RORγ Structural Plasticity and Druggability



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Plasticity of the RORγ Orthosteric Binding Site Affects Druggability
2.1. The Dynamics of H12
2.2. The Plasticity of the Orthosteric Binding Pocket on Accepting Modulators
3. The Plasticity of RORγ Allosteric Binding Sites Increases Druggability
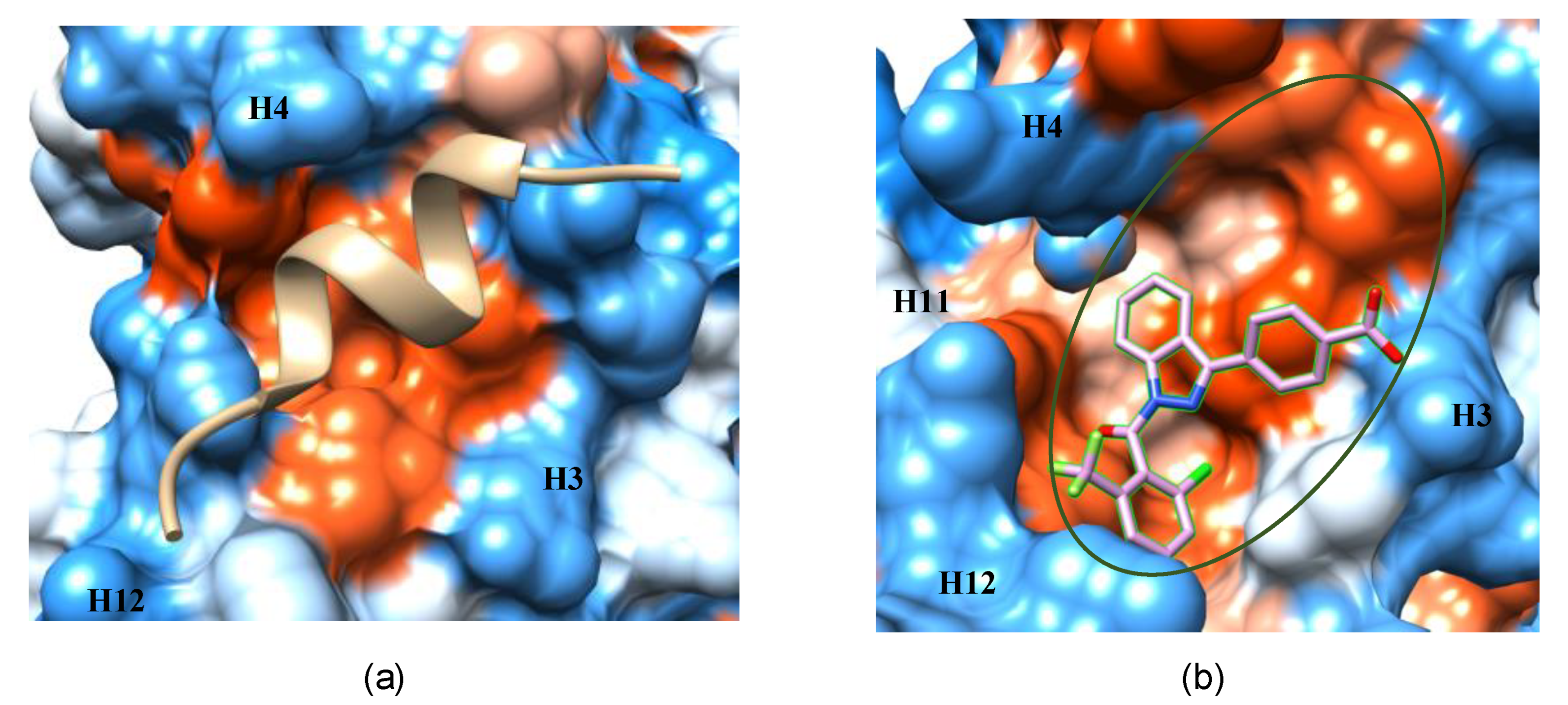
3.1. The AF-2 Allosteric Binding Site
3.2. An Allosteric Binding Site in the RORγ HD
3.3. Other Potential Allosteric Binding Sites
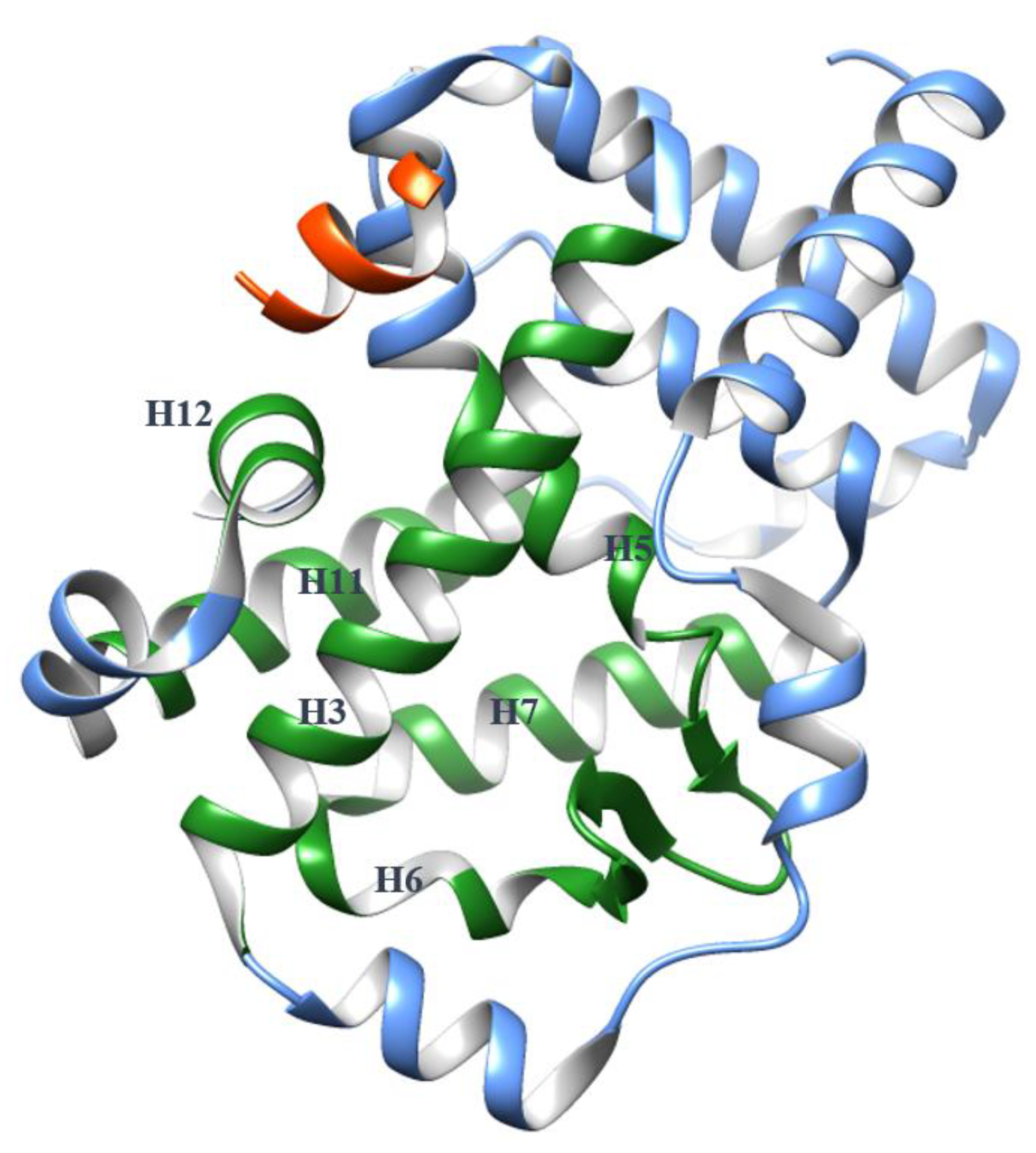
4. The Helices Acting as the Arm and Hand of the LBD
5. The Role of Protein Plasticity in the Computer-Aided Discovery of RORγ-Targeting Drugs
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RORγ | Retinoid-related orphan receptor γ |
| NR | Nuclear receptor |
| Th17 | T helper 17 |
| NTD | N-terminal domain |
| DBD | DNA-binding domain |
| HD | Hinge domain |
| LBD | Ligand-binding domain |
| CTD | C-terminal domain |
| PDB | Protein Data Bank |
| SAR | Structure-activity relationship |
| MD | Molecular dynamics |
| VdW | Van der Waals |
| VS | Virtual screening |
| RMSD | root-mean-square deviation |
| HTS | high-throughput screening |
| AF-2 | Activation function-2 |
| HRE | Hormone response element |
References
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Meissburger, B.; Ukropec, J.; Roeder, E.; Beaton, N.; Geiger, M.; Teupser, D.; Civan, B.; Langhans, W.; Nawroth, P.P.; Gasperikova, D.; et al. Adipogenesis and insulin sensitivity in obesity are regulated by retinoid-related orphan receptor gamma. EMBO Mol. Med. 2011, 3, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Kameswaran, V.; Zhang, Y.; Zheng, S.; Sun, J.; Wang, J.; DeVirgiliis, J.; Liou, H.-C.; Beg, A.A.; Chen, Y.H. The Th17 immune response is controlled by the Rel–RORγ–RORγT transcriptional axis. J. Exp. Med. 2011, 208, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, X.; Moisan, J.; Wang, Y.; Lesch, C.A.; Spooner, C.; Morgan, R.W.; Zawidzka, E.M.; Mertz, D.; Bousley, D.; et al. Synthetic RORγ agonists regulate multiple pathways to enhance antitumor immunity. OncoImmunology 2016, 5, e1254854. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Majchrzak, K.; Liu, X.; Wyatt, M.M.; Spooner, C.J.; Moisan, J.; Zou, W.; Carter, L.L.; Paulos, C.M. In vitro priming of adoptively transferred T cells with a RORγ agonist confers durable memory and stemness In vivo. Cancer Res. 2018, 78, 3888–3898. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis Is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Cui, G. TH9, TH17, and TH22 cell subsets and their main cytokine products in the pathogenesis of colorectal cancer. Front. Oncol. 2019, 9, 1002. [Google Scholar] [CrossRef]
