A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance

 , ,
, , 
Abstract
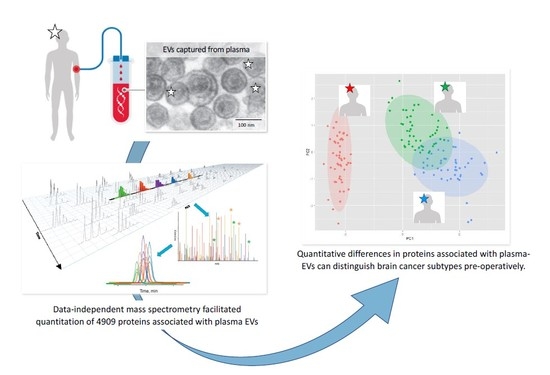
1. Introduction
2. Results
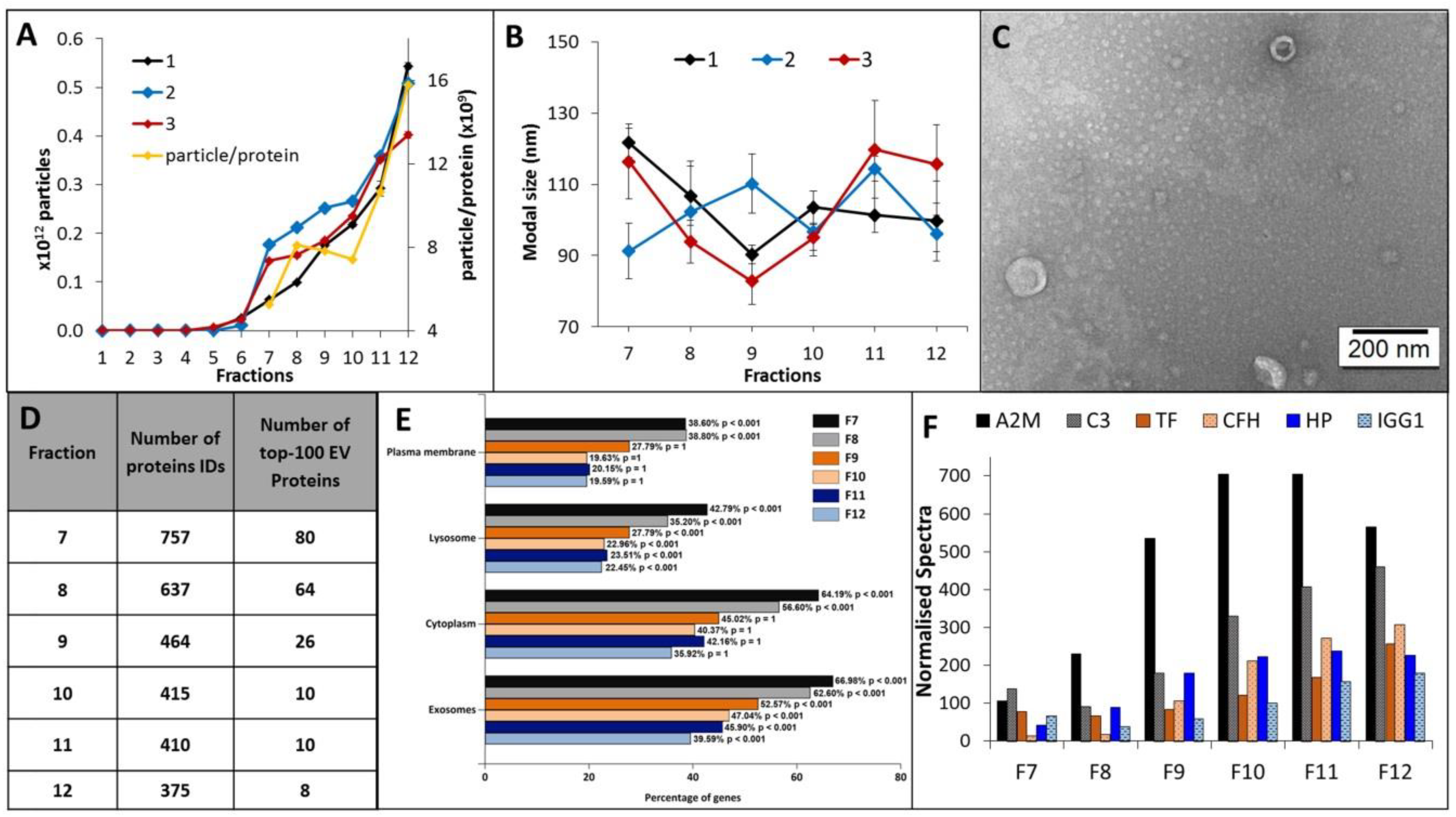
2.1. Characterisation of Plasma-EVs
2.2. Assembling a Spectral Library Tailored to Glioma SWATH Analysis
2.3. SWATH-MS Data Extraction, Quality and Reproducibility
2.4. SWATH-MS Based Characterisation of Glioma Plasma-EV Proteomes
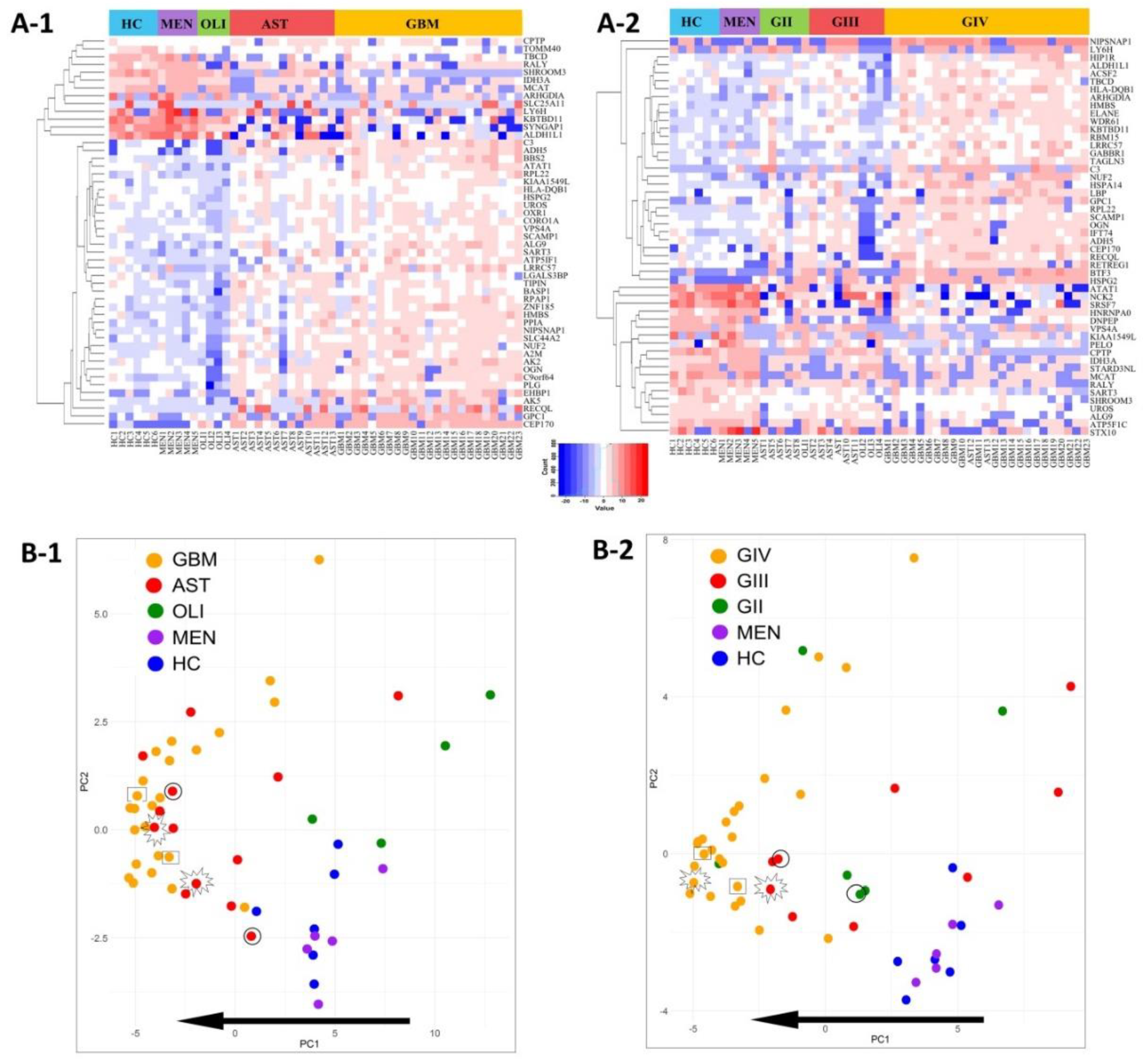
2.5. Glioma Plasma-EV Protein Profiles
2.6. Putative Plasma-EV Biomarkers for Distinguishing Glioma Subtypes
2.7. Comparisons to Previous GBM-EV Proteomics Studies
3. Discussion
3.1. SWATH Produces High-Quality Proteomic Data for Plasma-EV Biomarker Discovery
3.2. Plasma-EV Proteomes Reflect Glioma Subtype and Aggression
3.3. Links to Previous GBM-EV Studies
3.4. T-Complex Protein 1 Ring Complex (TRiC) Interactome May Indicate GBM Presence and/or Progression
4. Materials and Methods
4.1. Pre-Operative Plasma Specimens and Patient Cohorts
4.2. Plasma Processing and EV Isolation
4.3. EV Characterisation by Nanoparticle Tracking Analysis, Transmission Electron Microscopy and LC-MS/MS
4.4. Preparation of EV Proteomes for LC-MS/MS
4.5. Generating a Custom, Comprehensive Glioma Spectral Library
4.6. SWATH-MS Data Capture and Peak Extraction
4.7. Differential Expression (DE) Analysis and Statistics
4.8. In Silico Analysis
4.9. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| AST | Astrocytoma |
| CSF | Cerebrospinal fluid |
| DIA | Data independent acquisition |
| DE | Differential expression/differentially expressed |
| EV | Extracellular vesicle |
| FA | Formic acid |
| GBM | Glioblastoma |
| GIV | Grade IV |
| GII-III | Grade II–III |
| HC | Healthy control |
| HILIC | Hydrophilic liquid chromatography |
| HPLC | High performance liquid chromatography |
| ID | Identification |
| IDA | Information dependent acquisition |
| IDH | Isocitrate dehydrogenase |
| iRT | Indexed retention time |
| LC- | Liquid chromatography tandem mass spectrometry |
| MEN | Meningioma |
| MRI | Magnetic resonance imaging |
| MS | Mass spectrometry |
| Mut | Mutation |
| NA | Not applicable |
| NTA | Nanoparticle tracking analysis |
| PBS | Phosphate buffered saline |
| PCA | Principal component analysis |
| RANO | Response Assessment in Neuro-Oncology |
| RT | Room temperature |
