Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives



 , ,
, ,  and
and 
Abstract
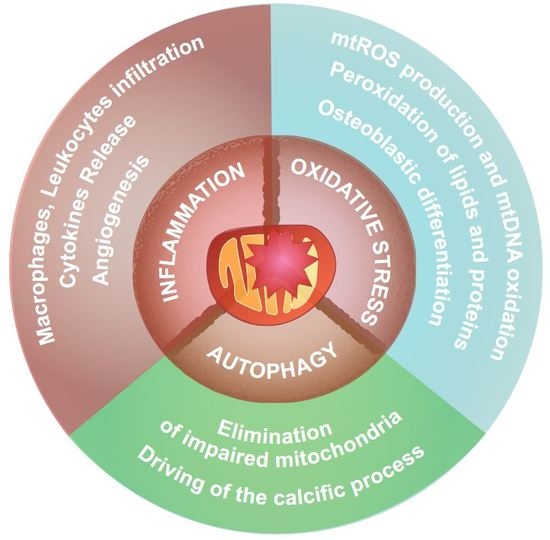
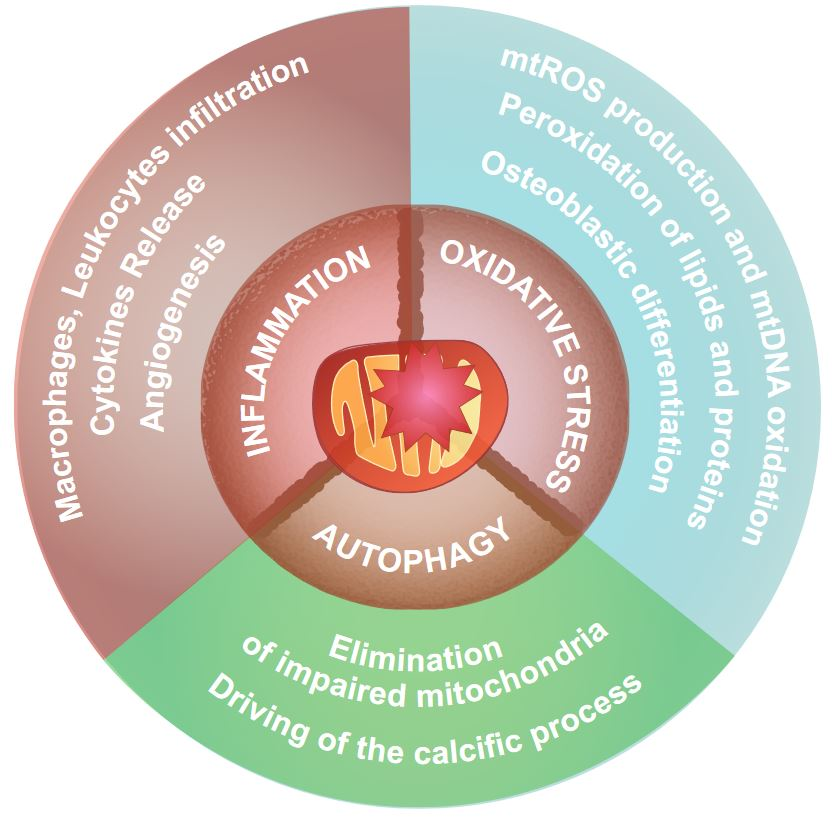{kind=link}
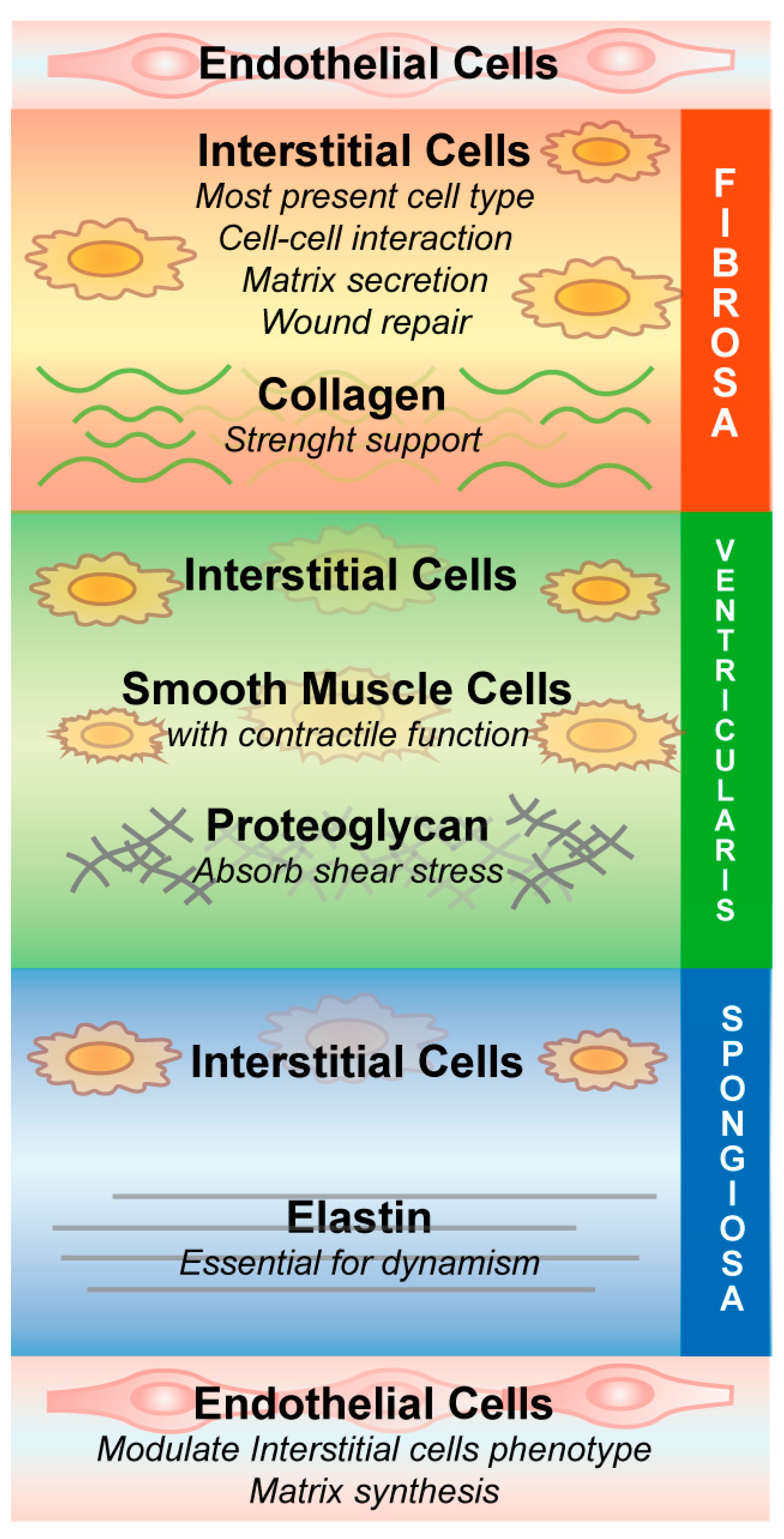{kind=link}
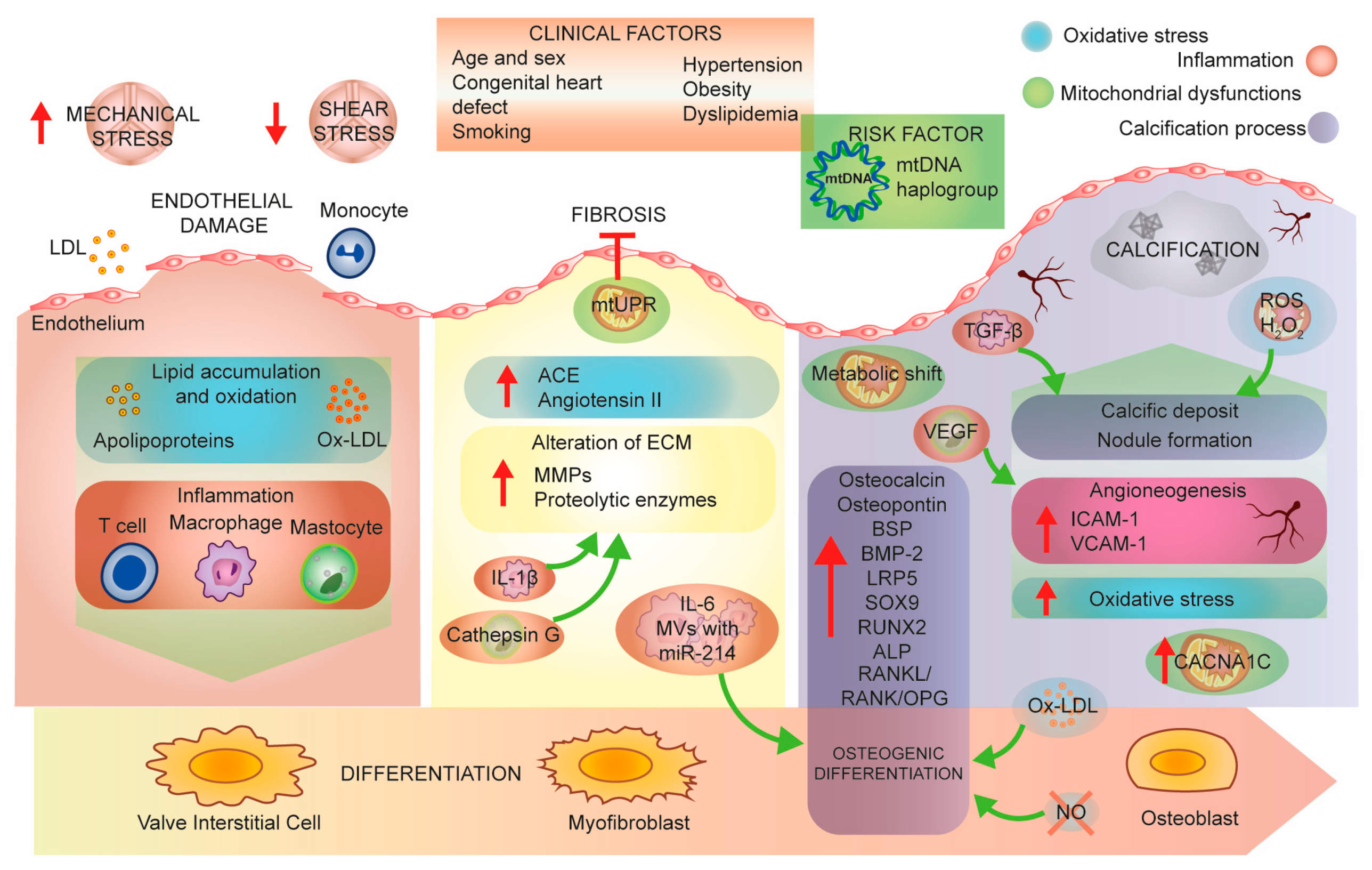{kind=link}
{kind=link}
1. Introduction
2. Heart Valve Stenosis Epidemiology, Diagnosis and Therapies
3. Aortic Stenosis Pathobiology, a Molecular Perspective
3.1. Mitochondrial Dysfunction
3.2. Mitochondria and Inflammation
3.3. Mitochondria and Oxidative Stress
3.4. Nitric Oxide Synthase
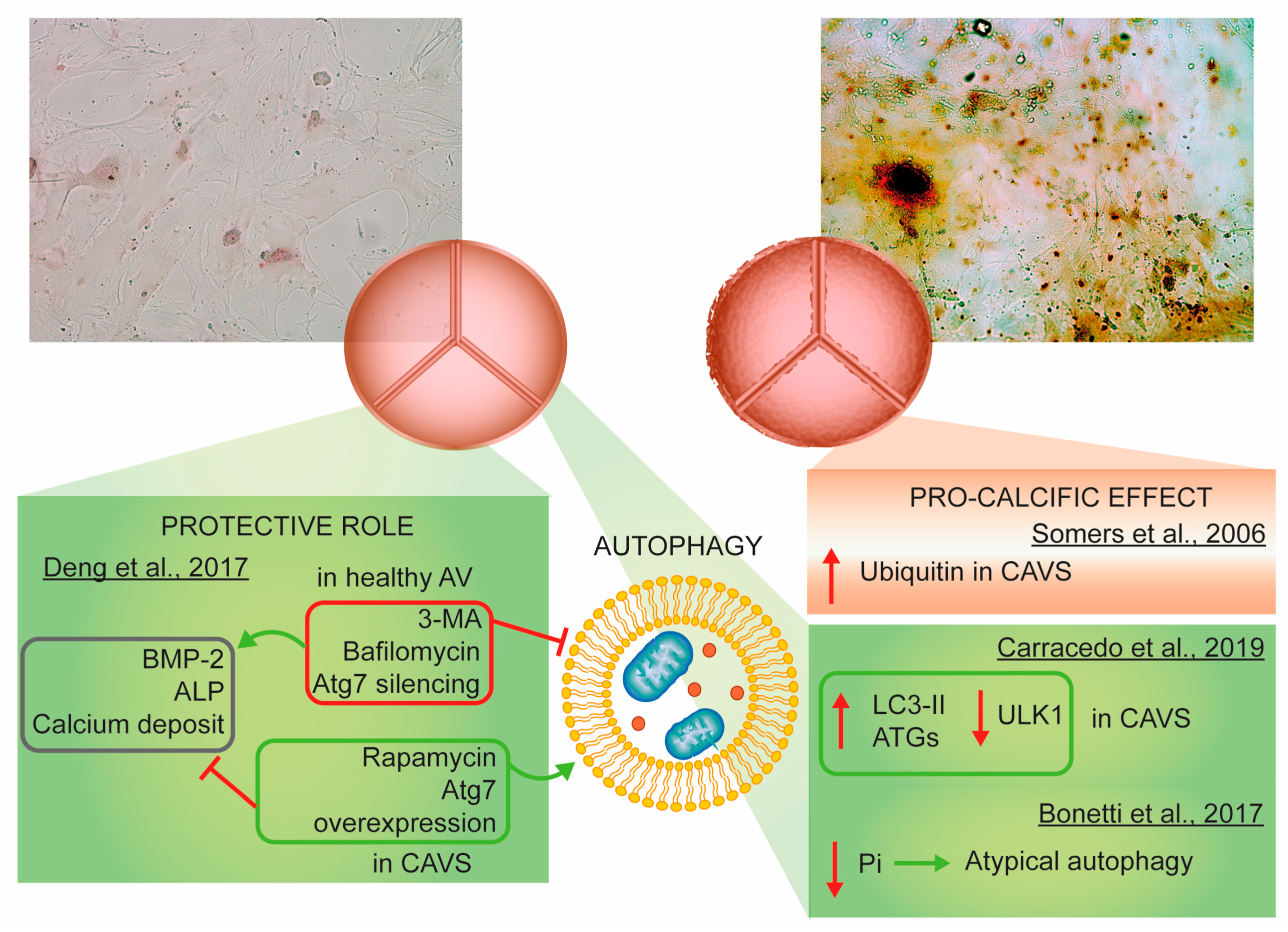
3.5. Autophagy
4. Efforts to Find Good Therapeutic Strategies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AS | Aortic stenosis |
| ECM | Extracellular matrix |
| VECs | Valvular endothelial cells |
| VICs | Valvular interstitial cells |
| MMPs | Matrix metalloproteinases |
| TIMPs | Tissue inhibitors of metalloproteinases |
| SMCs | Smooth muscle cells |
| CAVS | Calcific Aortic Valve Stenosis |
| AVR | Aortic valve replacement |
| BAVs | Bicuspid aortic valves |
| apo | apolipoproteins |
| Lp | lipoprotein |
| OxPL-apoB | Oxidized phospholipids-Lipoprotein(b) |
| HAVICs | Human Aortic Valvular Interstitial Cells |
| TNF-α | Tumor Necrosis Factor α |
| BSP | Bone sialoprotein |
| BMP-2 | Bone morphogenetic protein-2 |
| TGF-β | Transforming growth factor β |
| Lrp5 | Low-density lipoprotein receptor related protein 5 |
| SOX9 | SRY-box 9 |
| RUNX2 | Runt-related transcription factor 2 |
| ALP | Alkaline phosphatase |
| NOTCH1 | Notch homolog 1, translocation-associated |
| RhoA/ROCK | Ras homolog family member A/ Rho-associated protein kinase |
| ENPP1 | Ectonucleotide phosphodiesterase/pyrophosphatase-1 |
| RANKL | Receptor activator of NF-kappaB ligand |
| RANK | Receptor activator of nuclear factor κ |
| OPG | Osteoprotegerin |
| EVs | Extracellular vesicles |
| ROS | Reactive oxygen species |
| ΔΨm | Mitochondrial Membrane Potential |
| oxLDL | oxidized Low-Density Lipoproteins |
| mPTP | Mitochondrial Permeability Transition Pore |
| mtUPR | mitochondrial Unfolded Protein Response |
| FAT/CD36 | Fatty Acid Translocase |
| GLUT | Glucose Transporter |
| DRP1 | Dynamin-related protein 1 |
| VEGF | Vascular Endothelial Growth Factor |
| CACNA1C | Calcium Voltage-Gated Channel Subunit Alpha1 C |
| IL-1β | Interleukin 1 β |
| mtROS | Mitochondrial ROS |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| VDAC | Voltage-Dependent Anion Channel |
| Bcl-2 | B-cell lymphoma 2 |
| LC3 | Microtubule-Associated Protein 1 Light Chain 3 |
| LPS | Lipopolysaccharide |
| PET | Positron Emission Tomography |
| P2Y2R | P2Y purinoceptor 2 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| IFN-γ | Interferon gamma |
| MVs | Microvesicles |
| TWIST1 | Twist Family BHLH Transcription Factor 1 |
| Dlx5 | Distal-less homeobox 5 |
| TGF-β1 | Transforming growth factor beta 1 |
| BAFF | B cell-activating factor belonging to the TNF family |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| VCAM-1 | Vascular Cell Adhesion Molecule-1 |
| E-selectin | Endothelial selectin |
| miRNA | microRNA |
| CCL3 | C-C Motif Chemokine Ligand 3 |
| MM-LDL | Minimally Oxidized Low-Density Lipoprotein |
| CVCs | Calcifying Vascular Cells |
| mETC | Mitochondrial Electron Transport Chain |
| SOD | Superoxide Dismutase |
| CuZnSOD | Copper-Zinc-Superoxide Dismutase |
| MnSOD | Manganese-Dependent Superoxide Dismutase |
| ecSOD | Extracellular Superoxide Dismutase |
| NO | Nitric Oxide |
| eNOS | Endothelial Nitric Oxide Synthase |
| cGMP | cyclic Guanosine Monophosphate |
| DPP4 | Dipeptidyl Peptidase-4 |
| mTOR | mammalian Target Of Rapamycin |
| ULK1 | Unc-51-like kinase |
| Atg | Autophagy related |
| PI3K III | Class III phosphatidylinositol 3-kinase |
| PE | Phosphatidylethanolamine |
| 3-MA | 3-methyladenine |
| Pi | Inorganic Phosphate |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-coenzyme A |
| AVC | Aortic Valve Calcium |
| ACE | Angiotensin-converting enzyme |
| PCSK9 | Proprotein Convertase Subtilisin/Kexin type 9 |
References
- Yadgir, S.; Johnson, C.O.; Aboyans, V.; Adebayo, O.M.; Adedoyin, R.A.; Afarideh, M.; Alahdab, F.; Alashi, A.; Alipour, V.; Arabloo, J.; et al. Global, regional, and national burden of calcific aortic valve and degenerative mitral valve diseases, 1990–2017. Circulation 2020, 141, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, K.J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Mathieu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011, 124, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Schoen, F.J. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation 2008, 118, 1864–1880. [Google Scholar] [CrossRef] [PubMed]
