Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
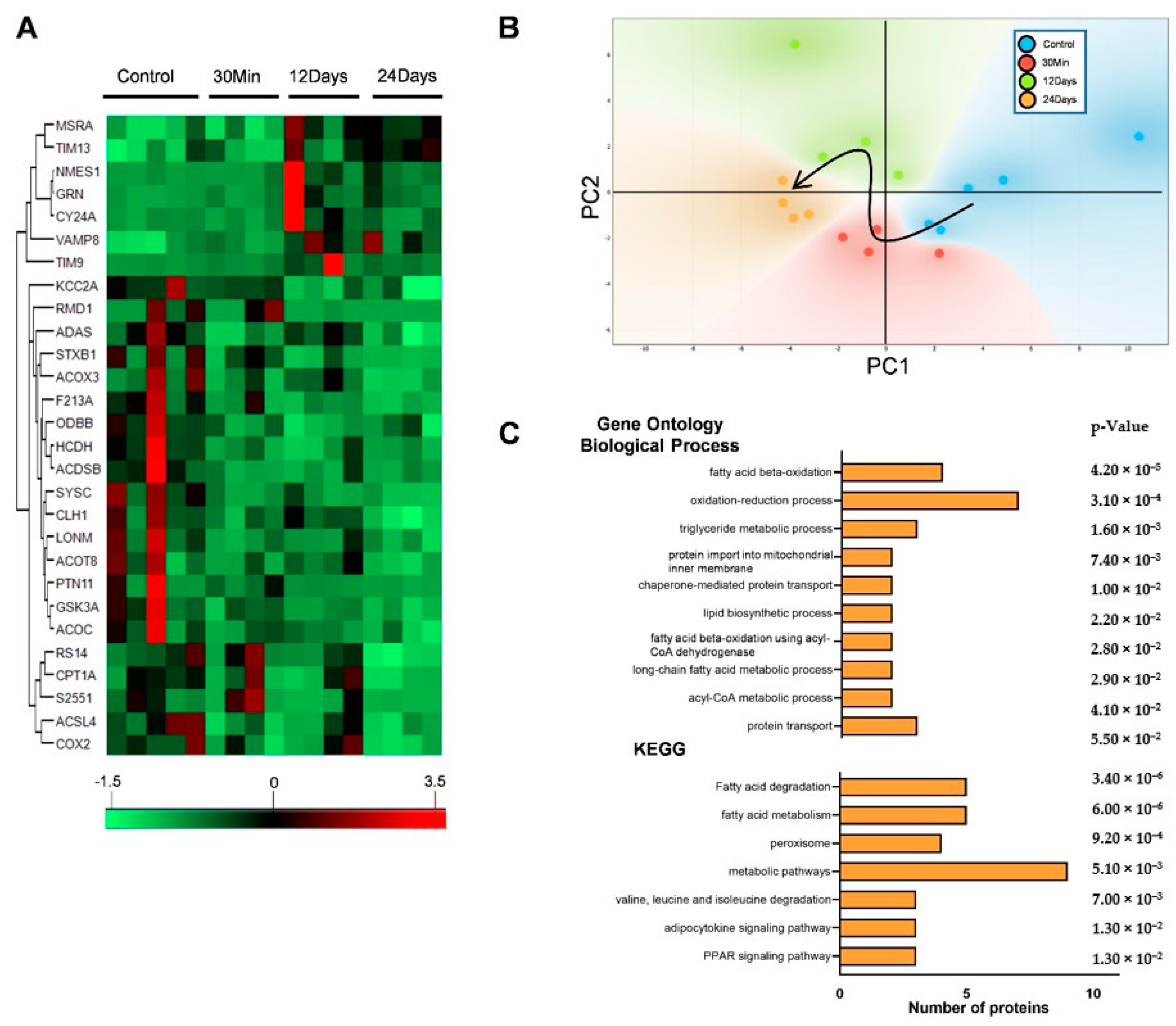
2.1. Complex III Inhibition Significantly Disrupted the Mitochondrial Proteome
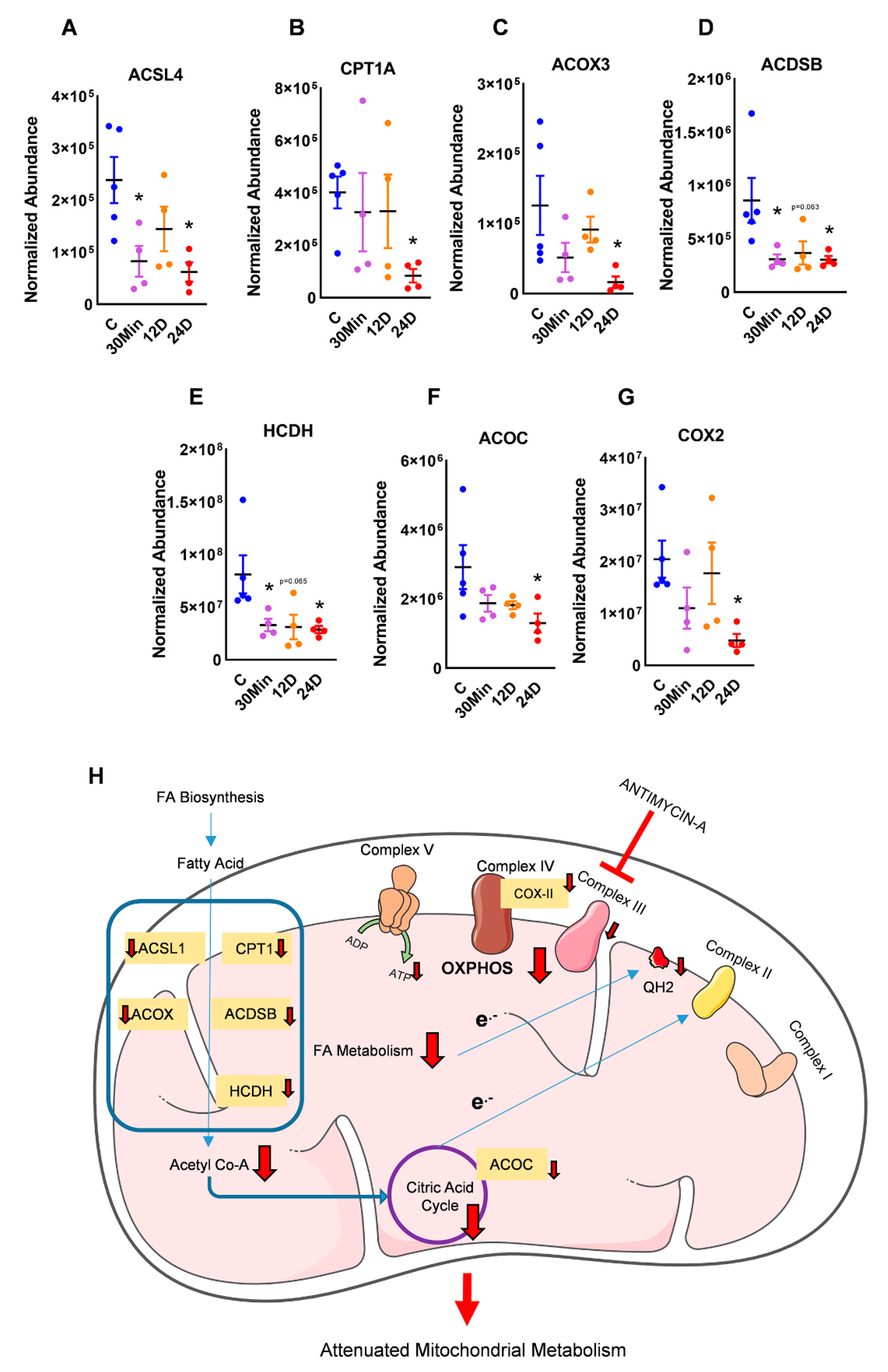
2.2. Complex III Inhibition Significantly Dysregulated Mitochondrial Fatty Acid and TCA Metabolism
2.3. Complex III Inhibition Impaired Amino Acid Metabolism and Protein Repair
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Mitochondrial Isolation
4.3. Mass Spectrometry
4.3.1. In-Solution Tryptic Digestion of Isolated Mitochondria
4.3.2. Mass Spectrometry and Data Processing
4.3.3. Label-Free Quantitative Proteomics
4.4. Bioinformatics Analysis
4.5. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal 2012, 16, 1150–1180. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Michelakis, E.D. The Metabolic Basis of Pulmonary Arterial Hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Nadege, B.; Patrick, L.; Rodrigue, R. Mitochondria: From bioenergetics to the metabolic regulation of carcinogenesis. Front. Biosci. 2009, 14, 4015–4034. [Google Scholar] [CrossRef]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and mechanism of mitochondrial electron transport chain. Biomed. J. 2018, 41, 9–20. [Google Scholar] [CrossRef]
- Rafikova, O.; Srivastava, A.; Desai, A.A.; Rafikov, R.; Tofovic, S.P. Recurrent inhibition of mitochondrial complex III induces chronic pulmonary vasoconstriction and glycolytic switch in the rat lung. Respir. Res. 2018, 19, 69. [Google Scholar] [CrossRef]
- Van Houten, B. Pulmonary Arterial hypertension is associated with oxidative stress–induced genome instability. Am. Thorac. Soc. 2015, 192. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. ET Biophys. Acta-Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed]
- King, N. Amino Acids and the Mitochondria. In Mitochondria: The Dynamic Organelle; Schaffer, S.W., Suleiman, M.S., Eds.; Springer: New York, NY, USA, 2007; pp. 151–166. [Google Scholar]
- Manoli, I.; Venditti, C.P. Disorders of branched chain amino acid metabolism. Transl. Sci. Rare. Dis. 2016, 1, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Wei, Y.-Y.; Yang, C.-C.; Liu, C.-J.; Yeh, L.-Y.; Chou, C.-H.; Chang, K.-W.; Lin, S.-C. miR-125b suppresses oral oncogenicity by targeting the anti-oxidative gene PRXL2A. Redox. Biol. 2019, 22, 101140. [Google Scholar] [CrossRef]
