The Smallest Isoform of Metridia longa Luciferase as a Fusion Partner for Hybrid Proteins



Abstract
1. Introduction
2. Results and Discussion
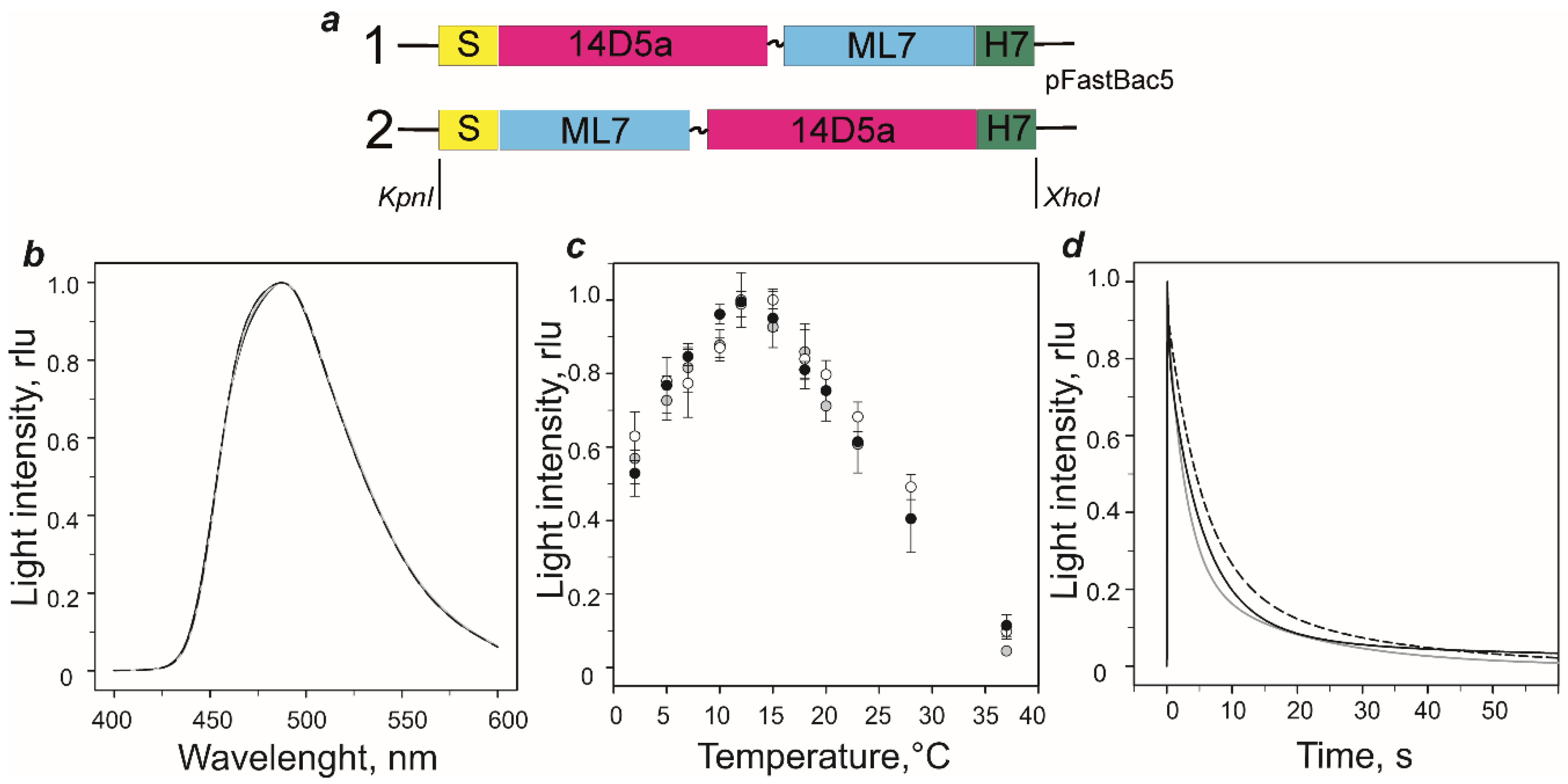
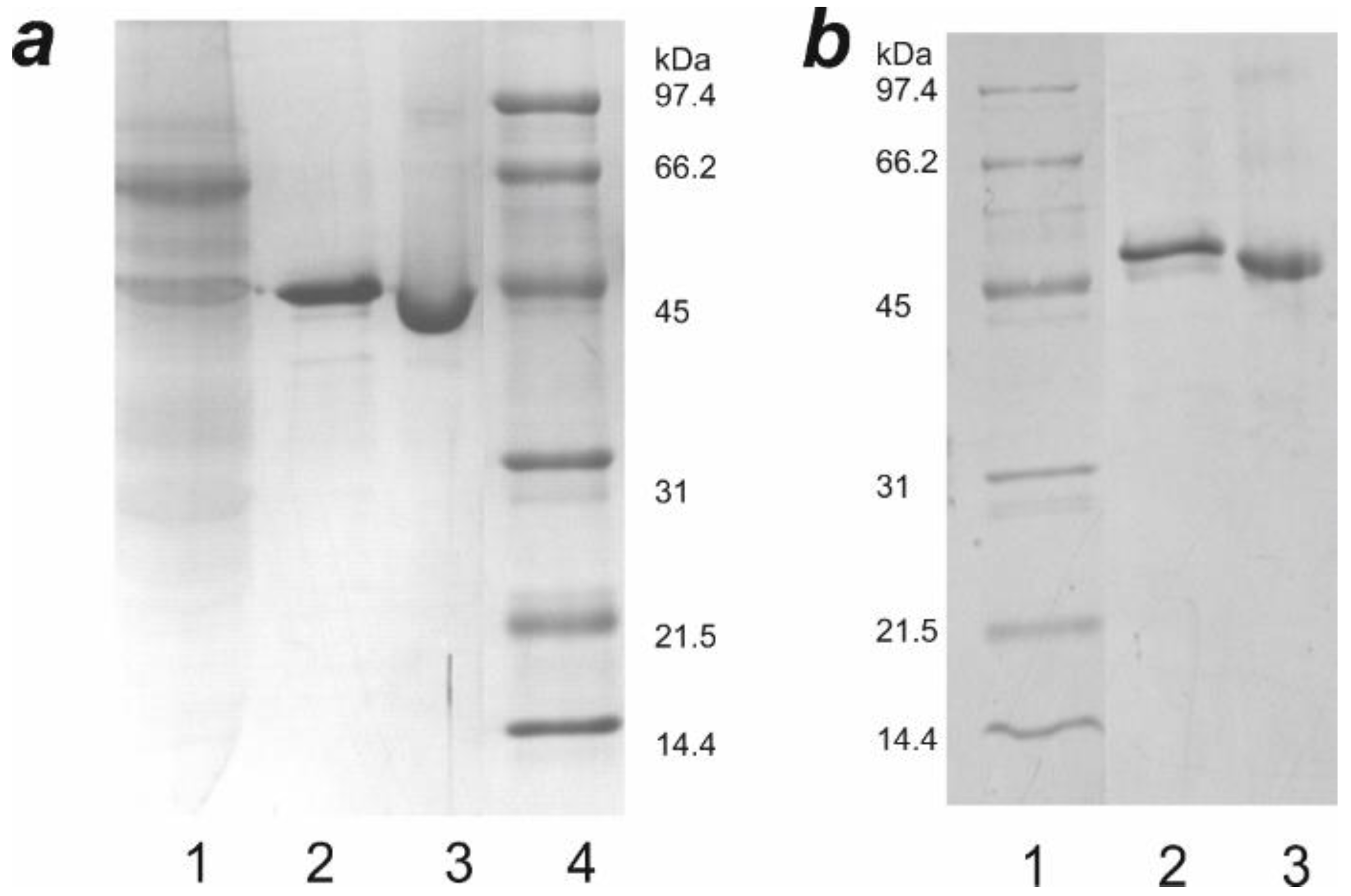
2.1. Genetic Constructs and Bioluminescent Properties of Hybrid Proteins
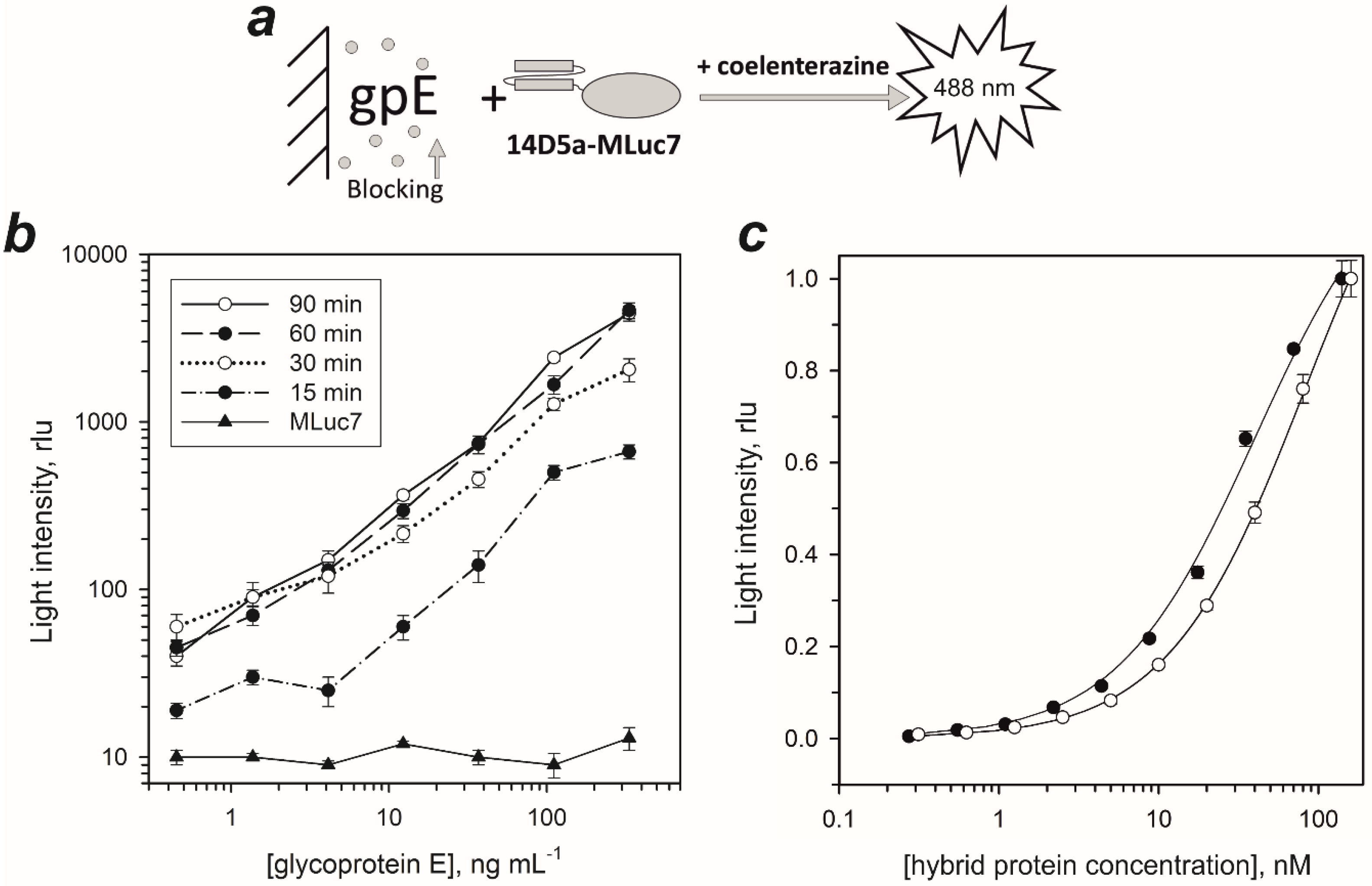
2.2. Dissociation Constants of Hybrid Proteins
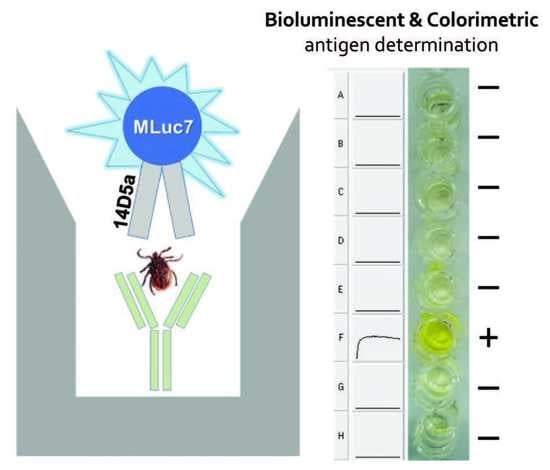
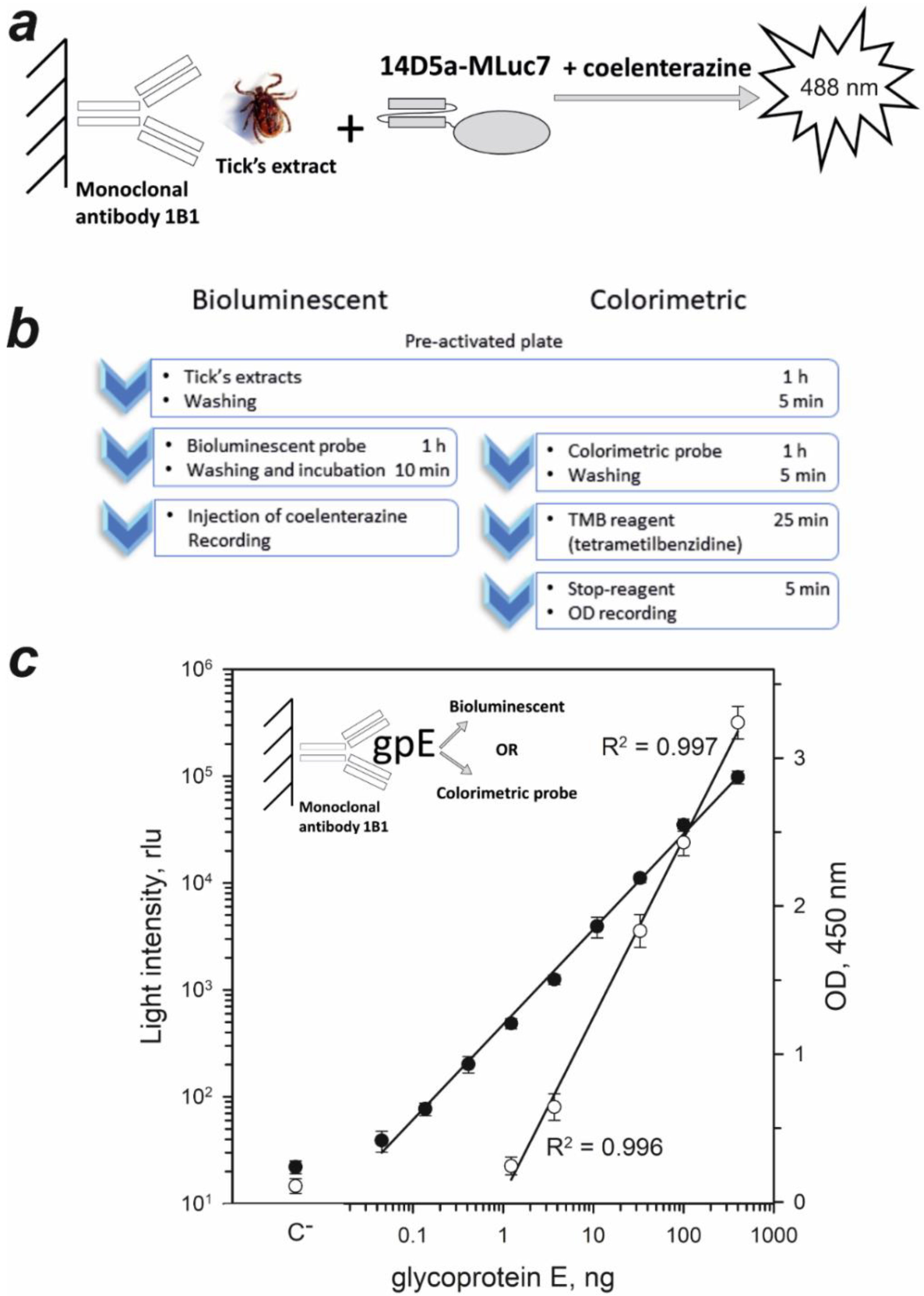
2.3. Sandwich-Type Immunoassay of TBEV Native gpE in Tick Extracts
3. Materials and Methods
3.1. Materials
3.2. Genetic Constructs
3.3. Expression and Purification of Hybrid Proteins
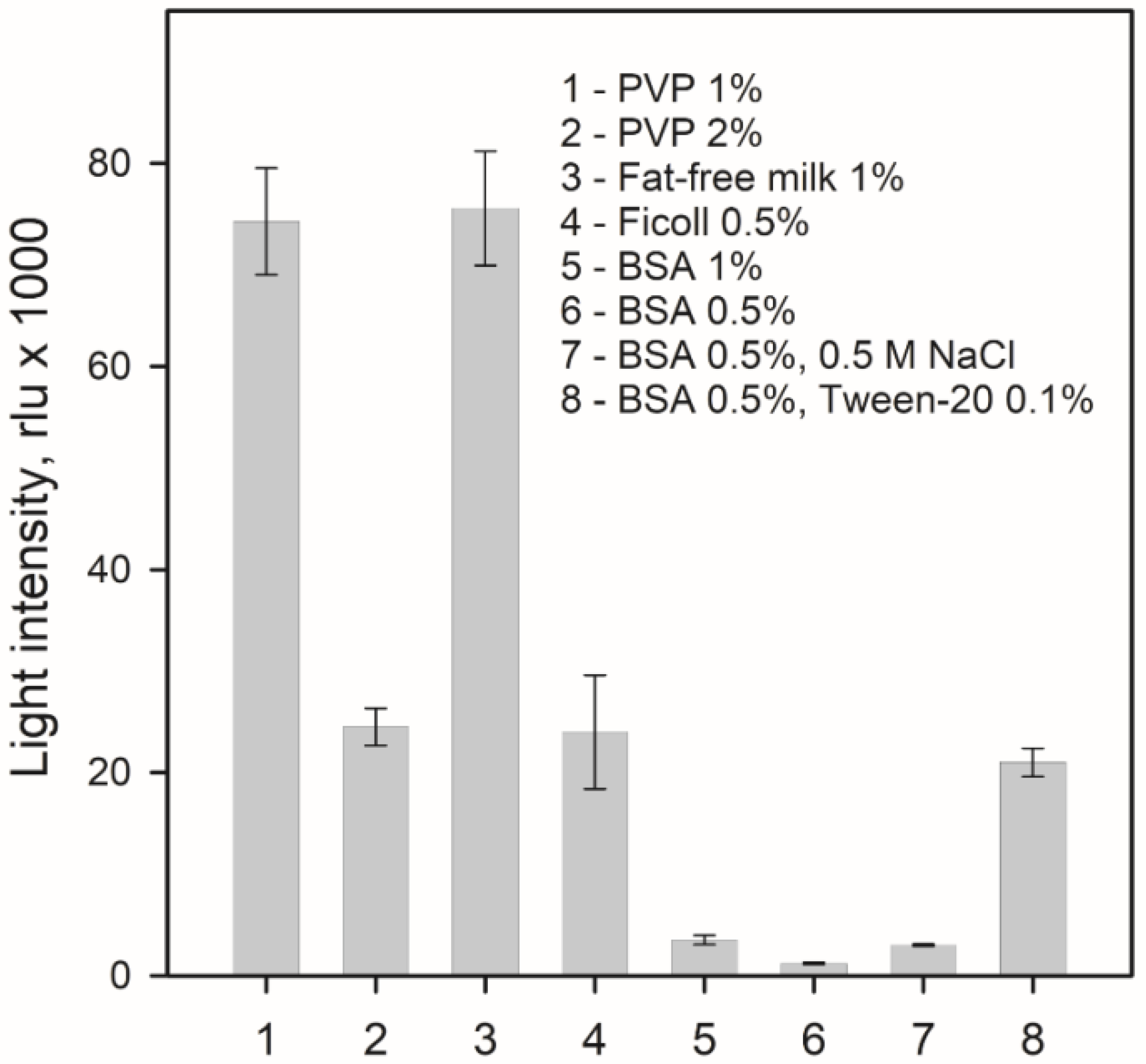
3.4. Bioluminescence Assay, Spectral Measurements, and Thermal Unfolding
3.5. Determination of Dissociation Constant
3.6. Sandwich-Type Bioluminescent Immunoassay
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MLuc7 | The shortest 16.5 kDa isoform of Metridia luciferase |
| TBEV | Tick-borne encephalitis virus |
| gpE | Glycoprotein E |
| scFv | Single-chain variable fragment |
| rlu | Relative light units |
Appendix A



References
- Lewis, J.C.; Daunert, S. Photoproteins as luminescent labels in binding assay. Fresenius J. Anal. Chem. 2000, 366, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Frank, L.A.; Krasitskaya, V.V. Application of enzyme bioluminescence for medical diagnostics. Adv. Biochem. Eng. Biotechnol. 2014, 144, 175–197. [Google Scholar] [PubMed]
- Smirnova, D.V.; Ugarova, N.N. Firefly luciferase-based fusion proteins and their applications in bioanalysis. Photochem. Photobiol. 2017, 93, 436–447. [Google Scholar] [CrossRef]