| SEC | Size exclusion chromatography |
| SWATH | Sequential window acquisition of all theoretic fragment ion spectra |
| TCEP | Tris(2-carboxyl) phosphine |
| TEAB | Tetraethylammonium bicarbonate |
| TEM | Transmission electron microscopy |
| TFA | Trifluoroacetic acid |
| TMZ | Temozolomide |
| TRiC | T-complex protein 1 ring complex |
| wt | Wild-type |
References
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Khasraw, M.; Lassman, A.B. Advances in the Treatment of Malignant Gliomas. Curr. Oncol. Rep. 2010, 12, 26–33. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C. Diffuse Gliomas for Nonneuropathologists: The New Integrated Molecular Diagnostics. Arch. Pathol. Lab. Med. 2018, 142, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgak, i.H.; Wiestler, O.D.; Cavenee, W.K. World Health Organization Histological Classification of Tumours of the Central Nervous System; International Agency for Research on Cancer: Lyon, France, 2016. [Google Scholar]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- De la Garza-Ramos, R.; Kerezoudis, P.; Tamargo, R.J.; Brem, H.; Huang, J.; Bydon, M. Surgical complications following malignant brain tumor surgery: An analysis of 2002–2011 data. Clin. Neurol. Neurosurg. 2016, 140, 6–10. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Hygino da Cruz, L.C., Jr.; Rodriguez, I.; Domingues, R.C.; Gasparetto, E.L.; Sorensen, A.G. Pseudoprogression and pseudoresponse: Imaging challenges in the assessment of posttreatment glioma. AJNR Am. J. Neuroradiol. 2011, 32, 1978–1985. [Google Scholar] [CrossRef]
- Abdulla, S.; Saada, J.; Johnson, G.; Jefferies, S.; Ajithkumar, T. Tumour progression or pseudoprogression? A review of post-treatment radiological appearances of glioblastoma. Clin. Radiol. 2015, 70, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.W.; Westerlaan, H.E.; Holtman, G.A.; Aden, K.M.; van Laar, P.J.; van der Hoorn, A. Incidence of Tumour Progression and Pseudoprogression in High-Grade Gliomas: A Systematic Review and Meta-Analysis. Clin. Neuroradiol. 2018, 28, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Smirniotopoulos, J.G.; Murphy, F.M.; Rushing, E.J.; Rees, J.H.; Schroeder, J.W. Patterns of Contrast Enhancement in the Brain and Meninges. RadioGraphics 2007, 27, 525–551. [Google Scholar] [CrossRef]