- Salazar, D.A.; Butler, V.J.; Argouarch, A.R.; Hsu, T.-Y.; Mason, A.; Nakamura, A.; McCurdy, H.; Cox, D.; Ng, R.; Pan, G.; et al. The progranulin cleavage products, granulins, exacerbate tdp-43 toxicity and increase tdp-43 levels. J. Neurosci. 2015, 35, 9315. [Google Scholar] [CrossRef]
- Ke, P.-Y. Mitophagy in the pathogenesis of liver diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef]
- Oku, M.; Maeda, Y.; Kagohashi, Y.; Kondo, T.; Yamada, M.; Fujimoto, T.; Sakai, Y. Evidence for ESCRT- and clathrin-dependent microautophagy. J. Cell Biol. 2017, 216, 3263–3274. [Google Scholar] [CrossRef]
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria—hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.; St. Croix, C.; et al. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohsen, A.-W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef] [PubMed]
- Acín-Pérez, R.; Bayona-Bafaluy, M.P.; Fernández-Silva, P.; Moreno-Loshuertos, R.; Pérez-Martos, A.; Bruno, C.; Moraes, C.T.; Enríquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Culley, M.K.; Chan, S.Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Investig. 2018, 128, 3704–3715. [Google Scholar] [CrossRef] [PubMed]
- Talati, M.; Hemnes, A. Fatty acid metabolism in pulmonary arterial hypertension: Role in right ventricular dysfunction and hypertrophy. Pulm. Circ. 2015, 5, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Cade, W.T.; Bohnert, K.L.; Peterson, L.R.; Patterson, B.W.; Bittel, A.J.; Okunade, A.L.; de las Fuentes, L.; Steger-May, K.; Bashir, A.; Schweitzer, G.G.; et al. Blunted fat oxidation upon submaximal exercise is partially compensated by enhanced glucose metabolism in children, adolescents, and young adults with Barth syndrome. J. Inherit. Metab. Dis. 2019, 42, 480–493. [Google Scholar] [CrossRef]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox. Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef]
- Guo, F.; He, H.; Fu, Z.-C.; Huang, S.; Chen, T.; Papasian, C.J.; Morse, L.R.; Xu, Y.; Battaglino, R.A.; Yang, X.-F.; et al. Adipocyte-derived PAMM suppresses macrophage inflammation by inhibiting MAPK signalling. Biochem. J. 2015, 472, 309–318. [Google Scholar] [CrossRef]
- Cantu, D.; Schaack, J.; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 2009, 4, e7095. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, W.; Xie, L.; Liu, T.; Liu, H.; Liu, B. Endoplasmic reticulum stress and pulmonary hypertension. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; Nasi, M.; De Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. ET Biophys. Acta-Bioenerg. 2016, 1857, 1300–1306. [Google Scholar] [CrossRef]
