Targeting Epigenetic Modifications in Uveal Melanoma



Abstract

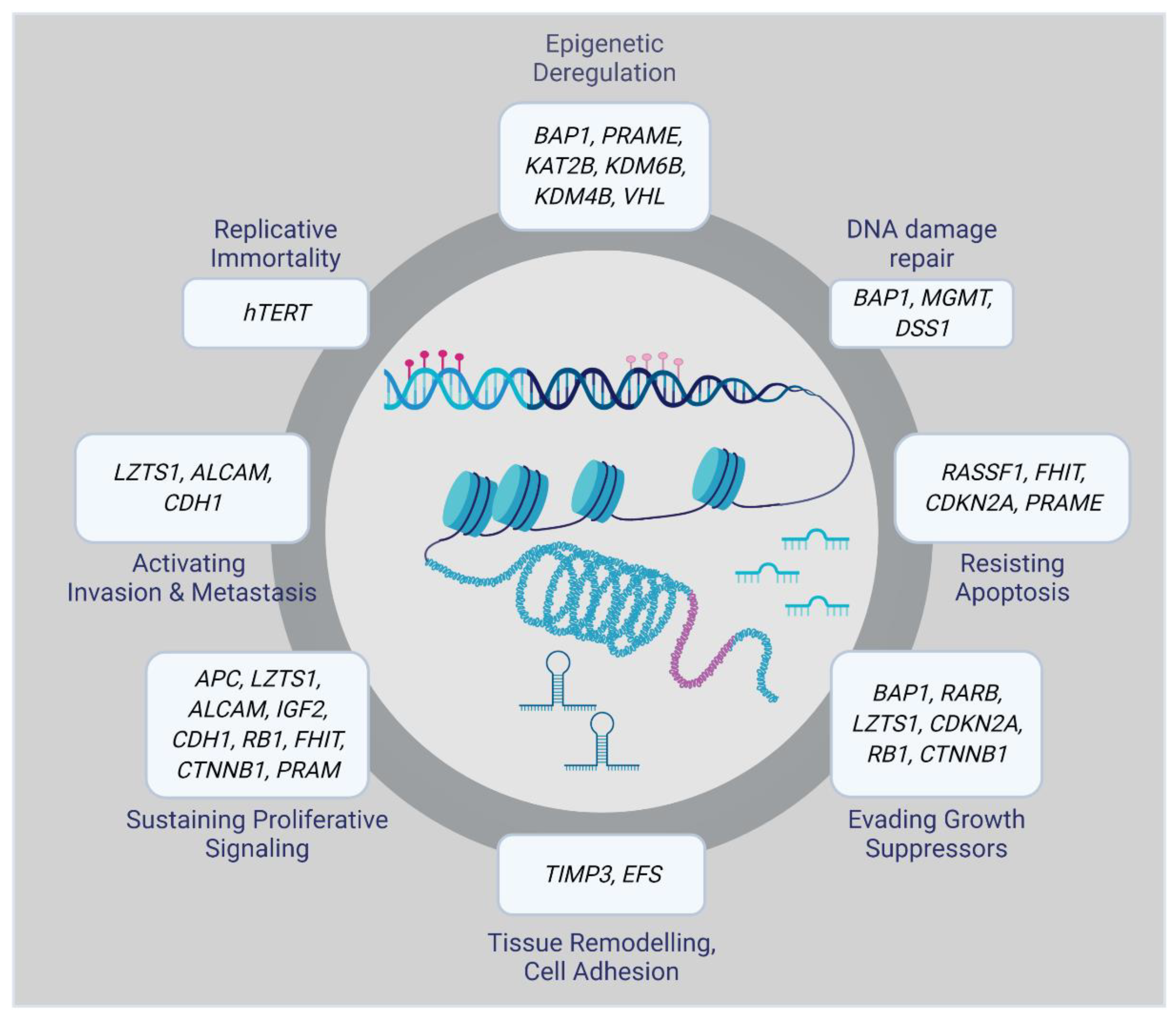
1. Biology and Molecular Subtypes of Uveal Melanoma
2. Role of Epigenetic Changes in UM Progression
2.1. DNA Methylation
2.2. Histone Modifications
2.3. miRNA-Based Epigenetic Mechanism
3. Current Therapeutic Approaches Available for UM
3.1. Primary UM Therapies
3.2. Metastatic UM Therapies
4. Potential of Epigenetic Therapies in UM
4.1. Inhibitors of DNA Methylation
4.2. Histone Deacetylase Inhibitors
4.3. Combination Therapy Using Epigenetic Drugs
4.4. Third Generation of Epigenetic Drugs
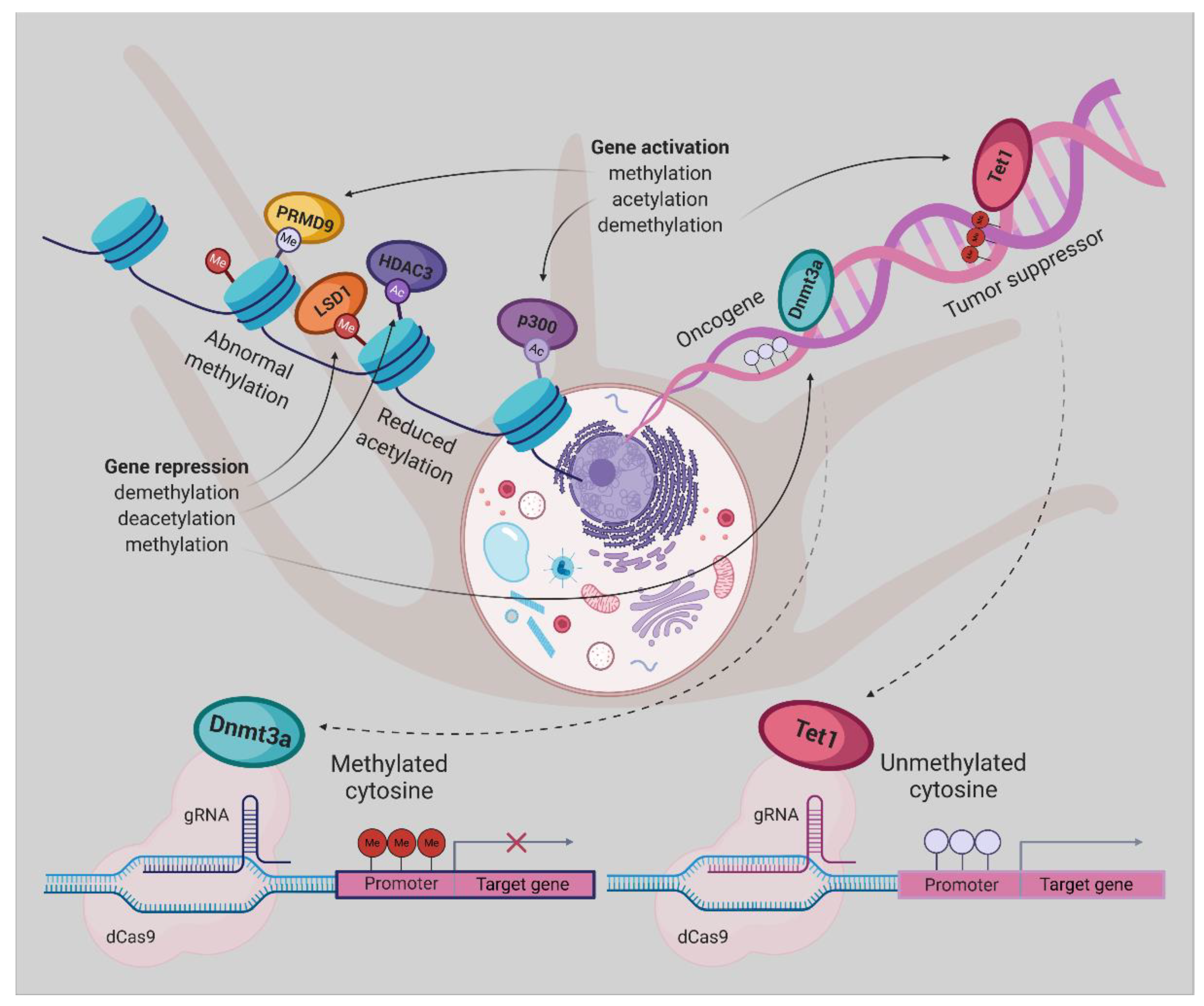
5. Epigenetic Editing
Epigenetic Editing Using CRISPR/dCas9
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| BAP1 | BRCA1 associated protein 1 |
| BET | Bromodomain and Extra-Terminal motif |
| BETi | Bromodomain and Extra-Terminal motif inhibitor |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase |
| BRD2,3,4 | Bromodomain Containing 2,3,4 |
| BRDT | Bromodomain Testis Associated |
| Cas9 | (CRISPR) associated protein 9 |
| CCND2 | Cyclin D2 |
| CDK4 | Cyclin Dependent Kinase 4 |
| CDK6 | Cyclin Dependent Kinase 6 |
| -CH3 | Methyl group |
| CpG | Cytosines followed by guanine residues |
| COMS | Collaborative Ocular Melanoma Study |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CTA | Cancer test antigens |
| CTLA-4 | Cytotoxic T-lymphocyte-associated antigen 4 |
| D3 | Disomy 3 |
| dCas9 | Nuclease-deficient Cas9 |
| DNA | Deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| DNMTi | DNMT inhibitor |
| DOT1Li | Histone H3K79 methyltransferase inhibitor |
| DSS1 | Deleted Split hand/Split foot 1 |
| EFS | Embryonal Fyn-Associated Substrate |
| EIF1AX | Eukaryotic Translation Initiation Factor 1A X-Linked |
| EHMT2 | Histone-lysine N-methyltransferase known as G9A |
| EZH2 | Enhancer of zeste homolog |
| GNAQ | G Protein Subunit Alpha Q |
| GNA11 | G Protein Subunit Alpha 11 |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HDACi | Histone deacetylase inhibitor |
| HDM | Histone demethylase |
| HDMi | Histone demethylase inhibitor |
| HIF1A | Hypoxia-inducible factor 1 alpha |
| HMT | Histone methyltransferase |
| HOXA5 | Homeobox A5 |
| H2A | Histone H2A |
| H3 | Histone 3 |
| H4 | Histone 4 |
| IDO1 | Indoleamine 2,3-Dioxygenase 1 |
| KDM1Ai | Lysine-specific histone demethylase 1A inhibitor |
| LSDi | Lysine-specific demethylase 1 inhibitor |
| MAPK | Mitogen-activated protein kinase |
| M3 | Monosomy 3 |
| Me | Methylation |
| MEK | Mitogen-activated protein kinase kinase |
| MITF | Melanocyte Inducing Transcription Factor |
| miRNA | MicroRNA |
| MYC | MYC Proto-Oncogene, BHLH Transcription Factor |
| NRAS | NRAS Proto-Oncogene, GTPase |
| OS | Overall survival |
| PD-1 | Programmed cell death-1 |
| PFS | Progression-free survival |
| PI3K | Phosphatidylinositol 3-kinase |
| PKC | Protein kinase C |
| PRMT | Protein arginine methyltransferase |
| PRMTi | Protein arginine methyltransferase inhibitor |
| PRAME | Preferentially expressed antigen in melanoma |
| p16, P16INK4A | Cyclin Dependent Kinase Inhibitor 2A |
| P14ARF | The alternate reading frame protein product of the CDKN2A locus |
| RASEF | RAS and EF-hand domain containing |
| RASSF1A | Ras association domain family member 1 |
| RNA | Ribonucleic acid |
| SAHA | Suberanilohydroxamic acid |
| SF3B1 | Splicing Factor 3b Subunit 1 |
| shRNA | Short hairpin RNA |
| TALE | Transcription activator-like effector |
| TET | Ten-eleven translocation protein |
| TIGIT | T-cell immunoreceptor with Ig and ITIM domains |
| TSA | Trichostatin A |
| TIMP3 | TIMP metallopeptidase inhibitor 3 |
| UM | Uveal melanoma |
| VEGF | Vascular endothelial growth factor |
| VPA | Valproic acid |
| ZBTB10 | Zinc finger and BTB domain containing 10 |
| ZFN | Zinc finger nuclease |
References