- Yetkin, E.; Waltenberger, J. Molecular and cellular mechanisms of aortic stenosis. Int. J. Cardiol. 2009, 135, 4–13. [Google Scholar] [CrossRef]
- Butcher, J.T.; Nerem, R.M. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: Effects of steady shear stress. Tissue Eng. 2006, 12, 905–915. [Google Scholar] [CrossRef]
- Simmons, C.A.; Grant, G.R.; Manduchi, E.; Davies, P.F. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ. Res. 2005, 96, 792–799. [Google Scholar] [CrossRef]
- Dreger, S.A.; Taylor, P.M.; Allen, S.P.; Yacoub, M.H. Profile and localization of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in human heart valves. J. Heart Valve Dis. 2002, 11, 875–880. [Google Scholar]
- Bertipaglia, B.; Ortolani, F.; Petrelli, L.; Gerosa, G.; Spina, M.; Pauletto, P.; Casarotto, D.; Marchini, M.; Sartore, S.; Vitalitate Exornatum Succedaneum Aorticum Labore Ingenioso Obtenibitur Project. Cell characterization of porcine aortic valve and decellularized leaflets repopulated with aortic valve interstitial cells: The VESALIO Project (Vitalitate Exornatum Succedaneum Aorticum Labore Ingenioso Obtenibitur). Ann. Thorac. Surg. 2003, 75, 1274–1282. [Google Scholar] [CrossRef]
- Latif, N.; Sarathchandra, P.; Chester, A.H.; Yacoub, M.H. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur. Heart J. 2015, 36, 1335–1345. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Shinde, S.; Kumar, P.; Mishra, K.; Patil, N. Defect in mitochondrial functions in damaged human mitral valve. Indian J. Clin. Biochem. 2006, 21, 156–160. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Larsson, S.C.; Wolk, A.; Back, M. Alcohol consumption, cigarette smoking and incidence of aortic valve stenosis. J. Intern. Med. 2017, 282, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Dweck, M.R.; Boon, N.A.; Newby, D.E. Calcific aortic stenosis: A disease of the valve and the myocardium. J. Am. Coll Cardiol. 2012, 60, 1854–1863. [Google Scholar] [CrossRef]
- Stewart, R.L.; Chan, K.L. Management of asymptomatic severe aortic stenosis. Curr. Cardiol. Rev. 2009, 5, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Spadaccio, C.; Alkhamees, K.; Al-Attar, N. Recent advances in aortic valve replacement. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.J.; Leon, M.B.; Thourani, V.H.; Makkar, R.; Kodali, S.K.; Russo, M.; Kapadia, S.R.; Malaisrie, S.C.; Cohen, D.J.; Pibarot, P.; et al. Transcatheter aortic-valve replacement with a balloon-expandable valve in low-risk patients. N. Engl. J. Med. 2019, 380, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Popma, J.J.; Deeb, G.M.; Yakubov, S.J.; Mumtaz, M.; Gada, H.; O’Hair, D.; Bajwa, T.; Heiser, J.C.; Merhi, W.; Kleiman, N.S.; et al. Transcatheter aortic-valve replacement with a self-expanding valve in low-risk patients. N. Engl. J. Med. 2019, 380, 1706–1715. [Google Scholar] [CrossRef]
- Dawson, L.; Huang, A.; Selkrig, L.; Shaw, J.A.; Stub, D.; Walton, A.; Duffy, S.J. Utility of balloon aortic valvuloplasty in the transcatheter aortic valve implantation era. Open Heart 2020, 7. [Google Scholar] [CrossRef]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the early lesion of ’degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef]
- Cote, N.; Mahmut, A.; Bosse, Y.; Couture, C.; Page, S.; Trahan, S.; Boulanger, M.C.; Fournier, D.; Pibarot, P.; Mathieu, P. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation 2013, 36, 573–581. [Google Scholar] [CrossRef]
- Hinton, R.B., Jr.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Lehti, S.; Kakela, R.; Horkko, S.; Kummu, O.; Helske-Suihko, S.; Kupari, M.; Werkkala, K.; Kovanen, P.T.; Oorni, K. Modified lipoprotein-derived lipid particles accumulate in human stenotic aortic valves. PLoS ONE 2013, 8, e65810. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D.; Reichenbach, D.D.; Marcovina, S.M.; Kuusisto, J.; Alpers, C.E.; Otto, C.M. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ’degenerative’ valvular aortic stenosis. Arter. Thromb. Vasc. Biol. 1996, 16, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Thyberg, J.; Nilsson, J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arter. Thromb. Vasc. Biol. 1999, 19, 1218–1222. [Google Scholar] [CrossRef]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bosse, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef]