- Fauber, B.P.; René, O.; de Leon Boenig, G.; Burton, B.; Deng, Y.; Eidenschenk, C.; Everett, C.; Gobbi, A.; Hymowitz, S.G.; Johnson, A.R.; et al. Reduction in lipophilicity improved the solubility, plasma–protein binding, and permeability of tertiary sulfonamide RORc inverse agonists. Bioorg. Med. Chem. Lett. 2014, 24, 3891–3897. [Google Scholar] [CrossRef]
- Sun, N.; Guo, H.; Wang, Y. Retinoic acid receptor-related orphan receptor gamma-t (RORγt) inhibitors in clinical development for the treatment of autoimmune diseases: A patent review (2016-present). Expert Opin. Ther. Pat. 2019, 29, 663–674. [Google Scholar] [CrossRef]
- Scheepstra, M.; Leysen, S.; van Almen, G.C.; Miller, J.R.; Piesvaux, J.; Kutilek, V.; van Eenennaam, H.; Zhang, H.; Barr, K.; Nagpal, S.; et al. Identification of an allosteric binding site for RORγt inhibition. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lao, C.; Zhou, X.; Chen, H.; Wei, F.; Huang, Z.; Bai, C. 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine derivatives as inhibitors of full-length RORγt. Bioorg. Chem. 2019, 90, 103077. [Google Scholar] [CrossRef] [PubMed]
- Meijer, F.A.; Leijten-van de Gevel, I.A.; de Vries, R.M.J.M.; Brunsveld, L. Allosteric small molecule modulators of nuclear receptors. Mol. Cell. Endocrinol. 2019, 485, 20–34. [Google Scholar] [CrossRef]
- Mittag, T.; Kay, L.E.; Forman-Kay, J.D. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.E.; Lindorff-Larsen, K. Structural heterogeneity and dynamics in protein evolution and design. Curr. Opin. Struc. Biol. 2018, 48, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A.; Forman-Kay, J.D. Probing the diverse landscape of protein flexibility and binding. Curr. Opin. Struc. Biol. 2012, 22, 643–650. [Google Scholar] [CrossRef]
- Pandya, V.B.; Kumar, S.; Sachchidanand; Sharma, R.; Desai, R.C. Combating autoimmune diseases with retinoic acid receptor-related orphan receptor-γ (RORγ or RORc) inhibitors: Hits and misses. J. Med. Chem. 2018, 61, 10976–10995. [Google Scholar] [CrossRef]
- Santori, F.R.; Huang, P.; van de Pavert, S.A.; Douglass, E.F.; Leaver, D.J.; Haubrich, B.A.; Keber, R.; Lorbek, G.; Konijn, T.; Rosales, B.N.; et al. Identification of natural RORγ ligands that regulate the development of lymphoid cells. Cell Metab. 2015, 21, 286–298. [Google Scholar] [CrossRef]
- Jin, L.; Martynowski, D.; Zheng, S.; Wada, T.; Xie, W.; Li, Y. Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORγ. Mol. Endocrinol. 2010, 24, 923–929. [Google Scholar] [CrossRef]