- Ramaiya, K.J.; Harbour, J.W. Current management of uveal melanoma. Expert Rev. Ophthalmol. 2007, 2, 939–946. [Google Scholar] [CrossRef]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A. Uveal melanoma. Nat. Rev. Dis. Primers 2020, 6, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.G.; Boldt, H.C. Ocular Melanoma: Advances in Diagnostic and Therapeutic Strategies; Future Medicine: London, UK, 2014. [Google Scholar]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Blum, E.S.; Yang, J.; Komatsubara, K.M.; Carvajal, R.D. Clinical management of uveal and conjunctival melanoma. Oncology (Williston Park) 2016, 30, 29–43. [Google Scholar]
- Khoja, L.; Atenafu, E.; Suciu, S.; Leyvraz, S.; Sato, T.; Marshall, E.; Keilholz, U.; Zimmer, L.; Patel, S.; Piperno-Neumann, S. Meta-Analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: An international rare cancers initiative (IRCI) ocular melanoma study. Ann. Oncol. 2019, 30, 1370–1380. [Google Scholar] [CrossRef]
- Shields, C.L.; Ganguly, A.; Bianciotto, C.G.; Turaka, K.; Tavallali, A.; Shields, J.A. Prognosis of uveal melanoma in 500 cases using genetic testing of fine-needle aspiration biopsy specimens. Ophthalmology 2011, 118, 396–401. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef]
- Versluis, M.; de Lange, M.J.; van Pelt, S.I.; Ruivenkamp, C.A.; Kroes, W.G.; Cao, J.; Jager, M.J.; Luyten, G.P.; van der Velden, P.A. Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PLoS ONE 2015, 10, e0116371. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Kalirai, H.; Dodson, A.; Faqir, S.; Damato, B.; Coupland, S. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing. Br. J. Cancer 2014, 111, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Char, D.H.; Augsburger, J.J.; Correa, Z.M.; Nudleman, E.; Aaberg, T.M., Jr.; Altaweel, M.M.; Bardenstein, D.S.; Finger, P.T. Collaborative Ocular Oncology Group report number 1: Prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology 2012, 119, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Field, M.G.; Harbour, J.W. Recent developments in prognostic and predictive testing in uveal melanoma. Curr. Opin. Ophthalmol. 2014, 25, 234. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220. e215. [Google Scholar] [CrossRef]
- Sharma, A.; Stei, M.; Fröhlich, H.; Holz, F.; Loeffler, K.; Herwig-Carl, M. Genetic and epigenetic insights into uveal melanoma. Clin. Genet. 2018, 93, 952–961. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Herlihy, N.; Dogrusöz, M.; Van Essen, T.H.; Harbour, J.W.; Van Der Velden, P.A.; Van Eggermond, M.C.; Haasnoot, G.W.; Van Den Elsen, P.J.; Jager, M.J. Skewed expression of the genes encoding epigenetic modifiers in high-risk uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1447–1458. [Google Scholar] [CrossRef]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; Van Der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Decatur, C.L.; Tarlan, B.; Oelschlager, K.M.; Stone, J.F.; Kuznetsov, J.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Epigenetic reprogramming and aberrant expression of PRAME are associated with increased metastatic risk in Class 1 and Class 2 uveal melanomas. Oncotarget 2016, 7, 59209. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. DSS1 promoter hypomethylation and overexpression predict poor prognosis in melanoma and squamous cell carcinoma patients. Hum. Pathol. 2017, 60, 137–146. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, P.A.; Metzelaar-Blok, J.A.; Bergman, W.; Monique, H.; Hurks, H.; Frants, R.R.; Gruis, N.A.; Jager, M.J. Promoter hypermethylation: A common cause of reduced p16INK4a expression in uveal melanoma. Cancer Res. 2001, 61, 5303–5306. [Google Scholar] [PubMed]
- Van der Velden, P.A.; Zuidervaart, W.; Hurks, M.H.; Pavey, S.; Ksander, B.R.; Krijgsman, E.; Frants, R.R.; Tensen, C.P.; Willemze, R.; Jager, M.J. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int. J. Cancer 2003, 106, 472–479. [Google Scholar] [CrossRef]
- Zeschnigk, M.; Tschentscher, F.; Lich, C.; Brandt, B.; Horsthemke, B.; Lohmann, D.R. Methylation analysis of several tumour suppressor genes shows a low frequency of methylation of CDKN2A and RARB in uveal melanomas. Int. J. Genom. 2003, 4, 329–336. [Google Scholar]
- Maat, W.; van der Velden, P.A.; Out-Luiting, C.; Plug, M.; Dirks-Mulder, A.; Jager, M.J.; Gruis, N.A. Epigenetic inactivation of RASSF1a in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 486–490. [Google Scholar] [CrossRef][Green Version]