- Yu, B.; Hafiane, A.; Thanassoulis, G.; Ott, L.; Filwood, N.; Cerruti, M.; Gourgas, O.; Shum-Tim, D.; Al Kindi, H.; de Varennes, B.; et al. Lipoprotein(a) Induces Human Aortic Valve Interstitial Cell Calcification. JACC. Basic Transl. Sci. 2017, 2, 358–371. [Google Scholar] [CrossRef]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef]
- Nsaibia, M.J.; Mahmut, A.; Boulanger, M.C.; Arsenault, B.J.; Bouchareb, R.; Simard, S.; Witztum, J.L.; Clavel, M.A.; Pibarot, P.; Bosse, Y.; et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J. Intern. Med. 2016, 280, 509–517. [Google Scholar] [CrossRef]
- Yu, B.; Khan, K.; Hamid, Q.; Mardini, A.; Siddique, A.; Aguilar-Gonzalez, L.P.; Makhoul, G.; Alaws, H.; Genest, J.; Thanassoulis, G.; et al. Pathological significance of lipoprotein(a) in aortic valve stenosis. Atherosclerosis 2018, 272, 168–174. [Google Scholar] [CrossRef]
- Capoulade, R.; Chan, K.L.; Mathieu, P.; Bosse, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Yang, X.; Witztum, J.L.; Arsenault, B.J.; et al. Autoantibodies and immune complexes to oxidation-specific epitopes and progression of aortic stenosis: Results from the ASTRONOMER trial. Atherosclerosis 2017, 260, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Despres, A.A.; Perrot, N.; Poulin, A.; Tastet, L.; Shen, M.; Chen, H.Y.; Bourgeois, R.; Trottier, M.; Tessier, M.; Guimond, J.; et al. Lipoprotein(a), oxidized phospholipids, and aortic valve microcalcification assessed by 18F-sodium fluoride positron emission tomography and computed tomography. CJC Open 2019, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.Y.; Chen, J.H.; Zhao, R.; Simmons, C.A. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arter. Thromb. Vasc. Biol. 2009, 29, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Jones, M.; Yamada, I.; Ferrans, V.J. Wound healing in the mitral valve. J. Heart Valve Dis. 2000, 9, 53–63. [Google Scholar]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kilic, R.; Sarikoc, A.; Pinol, R.; Hagl, S.; Lang, S.; et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, J.H. Involvement of immune cell network in aortic valve stenosis: Communication between valvular interstitial cells and immune cells. Immune Netw. 2016, 16, 26–32. [Google Scholar] [CrossRef]
- Xing, Y.; Warnock, J.N.; He, Z.; Hilbert, S.L.; Yoganathan, A.P. Cyclic pressure affects the biological properties of porcine aortic valve leaflets in a magnitude and frequency dependent manner. Ann. Biomed. Eng. 2004, 32, 1461–1470. [Google Scholar] [CrossRef]
- Balachandran, K.; Sucosky, P.; Jo, H.; Yoganathan, A.P. Elevated cyclic stretch alters matrix remodeling in aortic valve cusps: Implications for degenerative aortic valve disease. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 756–764. [Google Scholar] [CrossRef]
- Mohler, E.R., 3rd; Adam, L.P.; McClelland, P.; Graham, L.; Hathaway, D.R. Detection of osteopontin in calcified human aortic valves. Arter. Thromb. Vasc. Biol. 1997, 17, 547–552. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Kuusisto, J.; Reichenbach, D.D.; Ferguson, M.; Giachelli, C.; Alpers, C.E.; Otto, C.M. Osteopontin is expressed in human aortic valvular lesions. Circulation 1995, 92, 2163–2168. [Google Scholar] [CrossRef]
- Mohler, E.R., 3rd; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone formation and inflammation in cardiac valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Kaden, J.J.; Bickelhaupt, S.; Grobholz, R.; Vahl, C.F.; Hagl, S.; Brueckmann, M.; Haase, K.K.; Dempfle, C.E.; Borggrefe, M. Expression of bone sialoprotein and bone morphogenetic protein-2 in calcific aortic stenosis. J. Heart Valve Dis. 2004, 13, 560–566. [Google Scholar]
- Caira, F.C.; Stock, S.R.; Gleason, T.G.; McGee, E.C.; Huang, J.; Bonow, R.O.; Spelsberg, T.C.; McCarthy, P.M.; Rahimtoola, S.H.; Rajamannan, N.M. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J. Am. Coll. Cardiol. 2006, 47, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Subramaniam, M.; Rickard, D.; Stock, S.R.; Donovan, J.; Springett, M.; Orszulak, T.; Fullerton, D.A.; Tajik, A.J.; Bonow, R.O.; et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 2003, 107, 2181–2184. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Ferrari, R.; Rizzo, P. The Notch pathway: A novel target for myocardial remodelling therapy? Eur. Heart J. 2014, 35, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Miele, L.; Ferrari, R. The Notch pathway: A crossroad between the life and death of the endothelium. Eur. Heart J. 2013, 34, 2504–2509. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Hans, C.P.; Koenig, S.N.; Nichols, H.A.; Galindo, C.L.; Garner, H.R.; Merrill, W.H.; Hinton, R.B.; Garg, V. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS ONE 2011, 6, e27743. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, J.; Zhou, K.; Liao, X.; Zhou, X.; Shen, K. The methylation of Notch1 promoter mediates the osteogenesis differentiation in human aortic valve interstitial cells through Wnt/beta-catenin signaling. J. Cell Physiol. 2019, 234, 20366–20376. [Google Scholar] [CrossRef]