- Balaña, C.; Capellades, J.; Pineda, E.; Estival, A.; Puig, J.; Domenech, S.; Verger, E.; Pujol, T.; Martinez-García, M.; Oleaga, L.; et al. Pseudoprogression as an adverse event of glioblastoma therapy. Cancer Med. 2017, 6, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Alex, Y.L.; Jack, L.T.; Eyiyemisi, C.D.; Jie, L.; Ahmed, K.A.; Tore, E.; Alexander, O.V.; Veronica, L.C. Novel biomarker identification using metabolomic profiling to differentiate radiation necrosis and recurrent tumor following Gamma Knife radiosurgery. J. Neurosurg. JNS 2017, 127, 388–396. [Google Scholar]
- Zikou, A.; Sioka, C.; Alexiou, G.A.; Fotopoulos, A.; Voulgaris, S.; Argyropoulou, M.I. Radiation Necrosis, Pseudoprogression, Pseudoresponse, and Tumor Recurrence: Imaging Challenges for the Evaluation of Treated Gliomas. Contrast Media Mol. Imaging 2018, 2018. [Google Scholar] [CrossRef]
- Detsky, J.S.; Keith, J.; Conklin, J.; Symons, S.; Myrehaug, S.; Sahgal, A.; Heyn, C.C.; Soliman, H. Differentiating radiation necrosis from tumor progression in brain metastases treated with stereotactic radiotherapy: Utility of intravoxel incoherent motion perfusion MRI and correlation with histopathology. J. Neuro-Oncol. 2017, 134, 433–441. [Google Scholar] [CrossRef]
- Ellingson, B.M.; Wen, P.Y.; Cloughesy, T.F. Modified Criteria for Radiographic Response Assessment in Glioblastoma Clinical Trials. Neurotherapeutics 2017, 14, 307–320. [Google Scholar] [CrossRef]
- Hallal, S.; Ebrahimkhani, S.; Shivalingam, B.; Graeber, M.B.; Kaufman, K.L.; Buckland, M.E. The emerging clinical potential of circulating extracellular vesicles for non-invasive glioma diagnosis and disease monitoring. Brain Tumor Pathol. 2019, 36, 29–39. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; Zijl, S.; Reijneveld, J.C.; Wesseling, P.; Wurdinger, T. Liquid biopsies in patients with diffuse glioma. Acta Neuropathol. 2015, 129, 849–865. [Google Scholar] [CrossRef]
- Santiago-Dieppa, D.R.; Steinberg, J.; Gonda, D.; Cheung, V.J.; Carter, B.S.; Chen, C.C. Extracellular vesicles as a platform for ‘liquid biopsy’ in glioblastoma patients. Expert Rev. Mol. Diagn. 