- Merhavi, E.; Cohen, Y.; Avraham, B.C.R.; Frenkel, S.; Chowers, I.; Pe’er, J.; Goldenberg-Cohen, N. Promoter methylation status of multiple genes in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4403–4406. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moulin, A.P.; Clément, G.; Bosman, F.T.; Zografos, L.; Benhattar, J. Methylation of CpG island promoters in uveal melanoma. Br. J. Ophthalmol. 2008, 92, 281–285. [Google Scholar] [CrossRef]
- Calipel, A.; Abonnet, V.; Nicole, O.; Mascarelli, F.; Coupland, S.E.; Damato, B.; Mouriaux, F. Status of RASSF1A in uveal melanocytes and melanoma cells. Mol. Cancer Res. 2011, 9, 1187–1198. [Google Scholar] [CrossRef][Green Version]
- Pfeifer, G.P.; Yoon, J.-H.; Liu, L.; Tommasi, S.; Wilczynski, S.P.; Dammann, R. Methylation of the RASSF1A gene in human cancers. Biol. Chem. 2002, 383, 907–914. [Google Scholar] [CrossRef]
- Maat, W.; Beiboer, S.H.; Jager, M.J.; Luyten, G.P.; Gruis, N.A.; van der Velden, P.A. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1291–1298. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Beninati, C.; Biondo, C.; Teti, D.; Venza, I. Role of genetics and epigenetics in mucosal, uveal, and cutaneous melanomagenesis. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. -Anti-Cancer Agents) 2016, 16, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Field, M.G.; Kuznetsov, J.N.; Bussies, P.L.; Cai, L.Z.; Alawa, K.A.; Decatur, C.L.; Kurtenbach, S.; Harbour, J.W. BAP1 loss is associated with DNA methylomic repatterning in highly aggressive Class 2 uveal melanomas. Clin. Cancer Res. 2019, 25, 5663–5673. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Landreville, S.; Agapova, O.A.; Matatall, K.A.; Kneass, Z.T.; Onken, M.D.; Lee, R.S.; Bowcock, A.M.; Harbour, J.W. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin. Cancer Res. 2012, 18, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Matatall, K.A.; Agapova, O.A.; Onken, M.D.; Worley, L.A.; Bowcock, A.M.; Harbour, J.W. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer 2013, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, J.N.; Aguero, T.H.; Owens, D.A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; Rodriguez, D.A.; King, M.L.; Harbour, J.W. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci. Adv. 2019, 5, eaax1738. [Google Scholar] [CrossRef]
- Deng, S.; Calin, G.A.; Croce, C.M.; Coukos, G.; Zhang, L. Mechanisms of microRNA deregulation in human cancer. Cell Cycle 2008, 7, 2643–2646. [Google Scholar] [CrossRef]
- Li, Y.; Jia, R.; Ge, S. Role of epigenetics in uveal melanoma. Int. J. Biol. Sci. 2017, 13, 426. [Google Scholar] [CrossRef]
- Worley, L.A.; Long, M.D.; Onken, M.D.; Harbour, J.W. Micro-RNAs associated with metastasis in uveal melanoma identified by multiplexed microarray profiling. Melanoma Res. 2008, 18, 184–190. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Badhrinarayanan, N.; Biswas, J.; Krishnakumar, S. Analysis of chromosomal aberration (1, 3, and 8) and association of microRNAs in uveal melanoma. Mol. Vis. 2009, 15, 2146. [Google Scholar]
- Venza, M.; Dell’Aversana, C.; Visalli, M.; Altucci, L.; Teti, D.; Venza, I. Identification of microRNA expression patterns in cutaneous and uveal melanoma cell lines. Tumori J. 2014, 100, 4–7. [Google Scholar] [CrossRef]
- Sun, Q.; Cong, R.; Yan, H.; Gu, H.; Zeng, Y.; Liu, N.; Chen, J.; Wang, B. Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression. Oncol. Rep. 2009, 22, 563–567. [Google Scholar] [PubMed]
- Chen, X.; He, D.; Da Dong, X.; Dong, F.; Wang, J.; Wang, L.; Tang, J.; Hu, D.-N.; Yan, D.; Tu, L. MicroRNA-124a is epigenetically regulated and acts as a tumor suppressor by controlling multiple targets in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2248–2256. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.; Shen, H.; Lu, J.; Li, C.; Hu, D.-N.; Da Dong, X.; Yan, D.; Tu, L. Epigenetics, microRNAs, and carcinogenesis: Functional role of microRNA-137 in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Smolkova, B.; Horvathova, V.K.; Zmetakova, I.; Kalinkova, L.; Czanner, G.; Markova, A.; Furdova, A. Role of epigenetic deregulation in hematogenous dissemination of malignant uveal melanoma. Neoplasma 2018, 65, 840–854. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Shen, J.; Jiang, Y. MicroRNA dysregulation in uveal melanoma: A new player enters the game. Oncotarget 2015, 6, 4562. [Google Scholar] [CrossRef]