- Renaud, J.-P.; Rochel, N.; Ruff, M.; Vivat, V.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal structure of the RAR-γ ligand-binding domain bound to all- trans retinoic acid. Nature 1995, 378, 681–689. [Google Scholar] [CrossRef]
- Kallenberger, B.C.; Love, J.D.; Chatterjee, V.K.K.; Schwabe, J.W.R. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat. Struct. Biol. 2003, 10, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Anderson, M.; Collin, D.; Muegge, I.; Wan, J.; Brennan, D.; Kugler, S.; Terenzio, D.; Kennedy, C.; Lin, S.; et al. Structural studies unravel the active conformation of apo RORγt nuclear receptor and a common inverse agonism of two diverse classes of RORγt inhibitors. J. Biol. Chem. 2017, 292, 11618–11630. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Nomura, A.; Murase, K.; Doi, S.; Yamaguchi, K.; Hirata, K.; Shiozaki, M.; Hirashima, S.; Kotoku, M.; Yamaguchi, T.; et al. Ternary complex of human RORγ ligand-binding domain, inverse agonist and SMRT peptide shows a unique mechanism of corepressor recruitment. Genes Cells 2017, 22, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Strutzenberg, T.S.; Garcia-Ordonez, R.D.; Novick, S.J.; Park, H.; Chang, M.R.; Doebellin, C.; He, Y.; Patouret, R.; Kamenecka, T.M.; Griffin, P.R. HDX-MS reveals structural determinants for RORγ hyperactivation by synthetic agonists. eLife 2019, 8, e47172. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.I.; Xue, Y.; von Berg, S.; Aagaard, A.; McPheat, J.; Hansson, E.L.; Bernström, J.; Hansson, P.; Jirholt, J.; Grindebacke, H.; et al. Benzoxazepines achieve potent suppression of IL-17 release in human T-helper 17 (TH17) cells through an induced-fit binding mode to the nuclear receptor RORγ. ChemMedChem 2016, 11, 207–216. [Google Scholar] [CrossRef]
- Chao, J.; Enyedy, I.; Van Vloten, K.; Marcotte, D.; Guertin, K.; Hutchings, R.; Powell, N.; Jones, H.; Bohnert, T.; Peng, C.-C.; et al. Discovery of biaryl carboxylamides as potent RORγ inverse agonists. Bioorg. Med. Chem. Lett. 2015, 25, 2991–2997. [Google Scholar] [CrossRef]
- Marcotte, D.J.; Liu, Y.; Little, K.; Jones, J.H.; Powell, N.A.; Wildes, C.P.; Silvian, L.F.; Chodaparambil, J.V. Structural determinant for inducing RORgamma specific inverse agonism triggered by a synthetic benzoxazinone ligand. BMC Struct. Biol. 2016, 16, 7. [Google Scholar] [CrossRef]
- Sasaki, Y.; Odan, M.; Yamamoto, S.; Kida, S.; Ueyama, A.; Shimizu, M.; Haruna, T.; Watanabe, A.; Okuno, T. Discovery of a potent orally bioavailable retinoic acid receptor-related orphan receptor-gamma-t (RORγt) inhibitor, S18-000003. Bioorg. Med. Chem. Lett. 2018, 28, 3549–3553. [Google Scholar] [CrossRef]