2014, 14, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Song, K.S.; Park, Y.S.; Kang, Y.H.; Lee, Y.J.; Lee, K.R.; Kim, H.K.; Ryu, K.W.; Bae, J.M.; Kim, S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur. J. Cancer 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Zwicker, J.I.; Liebman, H.A.; Neuberg, D.; Lacroix, R.; Bauer, K.A.; Furie, B.C.; Furie, B. Tumor-Derived Tissue Factor–Bearing Microparticles Are Associated with Venous Thromboembolic Events in Malignancy. Clin. Cancer Res. 2009, 15, 6830–6840. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Alzate, O.; Dechkovskaia, A.M.; Keene, J.D.; Sampson, J.H.; Mitchell, D.A.; Bigner, D.D. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1541–1557. [Google Scholar] [CrossRef]
- Figueroa, J.M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro Oncol. 2017, 19, 1494–1502. [Google Scholar] [CrossRef]
- Ebrahimkhani, S.; Vafaee, F.; Hallal, S.; Wei, H.; Lee, M.Y.T.; Young, P.E.; Satgunaseelan, L.; Beadnall, H.; Barnett, M.H.; Shivalingam, B.; et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef]
- Hallal, S.; Russell, B.P.; Wei, H.; Lee, M.Y.T.; Toon, C.W.; Sy, J.; Shivalingam, B.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles from Neurosurgical Aspirates Identifies Chaperonin Containing TCP1 Subunit 6A as a Potential Glioblastoma Biomarker with Prognostic Significance. Proteomics 2019, 19, e1800157. [Google Scholar] [CrossRef]
- Mallawaaratchy, D.M.; Hallal, S.; Russell, B.; Ly, L.; Ebrahimkhani, S.; Wei, H.; Christopherson, R.I.; Buckland, M.E.; Kaufman, K.L. Comprehensive proteome profiling of glioblastoma-derived extracellular vesicles identifies markers for more aggressive disease. J. Neuro-Oncol. 2017, 131, 233–244. [Google Scholar] [CrossRef]
- Melki, I.; Tessandier, N.; Zufferey, A.; Boilard, E. Platelet microvesicles in health and disease. Platelets 2017, 28, 214–221. [Google Scholar] [CrossRef]
- Reategui, E.; van der Vos, K.E.; Lai, C.P.; Zeinali, M.; Atai, N.A.; Aldikacti, B.; Floyd, F.P., Jr.; Khankhel, A.H.; Thapar, V.; Hochberg, F.H.; et al. Engineered nanointerfaces for microfluidic isolation and molecular profiling of tumor-specific extracellular vesicles. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Cvjetkovic, A.; Jang, S.C.; Konecna, B.; Hoog, J.L.; Sihlbom, C.; Lasser, C.; Lotvall, J. Detailed Analysis of Protein Topology of Extracellular Vesicles-Evidence of Unconventional Membrane Protein Orientation. Sci. Rep. 2016, 8, 36338. [Google Scholar] [CrossRef] [PubMed]