- Markova, S.V.; Larionova, M.D.; Vysotski, E.S. Shining light on the secreted luciferases of marine copepods: Current knowledge and applications. Photochem. Photobiol. 2019, 95, 705–721. [Google Scholar] [CrossRef]
- Shimomura, O. Bioluminescence: Chemical Principles and Methods; World Scientific: Hackensack, NJ, USA, 2006. [Google Scholar]
- Head, J.F.; Inouye, S.; Teranishi, K.; Shimomura, O. The crystal structure of the photoprotein aequorin at 2.3 Å resolution. Nature 2000, 405, 372–376. [Google Scholar] [CrossRef]
- Liu, Z.J.; Vysotski, E.S.; Deng, L.; Lee, J.; Rose, J.; Wang, B.C. Atomic resolution structure of obelin: Soaking with calcium enhances electron density of the second oxygen atom substituted at the C2-position of coelenterazine. Biochem. Biophys. Res. Commun. 2003, 311, 433–439. [Google Scholar] [CrossRef]
- Vysotski, E.S.; Markova, S.V.; Frank, L.A. Calcium-regulated photoproteins of marine coelenterates. Mol. Biol. 2006, 40, 355–367. [Google Scholar] [CrossRef]
- Shimomura, O.; Johnson, F.H. Regeneration of the photoprotein aequorin. Nature 1975, 256, 236–238. [Google Scholar] [CrossRef]
- Viviani, V.R. The origin, diversity, and structure function relationships of insect luciferases. Cell. Mol. Life Sci. 2002, 59, 1833–1850. [Google Scholar] [CrossRef]
- Niwa, K.; Ichino, Y.; Ohmiya, Y. Quantum yield measurements of firefly bioluminescence reactions using a commercial luminometer. Chem. Lett. 2010, 39, 291–293. [Google Scholar] [CrossRef]
- Titushin, M.S.; Markova, S.V.; Frank, L.A.; Malikova, N.P.; Stepanyuk, G.A.; Lee, J.; Vysotski, E.S. Coelenterazine-binding protein of Renilla muelleri: cDNA cloning, overexpression, and characterization as a substrate of luciferase. Photochem. Photobiol. Sci. 2008, 7, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.C.; Hori, K.; Cormier, M.J. Purification and properties of Renilla reniformis luciferase. Biochemistry 1977, 16, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Loening, A.M.; Fenn, T.D.; Wu, A.M.; Gambhir, S.S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein. Eng. Des. Sel. 2006, 19, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Stepanyuk, G.A.; Unch, J.; Malikova, N.P.; Markova, S.V.; Lee, J.; Vysotski, E.S. Coelenterazine-v ligated to Ca2+-triggered coelenterazine-binding protein is a stable and efficient substrate of the red-shifted mutant of Renilla muelleri luciferase. Anal. Bioanal. Chem. 2010, 398, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.; Szent-Gyorgyi, C. Luciferases, Fluorescent Proteins, Nucleic Acids Encoding the Luciferases and Fluorescent Proteins and the Use Thereof in Diagnostics. WO9949019A2, 30 September 1999. [Google Scholar]
- Markova, S.V.; Golz, S.; Frank, L.A.; Kalthof, B.; Vysotski, E.S. Cloning and expression of cDNA for a luciferase from the marine copepod Metridia longa. A novel secreted bioluminescent reporter enzyme. J. Biol. Chem. 2004, 279, 3212–3217. [Google Scholar] [CrossRef]
- Borisova, V.V.; Frank, L.A.; Markova, S.V.; Burakova, L.P.; Vysotski, E.S. Recombinant Metridia luciferase isoforms: Expression, refolding and applicability for in vitro assay. Photochem. Photobiol. Sci. 2008, 7, 1025–1031. [Google Scholar] [CrossRef]
- Markova, S.V.; Larionova, M.D.; Burakova, L.P.; Vysotski, E.S. The smallest natural high-active luciferase: Cloning and characterization of novel 16.5-kDa luciferase from copepod Metridia longa. Biochem. Biophys. Res. Commun. 2015, 457, 77–82. [Google Scholar] [CrossRef]