- Melia, M.; Moy, C.S.; Reynolds, S.M.; Hayman, J.A.; Murray, T.G.; Hovland, K.R.; Earle, J.D.; Kurinij, N.; Dong, L.M.; Miskala, P.H. Quality of life after iodine 125 brachytherapy vs enucleation for choroidal melanoma: 5-year results from the Collaborative Ocular Melanoma Study: COMS QOLS Report No. 3. Arch. Ophthalmol. (Chicago Ill.: 1960) 2006, 124, 226–238. [Google Scholar]
- Furdova, A.; Sramka, M.; Chorvath, M.; Kralik, G.; Furda, R.; Gregus, M. Clinical experience of stereotactic radiosurgery at a linear accelerator for intraocular melanoma. Melanoma Res. 2017, 27, 463–468. [Google Scholar] [CrossRef]
- Puusaari, I.; Damato, B.; Kivelä, T. Transscleral local resection versus iodine brachytherapy for uveal melanomas that are large because of tumour height. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 522–533. [Google Scholar] [CrossRef]
- Bechrakis, N.E.; Petousis, V.; Willerding, G.; Krause, L.; Wachtlin, J.; Stroux, A.; Foerster, M.H. Ten-Year results of transscleral resection of large uveal melanomas: Local tumour control and metastatic rate. Br. J. Ophthalmol. 2010, 94, 460–466. [Google Scholar] [CrossRef]
- Kivelä, T.; Puusaari, I.; Damato, B. Transscleral resection versus iodine brachytherapy for choroidal malignant melanomas 6 mm or more in thickness: A matched case–Control study. Ophthalmology 2003, 110, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Furdova, A.; Sramka, M. Uveal Malignant Melanoma and Stereotactic Radiosurgery: Intraocular Uveal Melanoma and One-Day Session Stereotactic Radiosurgery at Linear Accelerator, 1st ed.; LAP LAMBERT Academic Publishing: Saarbrücken, Germany, 2014; p. 183. [Google Scholar]
- Furdova, A.; Strmen, P.; Waczulikova, I.; Chorvath, M.; Sramka, M.; Slezak, P. One-Day session LINAC–based stereotactic radiosurgery of posterior uveal melanoma. Eur. J. Ophthalmol. 2012, 22, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Damato, B. Progress in the management of patients with uveal melanoma. The 2012 Ashton Lecture. Eye 2012, 26, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Sagoo, M.S.; Shields, C.L.; Mashayekhi, A.; Freire, J.; Emrich, J.; Reiff, J.; Komarnicky, L.; Shields, J.A. Plaque radiotherapy for juxtapapillary choroidal melanoma: Tumor control in 650 consecutive cases. Ophthalmology 2011, 118, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.U.; Shields, C.L.; Bianciotto, C.G.; Iturralde, J.; Al-Dahmash, S.A.; Say, E.A.T.; Badal, J.; Mashayekhi, A.; Shields, J.A. Intravitreal bevacizumab at 4-month intervals for prevention of macular edema after plaque radiotherapy of uveal melanoma. Ophthalmology 2014, 121, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.K.; Quivey, J.M.; Daftari, I.K.; Weinberg, V.; Cole, T.B.; Patel, K.; Castro, J.R.; Phillips, T.L.; Char, D.H. Long-term Results of the UCSF-LBNL Randomized Trial: Charged Particle With Helium Ion Versus Iodine-125 Plaque Therapy for Choroidal and Ciliary Body Melanoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 376–383. [Google Scholar] [CrossRef]
- Gragoudas, E.; Li, W.; Goitein, M.; Lane, A.M.; Munzenrider, J.E.; Egan, K.M. Evidence-based estimates of outcome in patients irradiated for intraocular melanoma. Arch. Ophthalmol. 2002, 120, 1665–1671. [Google Scholar] [CrossRef]
- Turcotte, S.; Bergeron, D.; Rousseau, A.P.; Mouriaux, F. Primary transpupillary thermotherapy for choroidal indeterminate melanocytic lesions. Can. J. Ophthalmol. 2014, 49, 464–467. [Google Scholar] [CrossRef]