- Bota, D.A.; Davies, K.J.A. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef]
- Bender, T.; Lewrenz, I.; Franken, S.; Baitzel, C.; Voos, W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol. Biol. Cell 2011, 22, 541–554. [Google Scholar] [CrossRef]
- Rep, M.; van Dijl, J.M.; Suda, K.; Schatz, G.; Grivell, L.A.; Suzuki, C.K. Promotion of mitochondrial membrane complex assembly by a proteolytically inactive yeast Lon. Science 1996, 274, 103–106. [Google Scholar] [CrossRef]
- Quirós, P.M.; Español, Y.; Acín-Pérez, R.; Rodríguez, F.; Bárcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernández-García, M.S.; Fueyo, A.; et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef]
- Lee, H.J.; Chung, K.; Lee, H.; Lee, K.; Lim, J.H.; Song, J. Downregulation of mitochondrial lon protease impairs mitochondrial function and causes hepatic insulin resistance in human liver SK-HEP-1 cells. Diabetologia 2011, 54, 1437–1446. [Google Scholar] [CrossRef]
- Pareek, G.; Thomas, R.E.; Vincow, E.S.; Morris, D.R.; Pallanck, L.J. Lon protease inactivation in Drosophila causes unfolded protein stress and inhibition of mitochondrial translation. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef]
- Reddy, P.H. Misfolded proteins, mitochondrial dysfunction, and neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1167. [Google Scholar] [CrossRef][Green Version]
- Madamba, S.M.; Damri, K.N.; Dejean, L.M.; Peixoto, P.M. Mitochondrial Ion Channels in Cancer Transformation. Front. Oncol. 2015, 5, 120. [Google Scholar] [CrossRef][Green Version]
- Ryan, J.J.; Marsboom, G.; Fang, Y.-H.; Toth, P.T.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 865–878. [Google Scholar] [CrossRef]
- Horinokita, I.; Hayashi, H.; Oteki, R.; Mizumura, R.; Yamaguchi, T.; Usui, A.; Yuan, B.; Takagi, N. Involvement of progranulin and granulin expression in inflammatory responses after cerebral ischemia. Int. J. Mol. Sci. 2019, 20, 5210. [Google Scholar] [CrossRef]
- Chang, M.C.; Srinivasan, K.; Friedman, B.A.; Suto, E.; Modrusan, Z.; Lee, W.P.; Kaminker, J.S.; Hansen, D.V.; Sheng, M. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med. 2017, 214, 2611–2628. [Google Scholar] [CrossRef]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef]