- Larionova, M.D.; Markova, S.V.; Vysotski, E.S. The novel extremely psychrophilic luciferase from Metridia longa: Properties of a high-purity protein produced in insect cells. Biochem. Biophys. Res. Commun. 2017, 483, 772–778. [Google Scholar] [CrossRef]
- Markova, S.V.; Burakova, L.P.; Vysotski, E.S. High-active truncated luciferase of copepod Metridia longa. Biochem. Biophys. Res. Commun. 2012, 417, 98–103. [Google Scholar] [CrossRef]
- Inouye, S.; Sahara, Y. Identification of two catalytic domains in a luciferase secreted by the copepod Gaussia princeps. Biochem. Biophys. Res. Commun. 2008, 365, 96–101. [Google Scholar] [CrossRef]
- Larionova, M.D.; Markova, S.V.; Vysotski, E.S. Bioluminescent and structural features of native folded Gaussia luciferase. J. Photochem. Photobiol. B 2018, 183, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Markova, S.V.; Vysotski, E.S. Coelenterazine-dependent luciferases. Biochem. (Mosc.) 2015, 80, 714–732. [Google Scholar] [CrossRef]
- Tannous, B.A.; Teng, J. Secreted blood reporters: Insights and applications. Biotechnol. Adv. 2011, 29, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Verhaegent, M.; Christopoulos, T.K. Recombinant gaussia luciferase. Overexpression, purification, and analytical application of a bioluminescent reporter for DNA hybridization. Anal. Chem. 2002, 74, 4378–4385. [Google Scholar] [CrossRef]
- Moutsiopoulou, A.; Hunt, E.; Broyles, D.; Pereira, C.A.; Woodward, K.; Dikici, E.; Kaifer, A.; Daunert, S.; Deo, S.K. Bioorthogonal protein conjugation: Application to the development of a highly sensitive bioluminescent immunoassay for the detection of interferon-ɣ. Bioconjug. Chem. 2017, 28, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Ustinova, J.; Zusinaite, E.; Utt, M.; Metsküla, K.; Reimand, K.; Huchaiah, V.; Merits, A.; Uibo, R. Development of a luciferase-based system for the detection of ZnT8 autoantibodies. J. Immunol. Methods 2014, 405, 67–73. [Google Scholar] [CrossRef]