- Bouchareb, R.; Boulanger, M.C.; Fournier, D.; Pibarot, P.; Messaddeq, Y.; Mathieu, P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J. Mol. Cell Cardiol. 2014, 67, 49–59. [Google Scholar] [CrossRef]
- Bertazzo, S.; Gentleman, E.; Cloyd, K.L.; Chester, A.H.; Yacoub, M.H.; Stevens, M.M. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat. Mater. 2013, 12, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Sattler, A.M.; Schoppet, M.; Schaefer, J.R.; Hofbauer, L.C. Novel aspects on RANK ligand and osteoprotegerin in osteoporosis and vascular disease. Calcif. Tissue Int. 2004, 74, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Kaden, J.J.; Bickelhaupt, S.; Grobholz, R.; Haase, K.K.; Sarikoc, A.; Kilic, R.; Brueckmann, M.; Lang, S.; Zahn, I.; Vahl, C.; et al. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J. Mol. Cell. Cardiol. 2004, 36, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Krohn, J.B.; Hutcheson, J.D.; Martinez-Martinez, E.; Aikawa, E. Extracellular vesicles in cardiovascular calcification: Expanding current paradigms. J. Physiol. 2016, 594, 2895–2903. [Google Scholar] [CrossRef]
- Bakhshian Nik, A.; Hutcheson, J.D.; Aikawa, E. Extracellular vesicles as mediators of cardiovascular calcification. Front. Cardiovasc. Med. 2017, 4, 78. [Google Scholar] [CrossRef]
- Gardin, C.; Ferroni, L.; Latremouille, C.; Chachques, J.C.; Mitrecic, D.; Zavan, B. Recent Applications of Three Dimensional Printing in Cardiovascular Medicine. Cells 2020, 9, 742. [Google Scholar] [CrossRef]
- van der Valk, D.C.; van der Ven, C.F.T.; Blaser, M.C.; Grolman, J.M.; Wu, P.J.; Fenton, O.S.; Lee, L.H.; Tibbitt, M.W.; Andresen, J.L.; Wen, J.R.; et al. Engineering a 3D-bioprinted model of human heart valve disease using nanoindentation-based biomechanics. Nanomaterials 2018, 8, 296. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and reactive oxygen species in aging and age-related diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef]
- Pinton, P.; Leo, S.; Wieckowski, M.R.; Di Benedetto, G.; Rizzuto, R. Long-term modulation of mitochondrial Ca2+ signals by protein kinase C isozymes. J. Cell Biol. 2004, 165, 223–232. [Google Scholar] [CrossRef]
- Morciano, G.; Pedriali, G.; Sbano, L.; Iannitti, T.; Giorgi, C.; Pinton, P. Intersection of mitochondrial fission and fusion machinery with apoptotic pathways: Role of Mcl-1. Biol. Cell 2016, 108, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M. The retrograde response: When mitochondrial quality control is not enough. Biochim. Biophys. Acta. 2013, 1833, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium dynamics as a machine for decoding signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Rimessi, A.; Pedriali, G.; Vezzani, B.; Tarocco, A.; Marchi, S.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Interorganellar calcium signaling in the regulation of cell metabolism: A cancer perspective. Semin. Cell Dev. Biol. 2020, 98, 167–180. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef]
- Chen, J.; Mehta, J.L.; Haider, N.; Zhang, X.; Narula, J.; Li, D. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ. Res. 2004, 94, 370–376. [Google Scholar] [CrossRef]
- Mohler, E.R.; Sheridan, M.J.; Nichols, R.; Harvey, W.P.; Waller, B.F. Development and progression of aortic valve stenosis: Atherosclerosis risk factors—a causal relationship? A clinical morphologic study. Clin. Cardiol. 1991, 14, 995–999. [Google Scholar] [CrossRef]
- Dweck, M.R.; Jones, C.; Joshi, N.V.; Fletcher, A.M.; Richardson, H.; White, A.; Marsden, M.; Pessotto, R.; Clark, J.C.; Wallace, W.A.; et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation 2012, 125, 76–86. [Google Scholar] [CrossRef]
- Vindis, C.; Elbaz, M.; Escargueil-Blanc, I.; Auge, N.; Heniquez, A.; Thiers, J.C.; Negre-Salvayre, A.; Salvayre, R. Two distinct calcium-dependent mitochondrial pathways are involved in oxidized LDL-induced apoptosis. Arter. Thromb. Vasc. Biol. 2005, 25, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Ginghina, C.; Florian, A.; Beladan, C.; Iancu, M.; Calin, A.; Popescu, B.A.; Jurcut, R. Calcific aortic valve disease and aortic atherosclerosis--two faces of the same disease? Rom. J. Intern. Med. 2009, 47, 319–329. [Google Scholar] [PubMed]