- Binkley, E.; Triozzi, P.L.; Rybicki, L.; Achberger, S.; Aldrich, W.; Singh, A. A prospective trial of adjuvant therapy for high-Risk uveal melanoma: Assessing 5-Year survival outcomes. Br. J. Ophthalmol. 2020, 104, 524–528. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Rodrigues, M.J.; Servois, V.; Pierron, G.; Gastaud, L.; Negrier, S.; Levy-Gabriel, C.; Lumbroso, L.; Cassoux, N.; Bidard, F.-C.; et al. A randomized multicenter phase 3 trial of adjuvant fotemustine versus surveillance in high risk uveal melanoma (UM) patients (FOTEADJ). J. Clin. Oncol. 2017, 35, 9502. [Google Scholar] [CrossRef]
- Voelter, V.; Schalenbourg, A.; Pampallona, S.; Peters, S.; Halkic, N.; Denys, A.; Goitein, G.; Zografos, L.; Leyvraz, S. Adjuvant intra-Arterial hepatic fotemustine for high-Risk uveal melanoma patients. Melanoma Res. 2008, 18, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.M.; Egan, K.M.; Harmon, D.; Holbrook, A.; Munzenrider, J.E.; Gragoudas, E.S. Adjuvant interferon therapy for patients with uveal melanoma at high risk of metastasis. Ophthalmology 2009, 116, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- McLean, I.W.; Berd, D.; Mastrangelo, M.J.; Shields, J.A.; Davidorf, F.H.; Grever, M.; Makley, T.A.; Gamel, J.W. A randomized study of methanol-Extraction residue of bacille Calmette-Guerin as postsurgical adjuvant therapy of uveal melanoma. Am. J. Ophthalmol. 1990, 110, 522–526. [Google Scholar] [CrossRef]
- Fountain, E.; Bassett, R.L.; Cain, S.; Posada, L.; Gombos, D.S.; Hwu, P.; Bedikian, A.; Patel, S.P. Adjuvant Ipilimumab in High-Risk Uveal Melanoma. Cancers 2019, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, M.E.; Orloff, M.; Sato, R.; Chervoneva, I.; Shields, C.L.; Shields, J.A.; Mastrangelo, M.J.; Sato, T. Adjuvant Sunitinib in High-Risk Patients with Uveal Melanoma: Comparison with Institutional Controls. Ophthalmology 2018, 125, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef]
- Algazi, A.P.; Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.I. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016, 122, 3344–3353. [Google Scholar] [CrossRef]
- Khoja, L.; Atenafu, E.G.; Joshua, A.M.; Group, I.R.C.s.I.-O.M. Meta-Analysis of phase II trials in metastatic uveal melanoma (MUM) to determine progression-free (PFS) and overall survival (OS) benchmarks for future phase II trials: An irci-ocular melanoma initiative. Am. Soc. Clin. Oncol. 2016, 34 (Suppl. 15), 9567. [Google Scholar] [CrossRef]
- Willson, J.K.; Albert, D.M.; Diener-West, M.; McCaffrey, L.; Moy, C.S.; Scully, R.E. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the collaborative ocular melanoma study (coms) coms report no. 15. Arch Ophthalmol. 2001, 119, 670–676. [Google Scholar]
- Spagnolo, F.; Grosso, M.; Picasso, V.; Tornari, E.; Pesce, M.; Queirolo, P. Treatment of metastatic uveal melanoma with intravenous fotemustine. Melanoma Res. 2013, 23, 196–198. [Google Scholar] [CrossRef]
- Augsburger, J.J.; Corrêa, Z.M.; Shaikh, A.H. Effectiveness of treatments for metastatic uveal melanoma. Am. J. Ophthalmol. 2009, 148, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Schmittel, A.; Schmidt-Hieber, M.; Martus, P.; Bechrakis, N.; Schuster, R.; Siehl, J.; Foerster, M.; Thiel, E.; Keilholz, U. A randomized phase II trial of gemcitabine plus treosulfan versus treosulfan alone in patients with metastatic uveal melanoma. Ann. Oncol. 2006, 17, 1826–1829. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Caltabiano, G.; Queirolo, P. Uveal melanoma. Cancer Treat. Rev. 2012, 38, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Homsi, J.; Bedikian, A.Y.; Papadopoulos, N.E.; Kim, K.B.; Hwu, W.-J.; Mahoney, S.L.; Hwu, P. Phase 2 open-label study of weekly docosahexaenoic acid–paclitaxel in patients with metastatic uveal melanoma. Melanoma Res. 2010, 20, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.G.; Navrazhina, K.; Ding, F.; Bhatia, R.; Tsai, K.; Abbate, K.; Durden, B.; Eroglu, Z.; Bhatia, S.; Park, S.; et al. Ipilimumab plus nivolumab for patients with metastatic uveal melanoma: A multicenter, retrospective study. J. Immunother. Cancer 2020, 8, e000331. [Google Scholar] [CrossRef] [PubMed]
- Swaika, A.; Crozier, J.A.; Joseph, R.W. Vemurafenib: An evidence-based review of its clinical utility in the treatment of metastatic melanoma. Drug Des. Dev. Ther. 2014, 8, 775. [Google Scholar]