- Oyama, H.; Morita, I.; Kiguchi, Y.; Miyake, S.; Moriuchi, A.; Akisada, T.; Niwa, T.; Kobayashi, N. Gaussia luciferase as a genetic fusion partner with antibody fragments for sensitive immunoassay monitoring of clinical biomarkers. Anal. Chem. 2015, 87, 12387–12395. [Google Scholar] [CrossRef]
- Venisnik, K.M.; Olafsen, T.; Gambhir, S.S.; Wu, A.M. Fusion of Gaussia luciferase to an engineered anti-carcinoembryonic antigen (CEA) antibody for in vivo optical imaging. Mol. Imaging Biol. 2007, 9, 267–277. [Google Scholar] [CrossRef]
- Patel, K.G.; Ng, P.; Kuo, C.C.; Levy, S.; Levy, R.; Swartz, J.R. Cell-free production of Gaussia princeps luciferase-antibody fragment bioconjugates for ex vivo detection of tumor cells. Biochem. Biophys. Res. Commun. 2009, 390, 971–976. [Google Scholar] [CrossRef]
- Ng’ang´a, P.N.; Ebner, J.K.; Plessner, M.; Aktories, K.; Schmidt, G. Engineering Photorhabdus luminescens toxin complex (PTC) into a recombinant injection nanomachine. Life Sci. Alliance. 2019, 2, e20190048. [Google Scholar]
- Gopalakrishnan, R.; Matta, H.; Choi, S.; Natarajan, V.; Prins, R.; Gong, S.; Zenunovic, A.; Narasappa, N.N.; Patel, F.; Prakash, R.; et al. A novel luciferase-based assay for the detection of chimeric antigen receptors. Sci. Rep. 2019, 9, 1957. [Google Scholar] [CrossRef] [PubMed]