- Pendergrass, S.A.; Hayes, E.; Farina, G.; Lemaire, R.; Farber, H.W.; Whitfield, M.L.; Lafyatis, R. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS ONE 2010, 5, e12106. [Google Scholar] [CrossRef]
- Niihori, M.; Eccles, C.A.; Kurdyukov, S.; Zemskova, M.; Varghese, M.V.; Stepanova, A.A.; Galkin, A.; Rafikov, R.; Rafikova, O. Rats with a human mutation of nfu1 develop pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2019, 62, 231–242. [Google Scholar] [CrossRef]
- Kruse, R.; Krantz, J.; Barker, N.; Coletta, R.L.; Rafikov, R.; Luo, M.; Højlund, K.; Mandarino, L.J.; Langlais, P.R. Characterization of the CLASP2 protein interaction network identifies soga1 as a microtubule-associated protein. Mol. Cell. Proteom. 2017, 16, 1718–1735. [Google Scholar] [CrossRef]
- Parker, S.S.; Krantz, J.; Kwak, E.-A.; Barker, N.K.; Deer, C.G.; Lee, N.Y.; Mouneimne, G.; Langlais, P.R. Insulin induces microtubule stabilization and regulates the microtubule plus-end tracking protein network in adipocytes. Mol. Cell. Proteom. 2019, 18, 1363–1381. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Tyanova, S.; Cox, J. Perseus: A bioinformatics platform for integrative analysis of proteomics data in cancer research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
James, J.; Valuparampil Varghese, M.; Vasilyev, M.; Langlais, P.R.; Tofovic, S.P.; Rafikova, O.; Rafikov, R. Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape. Int. J. Mol. Sci. 2020, 21, 5683. https://doi.org/10.3390/ijms21165683
James J, Valuparampil Varghese M, Vasilyev M, Langlais PR, Tofovic SP, Rafikova O, Rafikov R. Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape. International Journal of Molecular Sciences. 2020; 21(16):5683. https://doi.org/10.3390/ijms21165683
Chicago/Turabian StyleJames, Joel, Mathews Valuparampil Varghese, Mikhail Vasilyev, Paul R. Langlais, Stevan P. Tofovic, Olga Rafikova, and Ruslan Rafikov. 2020. "Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape" International Journal of Molecular Sciences 21, no. 16: 5683. https://doi.org/10.3390/ijms21165683
APA StyleJames, J., Valuparampil Varghese, M., Vasilyev, M., Langlais, P. R., Tofovic, S. P., Rafikova, O., & Rafikov, R. (2020). Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape. International Journal of Molecular Sciences, 21(16), 5683. https://doi.org/10.3390/ijms21165683