- Trinh, V.A.; Davis, J.E.; Anderson, J.E.; Kim, K.B. Dabrafenib therapy for advanced melanoma. Ann. Pharmacother. 2014, 48, 519–529. [Google Scholar] [CrossRef]
- Cruz, F.; Rubin, B.P.; Wilson, D.; Town, A.; Schroeder, A.; Haley, A.; Bainbridge, T.; Heinrich, M.C.; Corless, C.L. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 2003, 63, 5761–5766. [Google Scholar]
- Park, J.J.; Diefenbach, R.J.; Byrne, N.; Kefford, R.; Long, G.V.; Scolyer, R.A.; Gray, E.; Carlino, M.S.; Rizos, H. Circulating tumor DNA (ctDNA) in patients (pts) with metastatic uveal melanoma (UM) treated with protein kinase C inhibitor (PKCi). J. Clin. Oncol. 2020, 38, e22054. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H. Effect of selumetinib vs chemotherapy on progression-Free survival in uveal melanoma: A randomized clinical trial. Jama 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Patel, S.P.; Kim, K.B.; Papadopoulos, N.E.; Hwu, W.-J.; Hwu, P.; Prieto, V.G.; Bar-Eli, M.; Zigler, M.; Dobroff, A.; Bronstein, Y. A phase II study of gefitinib in patients with metastatic melanoma. Melanoma Res. 2011, 21, 357. [Google Scholar] [CrossRef] [PubMed]
- Zeldis, J.; Heller, C.; Seidel, G.; Yuldasheva, N.; Stirling, D.; Shutack, Y.; Libutti, S. A randomized phase II trial comparing two doses of lenalidomide for the treatment of stage IV ocular melanoma. J. Clin. Oncol. 2009, 27, e20012. [Google Scholar]
- Reiriz, A.B.; Richter, M.F.; Fernandes, S.; Cancela, A.I.; Costa, T.D.; Di Leone, L.P.; Schwartsmann, G. Phase II study of thalidomide in patients with metastatic malignant melanoma. Melanoma Res. 2004, 14, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Guenterberg, K.D.; Grignol, V.P.; Relekar, K.V.; Varker, K.A.; Chen, H.X.; Kendra, K.L.; Olencki, T.E.; Carson III, W.E. A pilot study of bevacizumab and interferon-α2b in ocular melanoma. Am. J. Clin. Oncol. 2011, 34, 87. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Servois, V.; Bidard, F.-C.; Mariani, P.; Plancher, C.; Diallo, A.; Vago-Ady, N.; Desjardins, L. BEVATEM: Phase II study of bevacizumab (B) in combination with temozolomide (T) in patients (pts) with first-line metastatic uveal melanoma (MUM): Final results. Am. Soc. Clin. Oncol. 2013, 31 (Suppl. 15), 9057. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Frankel, P.; Margolin, K.A.; Christensen, S.; Ruel, C.; Shipe-Spotloe, J.; Gandara, D.R.; Chen, A.; Kirkwood, J.M. Aflibercept (VEGF Trap) in inoperable stage III or stage iv melanoma of cutaneous or uveal origin. Clin. Cancer Res. 2011, 17, 6574–6581. [Google Scholar] [CrossRef]
- Penel, N.; Delcambre, C.; Durando, X.; Clisant, S.; Hebbar, M.; Negrier, S.; Fournier, C.; Isambert, N.; Mascarelli, F.; Mouriaux, F. O-Mel-Inib: A Cancero-Pole Nord-Ouest multicenter phase II trial of high-Dose imatinib mesylate in metastatic uveal melanoma. Investig. New Drugs 2008, 26, 561–565. [Google Scholar] [CrossRef]
- Hofmann, U.B.; Kauczok-Vetter, C.S.; Houben, R.; Becker, J.C. Overexpression of the KIT/SCF in uveal melanoma does not translate into clinical efficacy of imatinib mesylate. Clin. Cancer Res. 2009, 15, 324–329. [Google Scholar] [CrossRef]
- Bhatia, S.; Moon, J.; Margolin, K.A.; Weber, J.S.; Lao, C.D.; Othus, M.; Aparicio, A.M.; Ribas, A.; Sondak, V.K. Phase II trial of sorafenib in combination with carboplatin and paclitaxel in patients with metastatic uveal melanoma: SWOG S0512. PLoS ONE 2012, 7, e48787. [Google Scholar] [CrossRef]
- Mahipal, A.; Tijani, L.; Chan, K.; Laudadio, M.; Mastrangelo, M.J.; Sato, T. A pilot study of sunitinib malate in patients with metastatic uveal melanoma. Melanoma Res. 2012, 22, 440–446. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Morel, D.; Almouzni, G.; Soria, J.-C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2017, 28, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Klisovic, D.D.; Katz, S.E.; Effron, D.; Klisovic, M.I.; Wickham, J.; Parthun, M.R.; Guimond, M.; Marcucci, G. Depsipeptide (FR901228) inhibits proliferation and induces apoptosis in primary and metastatic human uveal melanoma cell lines. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2390–2398. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klisovic, D.D.; Klisovic, M.I.; Effron, D.; Liu, S.; Marcucci, G.; Katz, S.E. Depsipeptide inhibits migration of primary and metastatic uveal melanoma cell lines in vitro: A potential strategy for uveal melanoma. Melanoma Res. 2005, 15, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhou, J.; Jin, B.; Pan, J. Class III-specific HDAC inhibitor Tenovin-6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Jansen, Y.J.; Verset, G.; Schats, K.; Van Dam, P.-J.; Seremet, T.; Kockx, M.; Van Laethem, J.-L.B.; Neyns, B. Phase I clinical trial of decitabine (5-aza-2′-deoxycytidine) administered by hepatic arterial infusion in patients with unresectable liver-predominant metastases. ESMO Open 2019, 4, e000464. [Google Scholar] [CrossRef]
- Gonçalves, J.; Emmons, M.F.; Faião-Flores, F.; Aplin, A.E.; Harbour, J.W.; Licht, J.D.; Wink, M.R.; Smalley, K.S. Decitabine limits escape from MEK inhibition in uveal melanoma. Pigment Cell Melanoma Res. 2020, 33, 507–514. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Jin, Y.; Jiang, S.; Pan, J. In vitro and in vivo anti-uveal melanoma activity of JSL-1, a novel HDAC inhibitor. Cancer Lett. 2017, 400, 47–60. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Biondo, C.; Lentini, M.; Catalano, T.; Teti, D.; Venza, I. Epigenetic regulation of p14ARF and p16INK4A expression in cutaneous and uveal melanoma. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2015, 1849, 247–256. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Ambrosini, G.; Sawle, A.D.; Musi, E.; Schwartz, G.K. BRD4-targeted therapy induces Myc-independent cytotoxicity in Gnaq/11-mutatant uveal melanoma cells. Oncotarget 2015, 6, 33397. [Google Scholar] [CrossRef] [PubMed]
- Plimack, E.R.; Stewart, D.J.; Issa, J.P. Combining epigenetic and cytotoxic therapy in the treatment of solid tumors. J. Clin. Oncol. 2007, 25, 4519–4521. [Google Scholar] [CrossRef] [PubMed]
- Coral, S.; Sigalotti, L.; Covre, A.; Nicolay, H.J.; Natali, P.G.; Maio, M. 5-AZA-2′-deoxycytidine in cancer immunotherapy: A mouse to man story. Cancer Res. 2007, 67, 2900–2901; author reply 2901–2902. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bi, G.; Jiang, G. The molecular mechanism of HDAC inhibitors in anticancer effects. Cell. Mol. Immunol. 2006, 3, 285–290. [Google Scholar]
- Xu, W.; Parmigiani, R.; Marks, P. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Dimitroulis, D.; Spartalis, E.; Margonis, G.-A.; Schizas, D.; Deskou, I.; Doula, C.; Magkouti, E. Targeting histone deacetylases in malignant melanoma: A future therapeutic agent or just great expectations? Anticancer Res. 2017, 37, 5355–5362. [Google Scholar] [PubMed]
- Moschos, M.M.; Dettoraki, M.; Androudi, S.; Kalogeropoulos, D.; Lavaris, A.; Garmpis, N.; Damaskos, C.; Garmpi, A.; Tsatsos, M. The role of histone deacetylase inhibitors in uveal melanoma: Current evidence. Anticancer Res. 2018, 38, 3817–3824. [Google Scholar] [CrossRef]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 5. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Tuscan, M.D.; Harbour, J.W. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J. Mol. Diagn. 2010, 12, 461–468. [Google Scholar] [CrossRef]
- Haas, N.; Quirt, I.; Hotte, S.; McWhirter, E.; Polintan, R.; Litwin, S.; Adams, P.; McBryan, T.; Wang, L.; Martin, L. Phase II trial of vorinostat in advanced melanoma. Investig. New Drugs 2014, 32, 526–534. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. Epidrugs in the immunotherapy of cutaneous and uveal melanoma. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. -Anti-Cancer Agents) 2017, 17, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Leon-Ferre, R.; Laux, D.; Deutsch, J.; Smith, B.J.; Frees, M.; Milhem, M. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother. Pharmacol. 2014, 74, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.-L.; Tian, M.; Li, X.; Li, J.-J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777. [Google Scholar] [CrossRef]
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cancer induction and suppression with transcriptional control and epigenome editing technologies. J. Hum. Genet. 2018, 63, 187–194. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487. [Google Scholar] [CrossRef]