- Markova, S.V.; Larionova, M.D.; Gorbunova, D.A.; Vysotski, E.S. The disulfide-rich Metridia luciferase refolded from E. coli inclusion bodies reveals the properties of a native folded enzyme produced in insect cells. J. Photochem. Photobiol. B 2017, 175, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ershova, A.S.; Gra, O.A.; Lyaschuk, A.M.; Grunina, T.M.; Tkachuk, A.P.; Bartov, M.S.; Savina, D.M.; Sergienko, O.V.; Galushkina, Z.M.; Gudov, V.P.; et al. Recombinant domains III of tick-borne encephalitis virus envelope protein in combination with dextran and CpGs induce immune response and partial protectiveness against TBE virus infection in mice. BMC Infect. Dis. 2016, 16, 544. [Google Scholar] [CrossRef] [PubMed]
- Larionova, M.D.; Markova, S.V.; Vysotski, E.S. Tyr72 and Tyr80 are involved in the formation of an active site of a luciferase of copepod Metridia longa. Photochem. Photobiol. 2017, 93, 503–510. [Google Scholar] [CrossRef]
- Burakova, L.P.; Kudryavtsev, A.N.; Stepanyuk, G.A.; Baykov, I.K.; Morozova, V.V.; Tikunova, N.V.; Dubova, M.A.; Lyapustin, V.N.; Yakimenko, V.V.; Frank, L.A. Bioluminescent detection probe for tick-borne encephalitis virus immunoassay. Anal. Bioanal. Chem. 2015, 408, 5417–5423. [Google Scholar] [CrossRef]
- Baykov, I.K.; Levanov, L.N.; Matveev, L.E.; Tikunova, N.V. Development of the single-chain antibodies against tick-borne encephalitis virus. In Proceedings of the conference “Genomics and proteomics in medicine”, Novosibirsk, Russia, 9–13 September 2009; p. 98. [Google Scholar]
- Liapustin, V.N.; Karganova, G.G.; Sobolev, S.G.; Gritsun, T.S. Preparative separation of the antigenic structures of the tick-borne encephalitis virus by electrophoresis in a liquid medium. Vopr. Virusol. 1988, 33, 98–102. (In Russian) [Google Scholar]