- Serrano-Teruel, M.E.; Garcia-Vieites, M.; Rego-Perez, I.; Domenech-Garcia, N.; Blanco-Garcia, F.; Cuenca-Castillo, J.J.; Bautista-Hernandez, V. Mitochondrial DNA haplogroups influence the risk of aortic stenosis. Asian Cardiovasc. Thorac. Ann. 2019, 27, 5–10. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; Lopez, B.; Gonzalez, A.; Ravassa, S.; Diez, J.; et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Heather, L.C.; Howell, N.J.; Emmanuel, Y.; Cole, M.A.; Frenneaux, M.P.; Pagano, D.; Clarke, K. Changes in cardiac substrate transporters and metabolic proteins mirror the metabolic shift in patients with aortic stenosis. PLoS ONE 2011, 6, e26326. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ. Res. 2017, 121, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ma, W.; Yang, Z.; Zhang, F.; Lu, L.; Ding, Z.; Ding, B.; Ha, T.; Gao, X.; Li, C. VEGF165 and angiopoietin-1 decreased myocardium infarct size through phosphatidylinositol-3 kinase and Bcl-2 pathways. Gene Ther. 2005, 12, 196–202. [Google Scholar] [CrossRef]
- Xu, X.H.; Xu, J.; Xue, L.; Cao, H.L.; Liu, X.; Chen, Y.J. VEGF attenuates development from cardiac hypertrophy to heart failure after aortic stenosis through mitochondrial mediated apoptosis and cardiomyocyte proliferation. J. Cardiothorac. Surg. 2011, 6, 54. [Google Scholar] [CrossRef]
- Hennessey, J.A.; Boczek, N.J.; Jiang, Y.H.; Miller, J.D.; Patrick, W.; Pfeiffer, R.; Sutphin, B.S.; Tester, D.J.; Barajas-Martinez, H.; Ackerman, M.J.; et al. A CACNA1C variant associated with reduced voltage-dependent inactivation, increased CaV1.2 channel window current, and arrhythmogenesis. PLoS ONE 2014, 9, e106982. [Google Scholar] [CrossRef]
- Guauque-Olarte, S.; Messika-Zeitoun, D.; Droit, A.; Lamontagne, M.; Tremblay-Marchand, J.; Lavoie-Charland, E.; Gaudreault, N.; Arsenault, B.J.; Dube, M.P.; Tardif, J.C.; et al. Calcium signaling pathway genes RUNX2 and CACNA1C are associated with calcific aortic valve disease. Circ. Cardiovasc. Genet. 2015, 8, 812–822. [Google Scholar] [CrossRef]
- Briasoulis, A.; Androulakis, E.; Christophides, T.; Tousoulis, D. The role of inflammation and cell death in the pathogenesis, progression and treatment of heart failure. Heart Fail. Rev. 2016, 21, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K.; Leducq Transatlantic Network on, A. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in cardiovascular diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- El Husseini, D.; Boulanger, M.C.; Mahmut, A.; Bouchareb, R.; Laflamme, M.H.; Fournier, D.; Pibarot, P.; Bosse, Y.; Mathieu, P. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: Implication for calcific aortic valve disease. J. Mol. Cell. Cardiol. 2014, 72, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qiao, W.; Zhang, W.; Li, F.; Shi, J.; Dong, N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J. Thorac. Cardiovasc. Surg. 2017, 153, 1318–1327.e1. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Wang, Y.; Zheng, D.D.; Xu, H.X.; Wang, T.; Pan, M.; Shi, J.H.; Zhu, J.H. M1 macrophages promote aortic valve calcification mediated by microRNA-214/TWIST1 pathway in valvular interstitial cells. Am. J. Transl. Res. 2016, 8, 5773–5783. [Google Scholar] [PubMed]
- Yu, Z.; Seya, K.; Daitoku, K.; Motomura, S.; Fukuda, I.; Furukawa, K. Tumor necrosis factor-alpha accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J. Pharmacol. Exp. Ther. 2011, 337, 16–23. [Google Scholar] [CrossRef]
- Kaden, J.J.; Kilic, R.; Sarikoc, A.; Hagl, S.; Lang, S.; Hoffmann, U.; Brueckmann, M.; Borggrefe, M. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: A potential regulatory mechanism of valvular calcification. Int. J. Mol. Med. 2005, 16, 869–872. [Google Scholar] [CrossRef]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Tran, H.T.; Kilic, R.; Sarikoc, A.; Brueckmann, M.; Vahl, C.; Hagl, S.; Haase, K.K.; et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis 2003, 170, 205–211. [Google Scholar] [CrossRef]
- Galis, Z.S.; Muszynski, M.; Sukhova, G.K.; Simon-Morrissey, E.; Libby, P. Enhanced expression of vascular matrix metalloproteinases induced in vitro by cytokines and in regions of human atherosclerotic lesions. Ann. NY Acad. Sci. 1995, 748, 501–507. [Google Scholar] [CrossRef]