- Saen-Oon, S.; Lozoya, E.; Segarra, V.; Guallar, V.; Soliva, R. Atomistic simulations shed new light on the activation mechanisms of RORγ and classify it as Type III nuclear hormone receptor regarding ligand-binding paths. Sci. Rep. 2019, 9, 17249. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Fauber, B.P.; de Leon Boenig, G.; Burton, B.; Eidenschenk, C.; Everett, C.; Gobbi, A.; Hymowitz, S.G.; Johnson, A.R.; Liimatta, M.; Lockey, P.; et al. Structure-based design of substituted hexafluoroisopropanol-arylsulfonamides as modulators of RORc. Bioorg. Med. Chem. Lett. 2013, 23, 6604–6609. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, M.B.; Fauber, B.P.; Cartwright, M.; Gaines, S.; Killen, J.C.; René, O.; Ward, S.I.; de Leon Boenig, G.; Deng, Y.; Eidenschenk, C.; et al. A reversed sulfonamide series of selective RORc inverse agonists. Bioorg. Med. Chem. Lett. 2014, 24, 5769–5776. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Liu, Q.; Cheng, Y.; Cai, W.; Ma, Y.; Yang, L.; Wu, Q.; Orband-Miller, L.A.; Zhou, L.; Xiang, Z.; et al. Discovery of tertiary amine and indole derivatives as potent RORγt inverse agonists. ACS Med. Chem. Lett. 2014, 5, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Nomura, A.; Doi, S.; Yamaguchi, K.; Hirata, K.; Shiozaki, M.; Maeda, K.; Hirashima, S.; Kotoku, M.; Yamaguchi, T.; et al. Ternary crystal structure of human RORγ ligand-binding-domain, an inhibitor and corepressor peptide provides a new insight into corepressor interaction. Sci. Rep. 2018, 8, 17374. [Google Scholar] [CrossRef]
- Kallen, J.; Izaac, A.; Be, C.; Arista, L.; Orain, D.; Kaupmann, K.; Guntermann, C.; Hoegenauer, K.; Hintermann, S. Structural states of RORγt: X-ray elucidation of molecular mechanisms and binding interactions for natural and synthetic compounds. ChemMedChem 2017, 12, 1014–1021. [Google Scholar] [CrossRef]
- Narjes, F.; Xue, Y.; von Berg, S.; Malmberg, J.; Llinas, A.; Olsson, R.I.; Jirholt, J.; Grindebacke, H.; Leffler, A.; Hossain, N.; et al. Potent and orally bioavailable inverse agonists of RORγt resulting from structure-based design. J. Med. Chem. 2018, 61, 7796–7813. [Google Scholar] [CrossRef]
- Zhang, H.; Lapointe, B.T.; Anthony, N.; Azevedo, R.; Cals, J.; Correll, C.C.; Daniels, M.; Deshmukh, S.; van Eenenaam, H.; Ferguson, H.; et al. Discovery of N-(Indazol-3-yl)piperidine-4-carboxylic acids as RORγt allosteric inhibitors for autoimmune diseases. ACS Med. Chem. Lett. 2020, 11, 114–119. [Google Scholar] [CrossRef]