- Tsekhanovskaya, N.A.; Matveev, L.E.; Rubin, S.G.; Karavanov, A.S.; Pressman, E.K. Epitope analysis of tick-borne encephalitis (TBE) complex viruses using monoclonal antibodies to envelope glycoprotein of TBE virus (persulcatus subtype). Virus Res. 1993, 30, 1–16. [Google Scholar] [CrossRef]
- Baykov, I.K.; Matveev, L.E.; Matveev, A.L.; Tikunova, N.V. Comparative analysis of variable domains of monoclonal antibodies against tick-born encephalitis virus. Sib. Med. J. 2012, 111, 30–33. (In Russian) [Google Scholar]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Eremeeva, E.V.; Markova, S.V.; Frank, L.A.; Visser, A.J.; van Berkel, W.J.; Vysotski, E.S. Bioluminescent and spectroscopic properties of His-Trp-Tyr triad mutants of obelin and aequorin. Photochem. Photobiol. Sci. 2013, 12, 1016–1024. [Google Scholar] [CrossRef]
- Permyakov, E.A.; Burstein, E.A. Some aspects of studies of thermal transitions in proteins by means of their intrinsic fluorescence. Biophys. Chem. 1984, 19, 265–271. [Google Scholar] [CrossRef]
- Bollen, Y.J.M.; Nabuurs, S.M.; van Berkel, W.J.H.; van Mierlo, C.P.M. Last in, first out: The role of cofactor binding in flavodoxin folding. J. Biol. Chem. 2005, 280, 7836–7844. [Google Scholar] [CrossRef] [PubMed]
- Eremeeva, E.V.; Markova, S.V.; Westphal, A.H.; Visser, A.J.; van Berkel, W.J.; Vysotski, E.S. The intrinsic fluorescence of apo-obelin and apo-aequorin and use of its quenching to characterize coelenterazine binding. FEBS Lett. 2009, 583, 1939–1944. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Peak Light Intensity, (rlu/mol, × 1011) | Biolumi- Nescence Spectrum Maximum (nm) | kdecay (s-1) | Free SH-Group | KD (nM) | |
|---|---|---|---|---|---|---|
| k1 | k2 | |||||
| MLuc7 | 1.85 ± 0.16 | 488 | 0.20 (0.90) ‡ ± 0.01 | 0.016 (0.10) ± 0.001 | 0.20 ± 0.09 | - |