- Siwik, D.A.; Chang, D.L.; Colucci, W.S. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ. Res. 2000, 86, 1259–1265. [Google Scholar] [CrossRef]
- Nagy, E.; Lei, Y.; Martinez-Martinez, E.; Body, S.C.; Schlotter, F.; Creager, M.; Assmann, A.; Khabbaz, K.; Libby, P.; Hansson, G.K.; et al. Interferon-gamma released by activated CD8(+) T lymphocytes impairs the calcium resorption potential of osteoclasts in calcified human aortic valves. Am. J. Pathol. 2017, 187, 1413–1425. [Google Scholar] [CrossRef]
- Jian, B.; Narula, N.; Li, Q.Y.; Mohler, E.R., 3rd; Levy, R.J. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann. Thorac. Surg. 2003, 75, 457–465. [Google Scholar] [CrossRef]
- Steiner, I.; Krbal, L.; Rozkos, T.; Harrer, J.; Laco, J. Calcific aortic valve stenosis: Immunohistochemical analysis of inflammatory infiltrate. Pathol. Res. Pract. 2012, 208, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.D.; Maurer, M.S.; Friedman, R.A.; Marboe, C.C.; Ruiz-Vazquez, E.M.; Ramakrishnan, R.; Schwartz, A.; Tilson, M.D.; Stewart, A.S.; Winchester, R. The lymphocytic infiltration in calcific aortic stenosis predominantly consists of clonally expanded T cells. J. Immunol. 2007, 178, 5329–5339. [Google Scholar] [CrossRef] [PubMed]
- Natorska, J.; Marek, G.; Sadowski, J.; Undas, A. Presence of B cells within aortic valves in patients with aortic stenosis: Relation to severity of the disease. J. Cardiol. 2016, 67, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Wypasek, E.; Natorska, J.; Grudzien, G.; Filip, G.; Sadowski, J.; Undas, A. Mast cells in human stenotic aortic valves are associated with the severity of stenosis. Inflammation 2013, 36, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Helske, S.; Syvaranta, S.; Kupari, M.; Lappalainen, J.; Laine, M.; Lommi, J.; Turto, H.; Mayranpaa, M.; Werkkala, K.; Kovanen, P.T.; et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur. Heart J. 2006, 27, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Syvaranta, S.; Helske, S.; Laine, M.; Lappalainen, J.; Kupari, M.; Mayranpaa, M.I.; Lindstedt, K.A.; Kovanen, P.T. Vascular endothelial growth factor-secreting mast cells and myofibroblasts: A novel self-perpetuating angiogenic pathway in aortic valve stenosis. Arter. Thromb. Vasc. Biol. 2010, 30, 1220–1227. [Google Scholar] [CrossRef]
- Mazzone, A.; Epistolato, M.C.; De Caterina, R.; Storti, S.; Vittorini, S.; Sbrana, S.; Gianetti, J.; Bevilacqua, S.; Glauber, M.; Biagini, A.; et al. Neoangiogenesis, T-lymphocyte infiltration, and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J. Am. Coll. Cardiol. 2004, 43, 1670–1676. [Google Scholar] [CrossRef]
- Ghaisas, N.K.; Foley, J.B.; O’Briain, D.S.; Crean, P.; Kelleher, D.; Walsh, M. Adhesion molecules in nonrheumatic aortic valve disease: Endothelial expression, serum levels and effects of valve replacement. J. Am. Coll. Cardiol. 2000, 36, 2257–2262. [Google Scholar] [CrossRef]
- Bosse, Y.; Miqdad, A.; Fournier, D.; Pepin, A.; Pibarot, P.; Mathieu, P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ. Cardiovasc. Genet. 2009, 2, 489–498. [Google Scholar] [CrossRef]
- Ohukainen, P.; Syvaranta, S.; Napankangas, J.; Rajamaki, K.; Taskinen, P.; Peltonen, T.; Helske-Suihko, S.; Kovanen, P.T.; Ruskoaho, H.; Rysa, J. MicroRNA-125b and chemokine CCL4 expression are associated with calcific aortic valve disease. Ann. Med. 2015, 47, 423–429. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.H.; Haendeler, J.; Galle, J.; Zeiher, A.M.; Dimmeler, S. Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation 1998, 98, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sinensky, M.S. 25-Hydroxycholesterol activates a cytochrome c release-mediated caspase cascade. Biochem. Biophys. Res. Commun. 2000, 278, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Bouchareb, R.; Boulanger, M.C. Innate and adaptive immunity in calcific aortic valve disease. J. Immunol. Res. 2015, 2015, 851945. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.; Pibarot, P.; Despres, J.P.; Mohty, D.; Cartier, A.; Arsenault, B.J.; Couture, C.; Mathieu, P. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart 2008, 94, 1175–1180. [Google Scholar] [CrossRef]
- Parhami, F.; Morrow, A.D.; Balucan, J.; Leitinger, N.; Watson, A.D.; Tintut, Y.; Berliner, J.A.; Demer, L.L. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arter. Thromb. Vasc. Biol. 1997, 17, 680–687. [Google Scholar] [CrossRef]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Pena-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef]