- De Vries, R.M.J.M.; Doveston, R.G.; Meijer, F.A.; Brunsveld, L. Elucidation of an allosteric mode of action for a thienopyrazole RORγt inverse agonist. ChemMedChem 2020, 15, 561–565. [Google Scholar] [CrossRef]
- Ouvry, G.; Bouix-Peter, C.; Ciesielski, F.; Chantalat, L.; Christin, O.; Comino, C.; Duvert, D.; Feret, C.; Harris, C.S.; Lamy, L.; et al. Discovery of phenoxyindazoles and phenylthioindazoles as RORγ inverse agonists. Bioorg. Med. Chem. Lett. 2016, 26, 5802–5808. [Google Scholar] [CrossRef]
- Meijer, F.A.; Doveston, R.G.; de Vries, R.M.J.M.; Vos, G.M.; Vos, A.A.A.; Leysen, S.; Scheepstra, M.; Ottmann, C.; Milroy, L.-G.; Brunsveld, L. Ligand-based design of allosteric retinoic acid receptor-related orphan receptor γt (RORγt) inverse agonists. J. Med. Chem. 2020, 63, 241–259. [Google Scholar] [CrossRef]
- Jiang, X.; Dulubova, I.; Reisman, S.A.; Hotema, M.; Lee, C.-Y.I.; Liu, L.; McCauley, L.; Trevino, I.; Ferguson, D.A.; Eken, Y.; et al. A novel series of cysteine-dependent, allosteric inverse agonists of the nuclear receptor RORγt. Bioorg. Med. Chem. Lett. 2020, 30, 126967. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fu, M.; Angeletti, R.H.; Siconolfi-Baez, L.; Reutens, A.T.; Albanese, C.; Lisanti, M.P.; Katzenellenbogen, B.S.; Kato, S.; Hopp, T.; et al. Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. J. Biol. Chem. 2001, 276, 18375–18383. [Google Scholar] [CrossRef] [PubMed]
- Clinckemalie, L.; Vanderschueren, D.; Boonen, S.; Claessens, F. The hinge region in androgen receptor control. Mol. Cell. Endocrinol. 2012, 358, 1–8. [Google Scholar] [CrossRef] [PubMed]
- McBroom, L.D.; Flock, G.; Giguère, V. The nonconserved hinge region and distinct amino-terminal domains of the ROR alpha orphan nuclear receptor isoforms are required for proper DNA bending and ROR alpha-DNA interactions. Mol. Cell. Biol. 1995, 15, 796–808. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Ban, F.; Dalal, K.; Li, H.; LeBlanc, E.; Rennie, P.S.; Cherkasov, A. Best practices of computer-aided drug discovery: Lessons learned from the development of a preclinical candidate for prostate cancer with a new mechanism of action. J. Chem. Inf. Model. 2017, 57, 1018–1028. [Google Scholar] [CrossRef]
- He, Z.; Ma, J.; Wang, R.; Zhang, J.; Huang, Z.; Wang, F.; Sen, S.; Rothenberg, E.V.; Sun, Z. A two-amino-acid substitution in the transcription factor RORγt disrupts its function in T H 17 differentiation but not in thymocyte development. Nat. Immunol. 2017, 18, 1128–1138. [Google Scholar] [CrossRef]
- Okada, S.; Markle, J.G.; Deenick, E.K.; Mele, F.; Averbuch, D.; Lagos, M.; Alzahrani, M.; Al-Muhsen, S.; Halwani, R.; Ma, C.S.; et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 2015, 349, 606–613. [Google Scholar] [CrossRef]
