incidence by naftopidil, an alpha1 -adrenoceptor antagonist and transforming growth factor-beta signaling inhibitor. Int. J. Urol. 2013, 20, 1220–1227. [Google Scholar] [CrossRef]
- Nakamura, A.; Matsunaga, W.; Gotoh, A. Autophagy induced by Naftopidil inhibits apoptosis of human gastric cancer cells. Anticancer Res. 2018, 38, 803–809. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Ishii, K.; Kanda, H.; Kato, M.; Miki, M.; Kajiwara, S.; Arima, K.; Shiraishi, T.; Sugimura, Y. Combination treatment with naftopidil increases the efficacy of radiotherapy in PC-3 human prostate cancer cells. J. Cancer Res. Clin. Oncol. 2017, 143, 933–939. [Google Scholar] [CrossRef]
- Ishii, K.; Matsuoka, I.; Kajiwara, S.; Sasaki, T.; Miki, M.; Kato, M.; Kanda, H.; Arima, K.; Shiraishi, T.; Sugimura, Y. Additive naftopidil treatment synergizes docetaxel-induced apoptosis in human prostate cancer cells. J. Cancer Res. Clin. Oncol. 2018, 144, 89–98. [Google Scholar] [CrossRef]
- Turanli, B.; Grøtli, M.; Boren, J.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Drug repositioning for effective prostate cancer treatment. Front. Physiol. 2018, 9, 500. [Google Scholar] [CrossRef]
- Besen, J.; Gan, S.D. A critical evaluation of clinical research study designs. J. Investig. Dermatol. 2014, 134, 1–4. [Google Scholar] [CrossRef]
- Batty, M.; Pugh, R.; Rathinam, I.; Simmonds, J.; Walker, E.; Forbes, A.; Anoopkumar-Dukie, S.; McDermott, C.M.; Spencer, B.; Christie, D.; et al. The role of alpha1-adrenoceptor antagonists in the treatment of prostate and other cancers. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Kyprianou, N.; Benning, C.M. Suppression of human prostate cancer cell growth by alpha1-adrenoceptor antagonists doxazosin and terazosin via induction of apoptosis. Cancer Res. 2000, 60, 4550–4555. [Google Scholar] [PubMed]
- Benning, C.M.; Kyprianou, N. Quinazoline-derived α1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an α1-Adrenoceptor-independent action. Cancer Res. 2002, 60, 4550–4555. [Google Scholar] [PubMed]
- Harris, A.M.; Warner, B.W.; Wilson, J.M.; Becker, A.; Rowland, R.G.; Conner, W.; Lane, M.; Kimbler, K.; Durbin, E.B.; Baron, A.T.; et al. Effect of alpha1-adrenoceptor antagonist exposure on prostate cancer incidence: An observational cohort study. J. Urol. 2007, 178, 2176–2180. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, C.L.; Sun, T.; Zhou, Z.; Niu, J.X.; Tian, X.M.; Yuan, M. Synthesis, biological evaluation and SAR of naftopidil-based arylpiperazine derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cao, Y.; Zhang, J.; Yu, S.; Zou, Y.; Chai, X.; Wu, Q.; Zhang, D.; Jiang, Y.; Sun, Q. Design, synthesis and antifungal activities of novel 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2011, 46, 3142–3148. [Google Scholar] [CrossRef] [PubMed]
- Bassetto, M.; Leyssen, P.; Neyts, J.; Yerukhimovich, M.M.; Frick, D.N.; Courtney-Smith, M.; Brancale, A. In silico identification, design and synthesis of novel piperazine-based antiviral agents targeting the hepatitis C virus helicase. Eur. J. Med. Chem. 2017, 125, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-X.; Li, Z.-G.; Xie, H.-Y.; Gao, J.-R.; Zou, J.-W. Quantitative structure–activity relationship analysis of aryl alkanol piperazine derivatives with antidepressant activities. Eur. J. Med. Chem. 2009, 44, 4367–4375. [Google Scholar] [CrossRef] [PubMed]