- Weiss, R.M.; Ohashi, M.; Miller, J.D.; Young, S.G.; Heistad, D.D. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation 2006, 114, 2065–2069. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.; Bassi, E.; Martinatti, M.K.; Lario, F.C.; Wosniak, J. Jr.; Pomerantzeff, P.M.; Laurindo, F.R. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arter. Thromb. Vasc. Biol. 2008, 28, 463–470. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Munzel, T.; Li, H. New therapeutic implications of endothelial nitric oxide synthase (eNOS) function/dysfunction in cardiovascular disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 2013, 60, 27–35. [Google Scholar] [CrossRef]
- Rattazzi, M.; Donato, M.; Bertacco, E.; Millioni, R.; Franchin, C.; Mortarino, C.; Faggin, E.; Nardin, C.; Scarpa, R.; Cinetto, F.; et al. l-Arginine prevents inflammatory and pro-calcific differentiation of interstitial aortic valve cells. Atherosclerosis 2020, 298, 27–35. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Subramaniam, M.; Stock, S.R.; Stone, N.J.; Springett, M.; Ignatiev, K.I.; McConnell, J.P.; Singh, R.J.; Bonow, R.O.; Spelsberg, T.C. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart 2005, 91, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.A.; Hua, X.; Mishra, K.; Murphy, G.A.; Rosenkranz, A.C.; Horowitz, J.D. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur. J. Pharmacol. 2009, 602, 28–35. [Google Scholar] [CrossRef]
- Choi, B.; Lee, S.; Kim, S.M.; Lee, E.J.; Lee, S.R.; Kim, D.H.; Jang, J.Y.; Kang, S.W.; Lee, K.U.; Chang, E.J.; et al. Dipeptidyl peptidase-4 induces aortic valve calcification by inhibiting insulin-like growth factor-1 signaling in valvular interstitial cells. Circulation 2017, 135, 1935–1950. [Google Scholar] [CrossRef]
- Varennes, O.; Mary, A.; Bricca, G.; Kamel, S.; Bellien, J. Dipeptidyl peptidase-4 inhibition prevents vascular calcification by potentiating the insulin-like growth factor-1 signaling pathway. JACC. Basic Transl. Sci. 2019, 4, 113–115. [Google Scholar] [CrossRef]
- Liu, M.; Luo, M.; Sun, H.; Ni, B.; Shao, Y. Integrated bioinformatics analysis predicts the key genes involved in aortic valve calcification: From hemodynamic changes to extracellular remodeling. Tohoku J. Exp. Med. 2017, 243, 263–273. [Google Scholar] [CrossRef]
- Lee, T.C.; Zhao, Y.D.; Courtman, D.W.; Stewart, D.J. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation 2000, 101, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- El Accaoui, R.N.; Gould, S.T.; Hajj, G.P.; Chu, Y.; Davis, M.K.; Kraft, D.C.; Lund, D.D.; Brooks, R.M.; Doshi, H.; Zimmerman, K.A.; et al. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Aicher, D.; Urbich, C.; Zeiher, A.; Dimmeler, S.; Schafers, H.J. Endothelial nitric oxide synthase in bicuspid aortic valve disease. Ann. Thorac. Surg. 2007, 83, 1290–1294. [Google Scholar] [CrossRef] [PubMed]
- Doria, A.; Gatto, M.; Punzi, L. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and autophagy in the heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef]
- Weng, L.Q.; Zhang, W.B.; Ye, Y.; Yin, P.P.; Yuan, J.; Wang, X.X.; Kang, L.; Jiang, S.S.; You, J.Y.; Wu, J.; et al. Aliskiren ameliorates pressure overload-induced heart hypertrophy and fibrosis in mice. Acta. Pharmacol. Sin. 2014, 35, 1005–1014. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Martinet, W.; Schrijvers, D.M.; Timmermans, J.P.; Bult, H. Interactions between cell death induced by statins and 7-ketocholesterol in rabbit aorta smooth muscle cells. Br. J. Pharmacol. 2008, 154, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef] [PubMed]
- Somers, P.; Knaapen, M.; Kockx, M.; van Cauwelaert, P.; Bortier, H.; Mistiaen, W. Histological evaluation of autophagic cell death in calcified aortic valve stenosis. J. Heart Valve Dis. 2006, 15, 43–47. [Google Scholar] [PubMed]
- Deng, X.S.; Meng, X.; Venardos, N.; Song, R.; Yamanaka, K.; Fullerton, D.; Jaggers, J. Autophagy negatively regulates pro-osteogenic activity in human aortic valve interstitial cells. J. Surg. Res. 2017, 218, 285–291. [Google Scholar] [CrossRef]
- Carracedo, M.; Persson, O.; Saliba-Gustafsson, P.; Artiach, G.; Ehrenborg, E.; Eriksson, P.; Franco-Cereceda, A.; Back, M. Upregulated autophagy in calcific aortic valve stenosis confers protection of valvular interstitial cells. Int. J. Mol. Sci. 2019, 20, 1486. [Google Scholar] [CrossRef]