- He, Z.; Zhang, J.; Huang, Z.; Du, Q.; Li, N.; Zhang, Q.; Chen, Y.; Sun, Z. Sumoylation of RORγt regulates T H 17 differentiation and thymocyte development. Nat. Commun. 2018, 9, 4870. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, X.; Jin, X.; Song, Y.; Li, J.; Luo, X.; Song, M.; Yan, W.; Song, H.; Xu, Y. Discovery of 2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide derivatives as new RORγ inhibitors using virtual screening, synthesis and biological evaluation. Eur. J. Med. Chem. 2014, 78, 431–441. [Google Scholar] [CrossRef]
- Song, Y.; Xue, X.; Wu, X.; Wang, R.; Xing, Y.; Yan, W.; Zhou, Y.; Qian, C.-N.; Zhang, Y.; Xu, Y. Identification of N-phenyl-2-(N-phenylphenylsulfonamido)acetamides as new RORγ inverse agonists: Virtual screening, structure-based optimization, and biological evaluation. Eur. J. Med. Chem. 2016, 116, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ng, H.L. Deep neural network affinity model for BACE inhibitors in D3R Grand Challenge 4. J. Comput. Aided Mol. Des. 2020, 34, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yuan, C.; Ma, X.; Wang, Y.; Gu, X.; Fu, W. Molecular mechanism of action of RORγt agonists and inverse agonists: Insights from molecular dynamics simulation. Molecules 2018, 23, 3181. [Google Scholar] [CrossRef]
- Ng, H.L. Simulations reveal increased fluctuations in estrogen receptor-alpha conformation upon antagonist binding. J. Mol. Graph. Model. 2016, 69, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Smith, R.J.; Jetten, A.M. ROR gamma: The third member of ROR/RZR orphan receptor subfamily that Is highly expressed in skeletal muscle. Biochem. Biophys. Res. Commun. 1994, 205, 1976–1983. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Bolin, S.; Miller, H.; Ng, H.L. RORγ Structural Plasticity and Druggability. Int. J. Mol. Sci. 2020, 21, 5329. https://doi.org/10.3390/ijms21155329
Huang M, Bolin S, Miller H, Ng HL. RORγ Structural Plasticity and Druggability. International Journal of Molecular Sciences. 2020; 21(15):5329. https://doi.org/10.3390/ijms21155329
Chicago/Turabian StyleHuang, Mian, Shelby Bolin, Hannah Miller, and Ho Leung Ng. 2020. "RORγ Structural Plasticity and Druggability" International Journal of Molecular Sciences 21, no. 15: 5329. https://doi.org/10.3390/ijms21155329
APA StyleHuang, M., Bolin, S., Miller, H., & Ng, H. L. (2020). RORγ Structural Plasticity and Druggability. International Journal of Molecular Sciences, 21(15), 5329. https://doi.org/10.3390/ijms21155329