| 14D5a-MLuc7 | 1.83 ± 0.12 | 487 | 0.26 (0.73) ± 0.05 | 0.057 (0.27) ± 0.001 | 0.41 ± 0.11 | 36.2 ± 0.3 |
| MLuc7-14D5a | 1.80 ± 0.21 | 488 | 0.19 (0.74) ± 0.02 | 0.040 (0.26) ± 0.001 | 0.30 ± 0.07 | 87.6 ± 0.7 |
| No. | Primer Sequences † |
|---|---|
| 1 | F 5′-GGCGCGGTACCATGGATATCAAATTTATTTTTG-3′ |
| 3 | F 5′-GTCTTGCTGGAGATCGTGGATCCGGTGGTGCCGAGGTGCAGCTG-3′ |
| 4 | R 5′-ATGATGATGACCTTGAAAGTACAAGTTCTCACGTTTGATTTCCAGC-3′ |
| 5 | R 5′-TACTCGAGTCATTAGTGATGGTGATGGTGATGATGACCTTGAAAG-3′ |
| 6 | R 5′-CCAGCTGCACCTCGGCAGCCTGGACCAATGCAA-3′ |
| 8 | R 5′-ATTGTTTACAGTAGGGTTGCCGGATCCACGTTTGATTTCCAGC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larionova, M.D.; Markova, S.V.; Tikunova, N.V.; Vysotski, E.S. The Smallest Isoform of Metridia longa Luciferase as a Fusion Partner for Hybrid Proteins. Int. J. Mol. Sci. 2020, 21, 4971. https://doi.org/10.3390/ijms21144971
Larionova MD, Markova SV, Tikunova NV, Vysotski ES. The Smallest Isoform of Metridia longa Luciferase as a Fusion Partner for Hybrid Proteins. International Journal of Molecular Sciences. 2020; 21(14):4971. https://doi.org/10.3390/ijms21144971
Chicago/Turabian StyleLarionova, Marina D., Svetlana V. Markova, Nina V. Tikunova, and Eugene S. Vysotski. 2020. "The Smallest Isoform of Metridia longa Luciferase as a Fusion Partner for Hybrid Proteins" International Journal of Molecular Sciences 21, no. 14: 4971. https://doi.org/10.3390/ijms21144971
APA StyleLarionova, M. D., Markova, S. V., Tikunova, N. V., & Vysotski, E. S. (2020). The Smallest Isoform of Metridia longa Luciferase as a Fusion Partner for Hybrid Proteins. International Journal of Molecular Sciences, 21(14), 4971. https://doi.org/10.3390/ijms21144971