- Fytas, C.; Zoidis, G.; Tsotinis, A.; Fytas, G.; Khan, M.A.; Akhtar, S.; Rahman, K.M.; Thurston, D.E. Novel 1-(2-aryl-2-adamantyl)piperazine derivatives with antiproliferative activity. Eur. J. Med. Chem. 2015, 93, 281–290. [Google Scholar] [CrossRef]
- Trivedi, P.; Adhikari, N.; Amin, S.A.; Bobde, Y.; Ganesh, R.; Jha, T.; Ghosh, B. Design, synthesis, biological evaluation and molecular docking study of arylcarboxamido piperidine and piperazine-based hydroxamates as potential HDAC8 inhibitors with promising anticancer activity. Eur. J. Pharm. Sci. 2019, 138, 105046. [Google Scholar] [CrossRef]
- Chen, H.; Xu, B.-B.; Sun, T.; Zhou, Z.; Ya, H.-Y.; Yuan, M. Synthesis and antitumor activity of novel arylpiperazine derivatives containing the saccharin moiety. Molecules 2017, 22, 1857. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; He, F.; Huang, M.; Liu, X.; Xiong, Y.; Huang, Y.; Zhu, L.; Yang, Y.; Xu, X.; Yuan, M. Novel naftopidil-related derivatives and their biological effects as alpha1-adrenoceptors antagonists and antiproliferative agents. Eur. J. Med. Chem. 2015, 96, 83–91. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Ho, C.-T.; Chang, Y.-J.; Yang, L.-X.; Wei, P.-L.; Liu, T.-Z.; Liu, J.-J. A novel microtubule-disrupting agent induces endoplasmic reticular stress-mediated cell death in human hepatocellular carcinoma cells. PLoS ONE 2015, 10, e0136340. [Google Scholar] [CrossRef]
- Chopra, A.; Anderson, A.; Giardina, C. Novel piperazine-based compounds inhibit microtubule dynamics and sensitize colon cancer cells to tumor necrosis factor-induced apoptosis. J. Biol. Chem. 2014, 289, 2978–2991. [Google Scholar] [CrossRef]
- Gayvert, K.M.; Dardenne, E.; Cheung, C.; Boland, M.R.; Lorberbaum, T.; Wanjala, J.; Chen, Y.; Rubin, M.A.; Tatonetti, N.P.; Rickman, D.S.; et al. A Computational drug repositioning approach for targeting oncogenic transcription factors. Cell Rep. 2016, 15, 2348–2356. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; van den Heuvel, A.P.J.; Prabhu, V.V.; Zhang, S.; El-Deiry, W.S. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Yamaguchi, K.; Yamamoto, M.; Kurioka, R.; Kino, Y.; Matsunaga, W.; Nakao, S.; Fukuhara, H.; Tanaka, A.; Gotoh, A.; et al. Identification of HUHS190, a human naftopidil metabolite, as a novel anti-bladder cancer drug. Bioorg. Med. Chem. Lett. 2020, 30, 126744. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-J.; Zhang, Z.-H.; He, F.; Liu, X.-W.; Xu, X.-J.; Dai, L.-J.; Liu, Q.-M.; Yuan, M. Novel naftopidil derivatives containing methyl phenylacetate and their blocking effects on α1D/1A-adrenoreceptor subtypes. Bioorg. Med. Chem. Lett. 2018, 28, 547–551. [Google Scholar] [CrossRef]
- Chen, H.; Qian, Y.; Jia, H.; Yu, Y.; Zhang, H.; Shen, J.; Zhao, S. Synthesis and pharmacological evaluation of naftopidil-based arylpiperazine derivatives containing the bromophenol moiety. Pharm. Rep. 2020, PR, 1–11. [Google Scholar] [CrossRef]