- Bonetti, A.; Della Mora, A.; Contin, M.; Gregoraci, G.; Tubaro, F.; Marchini, M.; Ortolani, F. Survival-related autophagic activity versus procalcific death in cultured aortic valve interstitial cells treated with critical normophosphatemic-like phosphate concentrations. J. Histochem. Cytochem. 2017, 65, 125–138. [Google Scholar] [CrossRef]
- Shavelle, D.M.; Takasu, J.; Budoff, M.J.; Mao, S.; Zhao, X.Q.; O’Brien, K.D. HMG CoA reductase inhibitor (statin) and aortic valve calcium. Lancet 2002, 359, 1125–1126. [Google Scholar] [CrossRef]
- Dichtl, W.; Alber, H.F.; Feuchtner, G.M.; Hintringer, F.; Reinthaler, M.; Bartel, T.; Sussenbacher, A.; Grander, W.; Ulmer, H.; Pachinger, O.; et al. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg). Am. J. Cardiol. 2008, 102, 743–748. [Google Scholar] [CrossRef]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J.; ASTRONOMER Investigators. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A.; Scottish Aortic Stenosis and Lipid Lowering Trial, Impact on Regression (SALTIRE) Investigators. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef]
- Rossebo, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Barwolf, C.; Holme, I.; Kesaniemi, Y.A.; et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D.; Probstfield, J.L.; Caulfield, M.T.; Nasir, K.; Takasu, J.; Shavelle, D.M.; Wu, A.H.; Zhao, X.Q.; Budoff, M.J. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch. Intern. Med. 2005, 165, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Rosenhek, R.; Rader, F.; Loho, N.; Gabriel, H.; Heger, M.; Klaar, U.; Schemper, M.; Binder, T.; Maurer, G.; Baumgartner, H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004, 110, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, Y.; Yagmur, C.; Tekin, G.O.; Yagmur, J.; Topal, E.; Kekilli, E.; Turhan, H.; Kosar, F.; Yetkin, E. Aortic valve calcification: Association with bone mineral density and cardiovascular risk factors. Coron. Artery Dis. 2005, 16, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Persy, V.; D’Haese, P. Vascular calcification and bone disease: The calcification paradox. Trends Mol. Med. 2009, 15, 405–416. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Bisphosphonates alendronate and ibandronate inhibit artery calcification at doses comparable to those that inhibit bone resorption. Arter. Thromb. Vasc. Biol. 2001, 21, 817–824. [Google Scholar] [CrossRef]
- Rapoport, H.S.; Connolly, J.M.; Fulmer, J.; Dai, N.; Murti, B.H.; Gorman, R.C.; Gorman, J.H.; Alferiev, I.; Levy, R.J. Mechanisms of the in vivo inhibition of calcification of bioprosthetic porcine aortic valve cusps and aortic wall with triglycidylamine/mercapto bisphosphonate. Biomaterials 2007, 28, 690–699. [Google Scholar] [CrossRef][Green Version]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in aortic stenosis: The skeleton key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef]
- Kleinauskiene, R.; Jonkaitiene, R. Degenerative aortic stenosis, dyslipidemia and possibilities of medical treatment. Medicina 2018, 54, 24. [Google Scholar] [CrossRef]
- Cote, N.; El Husseini, D.; Pepin, A.; Bouvet, C.; Gilbert, L.A.; Audet, A.; Fournier, D.; Pibarot, P.; Moreau, P.; Mathieu, P. Inhibition of ectonucleotidase with ARL67156 prevents the development of calcific aortic valve disease in warfarin-treated rats. Eur. J. Pharmacol. 2012, 689, 139–146. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedriali, G.; Morciano, G.; Patergnani, S.; Cimaglia, P.; Morelli, C.; Mikus, E.; Ferrari, R.; Gasbarro, V.; Giorgi, C.; Wieckowski, M.R.; et al. Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. Int. J. Mol. Sci. 2020, 21, 4899. https://doi.org/10.3390/ijms21144899
Pedriali G, Morciano G, Patergnani S, Cimaglia P, Morelli C, Mikus E, Ferrari R, Gasbarro V, Giorgi C, Wieckowski MR, et al. Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. International Journal of Molecular Sciences. 2020; 21(14):4899. https://doi.org/10.3390/ijms21144899
Chicago/Turabian StylePedriali, Gaia, Giampaolo Morciano, Simone Patergnani, Paolo Cimaglia, Cristina Morelli, Elisa Mikus, Roberto Ferrari, Vincenzo Gasbarro, Carlotta Giorgi, Mariusz R. Wieckowski, and et al. 2020. "Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives" International Journal of Molecular Sciences 21, no. 14: 4899. https://doi.org/10.3390/ijms21144899
APA StylePedriali, G., Morciano, G., Patergnani, S., Cimaglia, P., Morelli, C., Mikus, E., Ferrari, R., Gasbarro, V., Giorgi, C., Wieckowski, M. R., & Pinton, P. (2020). Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. International Journal of Molecular Sciences, 21(14), 4899. https://doi.org/10.3390/ijms21144899