- Akram, F.; Haq, I.U.; Ahmed, Z.; Khan, H.; Ali, M. CRISPR-Cas9, A Promising Therapeutic Tool for Cancer Therapy: A Review. Protein Pept. Lett. 2020. [Google Scholar] [CrossRef]
- Moses, C.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Hallmarks of cancer: The CRISPR generation. Eur. J. Cancer 2018, 93, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247. e217. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells 2020, 9, 993. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug name | Function | Preclinical Model | References |
|---|---|---|---|
| Valproic acid | ↑ Proliferation ↓ G1 cell cycle arrest ↓ Clonogenicity Shift Class 2 gene expression profile → Class 1 | Primary UM cells, 92.1, OCM1A, Mel202, NOD SCID gamma mice | [35] |
| Trichostatin A | ↓ Cell growth ↓ Migration and invasion ↓ Proliferation ↓ G1 cell cycle arrest | M619, C918, OCM-1, MUM-2b, -2c | [35,95] |
| Depsipeptide | ↓ Cell growth ↑ Apoptosis ↓ Migration | M619, C918, OCM-1, MUM-2b, -2c | [95,96] |
| Tenovin-6 | ↓ Growth of UM cells ↑ P53 expression ↑ Apoptosis ↓ Viability of UM cells ↓ β-catenin ↓ Cancer stem cells Blocks WNT/β-catenin signaling | 92.1, Mel 270, Omm 1, Omm 2.3 | [97] |
| Panobinostat | ↑ Morphological differentiation G1 cell-cycle arrest ↑ Melanocytic gene-expression ↓ Fraction of viable UM cells Shift to a more differentiated phenotype | 92-1, OCM1A, Mel202, NOD SCID gamma mice | [35] |
| Decitabine | ↑ Anti-proliferative activity of trametinib ↑ Cell survival ↑ CDK inhibitor expression ↑ Pro-apoptotic BCL-2-like protein 11 (BIM) ↓ Growth of UM cell in xenografts ↑ Activity of MEKi | 92-1, Mel270, MP41, Mel202, Mel290, CBySmn.CB17-Prdkc scid/j mice, Phase I clinical study | [98,99] |
| JSL-1 | ↓ Migration and invasion ↑ Expression of pro-apoptotic BH3 gene ↓ Growth of UM xenografts ↓ Cell proliferation | 92-1, Mel270, OMM1, OMM2.3, NOD-SCID mice | [100] |
| Vorinostat | ↑ P14ARF expression ↓ UM cell growth ↓ Migration and invasion Shift Class 2 gene expression profile → Class 1 | OCM-1, OCM3, 92-1, OMM-2.5, UMel-1, UMel-2 | [101] |
| BRD4 inhibitors | ↓ MYC and MYC-dependent genes ↓ Tumor growth ↑ Apoptosis | OMM1.3, Mel270, Mel202, SCID-beige and Vk*myc mice | [102,103] |
| Drug Name | Recruitment Status | Phase | Dose | Regimen | Estimated Enrollment | Cancer Type | Clinical Trial Identifier |
|---|---|---|---|---|---|---|---|
| Vorinostat | Withdrawn | I | 400 mg | Once a day for 15 days | 10 | UM | NCT03022565 |
| Vorinostat | Suspended | II | Twice a day 3 days weekly for 4 weeks | 40 | UM | NCT01587352 | |
| Vorinostat | Completed | II | Once a day for 4 weeks | 32 | UM | NCT00121225 | |
| Entinostat | Active, not recruiting | II | 5 mg | Once a day for a maximum of 24 weeks | 29 | UM | NCT02697630 |
| Entinostat | Completed | II | Once a day, repeat every 2 weeks/once a day, repeat every 6 weeks | 75 | Choroid melanoma $ | NCT00020579 | |
| Valproic Acid | Recruiting | II | Daily for 6 months | 150 | UM | NCT02068586 | |
| PLX2853 | Recruiting | I/II | Dose Escalation/ Expansion | 166 | UM * | NCT03297424 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chokhachi Baradaran, P.; Kozovska, Z.; Furdova, A.; Smolkova, B. Targeting Epigenetic Modifications in Uveal Melanoma. Int. J. Mol. Sci. 2020, 21, 5314. https://doi.org/10.3390/ijms21155314
Chokhachi Baradaran P, Kozovska Z, Furdova A, Smolkova B. Targeting Epigenetic Modifications in Uveal Melanoma. International Journal of Molecular Sciences. 2020; 21(15):5314. https://doi.org/10.3390/ijms21155314
Chicago/Turabian StyleChokhachi Baradaran, Pooneh, Zuzana Kozovska, Alena Furdova, and Bozena Smolkova. 2020. "Targeting Epigenetic Modifications in Uveal Melanoma" International Journal of Molecular Sciences 21, no. 15: 5314. https://doi.org/10.3390/ijms21155314
APA StyleChokhachi Baradaran, P., Kozovska, Z., Furdova, A., & Smolkova, B. (2020). Targeting Epigenetic Modifications in Uveal Melanoma. International Journal of Molecular Sciences, 21(15), 5314. https://doi.org/10.3390/ijms21155314