- Kaku, Y.; Nagaya, H.; Tsuchiya, A.; Kanno, T.; Gotoh, A.; Tanaka, A.; Shimizu, T.; Nakao, S.; Tabata, C.; Nakano, T.; et al. Newly synthesized anticancer drug HUHS1015 is effective on malignant pleural mesothelioma. Cancer Sci. 2014, 105, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, T.; Kanno, T.; Tsuchiya, A.; Kaku, Y.; Shimizu, T.; Tanaka, A. 1-[2-(2-methoxyphenylamino) ethylamino]-3-(naphthalene-1- yloxy) propan-2-ol may be a promising anticancer drug. Molecules 2014, 19, 21462–21472. [Google Scholar] [CrossRef]
- Kanno, T.; Tanaka, A.; Shimizu, T.; Nakano, T.; Nishizaki, T. 1-[2-(2-methoxyphenylamino) ethylamino]-3-(naphthalene-1-yloxy) propan-2-ol as a potential anticancer drug. Pharmacology 2013, 91, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Tsuchiya, A.; Kanno, T.; Nakao, S.; Shimizu, T.; Tanaka, A.; Nishizaki, T. The newly synthesized anticancer drug HUHS1015 is useful for treatment of human gastric cancer. Cancer Chemother Pharm. 2015, 75, 527–535. [Google Scholar] [CrossRef]
- Kaku, Y.; Tsuchiya, A.; Kanno, T.; Nishizaki, T. HUHS1015 induces necroptosis and caspase-independent apoptosis of MKN28 human gastric cancer cells in association with AMID accumulation in the nucleus. Anticancer Agents Med. Chem. 2015, 15, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Tsuchiya, A.; Shimizu, T.; Tanaka, A.; Nishizaki, T. HUHS1015 suppresses colonic cancer growth by inducing necrosis and apoptosis in association with mitochondrial damage. Anticancer Res. 2016, 36, 39–48. [Google Scholar] [PubMed]
- Quaglia, W.; Santoni, G.; Pigini, M.; Piergentili, A.; Gentili, F.; Buccioni, M.; Mosca, M.; Lucciarini, R.; Amantini, C.; Nabissi, M.I.; et al. Structure−activity relationships in 1,4-benzodioxan-related compounds. 8.{2-[2-(4-chlorobenzyloxy) phenoxy] ethyl}-[2-(2,6-dimethoxyphenoxy) ethyl] amine (clopenphendioxan) as a tool to highlight the involvement of α 1D- and α 1B- adrenoreceptor subtypes in the regulation of human PC-3 prostate cancer cell apoptosis and proliferation. J. Med. Chem. 2005, 48, 7750–7763. [Google Scholar] [CrossRef]
- Colciago, A.; Mornati, O.; Ferri, N.; Castelnovo, L.F.; Fumagalli, L.; Bolchi, C.; Pallavicini, M.; Valoti, E.; Negri-Cesi, P. A selective alpha1D-adrenoreceptor antagonist inhibits human prostate cancer cell proliferation and motility “in vitro”. Pharm. Res. 2016, 103, 215–226. [Google Scholar] [CrossRef]
- Thebault, S.; Roudbaraki, M.; Sydorenko, V.; Shuba, Y.; Lemonnier, L.; Slomianny, C.; Dewailly, E.; Bonnal, J.-L.; Mauroy, B.; Skryma, R.; et al. Alpha1-adrenergic receptors activate Ca(2+)-permeable cationic channels in prostate cancer epithelial cells. J. Clin. Investig. 2003, 111, 1691–1701. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Liberati, S.; Cardinali, C.; Farfariello, V.; Tomassoni, D.; Quaglia, W.; Piergentili, A.; Bonifazi, A.; et al. Cross-talk between alpha1D-adrenoceptors and transient receptor potential vanilloid type 1 triggers prostate cancer cell proliferation. BMC Cancer 2014, 14, 921. [Google Scholar] [CrossRef]
- Castiglione, F.; Benigni, F.; Briganti, A.; Salonia, A.; Villa, L.; Nini, A.; Di Trapani, E.; Capitanio, U.; Hedlund, P.; Montorsi, F. Naftopidil for the treatment of benign prostate hyperplasia: A systematic review. Curr. Med. Res. Opin. 2014, 30, 719–732. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).