- Redzic, J.S.; Ung, T.H.; Graner, M.W. Glioblastoma extracellular vesicles: Reservoirs of potential biomarkers. Pharm. Pers. Med. 2014, 7, 65–77. [Google Scholar]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 846–867. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, D.; Kirchner, B.; Hermann, S.; Märte, M.; Wurmser, C.; Brandes, F.; Kotschote, S.; Bonin, M.; Steinlein, O.K.; Pfaffl, M.W.; et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J. Extracell. Vesicles 2018, 7, 1481321. [Google Scholar] [CrossRef]
- Prakash, A.; Peterman, S.; Ahmad, S.; Sarracino, D.; Frewen, B.; Vogelsang, M.; Byram, G.; Krastins, B.; Vadali, G.; Lopez, M. Hybrid data acquisition and processing strategies with increased throughput and selectivity: pSMART analysis for global qualitative and quantitative analysis. J. Proteome Res. 2014, 13, 5415–5430. [Google Scholar] [CrossRef]
- Hathout, Y. Proteomic methods for biomarker discovery and validation. Are we there yet? Expert Rev. Proteom. 2015, 12, 329–331. [Google Scholar] [CrossRef][Green Version]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef]
- Miyauchi, E.; Furuta, T.; Ohtsuki, S.; Tachikawa, M.; Uchida, Y.; Sabit, H.; Obuchi, W.; Baba, T.; Watanabe, M.; Terasaki, T.; et al. Identification of blood biomarkers in glioblastoma by SWATH mass spectrometry and quantitative targeted absolute proteomics. PLoS ONE 2018, 13, e0193799. [Google Scholar] [CrossRef]
- Collins, B.C.; Gillet, L.C.; Rosenberger, G.; Rost, H.L.; Vichalkovski, A.; Gstaiger, M.; Aebersold, R. Quantifying protein interaction dynamics by SWATH mass spectrometry: Application to the 14-3-3 system. Nat. Methods 2013, 10, 1246–1253. [Google Scholar] [CrossRef]
- Croft, N.P.; de Verteuil, D.A.; Smith, S.A.; Wong, Y.C.; Schittenhelm, R.B.; Tscharke, D.C.; Purcell, A.W. Simultaneous Quantification of Viral Antigen Expression Kinetics Using Data-Independent (DIA) Mass Spectrometry. Mol. Cell. Proteom. 2015, 14, 1361–1372. [Google Scholar] [CrossRef]
- Schubert, O.T.; Gillet, L.C.; Collins, B.C.; Navarro, P.; Rosenberger, G.; Wolski, W.E.; Lam, H.; Amodei, D.; Mallick, P.; MacLean, B.; et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat. Protoc. 2015, 10, 426–441. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, X.; Chen, S.; Zhang, Y.; Zhi, F.; Wang, J.; Li, L.; Zhou, X.; Li, N.; Pan, H.; et al. Identification of seven serum microRNAs from a genome-wide serum microRNA expression profile as potential noninvasive biomarkers for malignant astrocytomas. Int. J. Cancer 2013, 132, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-S.; Gho, Y.S. Isolation of extracellular vesicles for proteomic profiling. In Methods in Molecular Biology; Clifton, N.J., Ed.; Humana Press: New York, NY, USA, 2015; p. 167. [Google Scholar]
- Choi, D.; Montermini, L.; Kim, D.K.; Meehan, B.; Roth, F.P.; Rak, J. The Impact of Oncogenic EGFRvIII on the Proteome of Extracellular Vesicles Released from Glioblastoma Cells. Mol. Cell. Proteom. 2018, 17, 1948–1964. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, S.; Belov, A.M.; Ghiran, I.; Murthy, S.K.; Frank, D.A.; Ivanov, A.R. Mass-Spectrometry-Based Molecular Characterization of Extracellular Vesicles: Lipidomics and Proteomics. J. Proteome Res. 2015, 14, 2367–2384. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Baffour, S.; Adjei, J.; Aryeh, C.; Kyeremeh, R.; Kyei, F.; Seidu, M.A. Understanding the biosynthesis of platelets-derived extracellular vesicles. Immun. Inflamm. Dis. 2015, 3, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Wang, C.; Lu, X.; Zhang, C.; Zhou, Z.; Chen, X.; Zhang, C.-Y.; Zen, K.; Zhang, C. Comparison of commercial exosome isolation kits for circulating exosomal microRNA profiling. Anal. Bioanal. Chem. 2018, 410, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Takov, K.; Yellon, D.M.; Davidson, S.M. Comparison of small extracellular vesicles isolated from plasma by ultracentrifugation or size-exclusion chromatography: Yield, purity and functional potential. J. Extracell. Vesicles 2018, 8, 1560809. [Google Scholar] [CrossRef]
- An, M.; Wu, J.; Zhu, J.; Lubman, D.M. Comparison of an Optimized Ultracentrifugation Method versus Size-Exclusion Chromatography for Isolation of Exosomes from Human Serum. J. Proteome Res. 2018, 17, 3599–3605. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Escher, C.; Reiter, L.; MacLean, B.; Ossola, R.; Herzog, F.; Chilton, J.; MacCoss, M.J.; Rinner, O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 2012, 12, 1111–1121. [Google Scholar] [CrossRef]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Messenger, S.W.; Woo, S.S.; Sun, Z.; Martin, T.F.J. A Ca2+-stimulated exosome release pathway in cancer cells is regulated by Munc13-4. J. Cell Biol. 2018, 217, 2877–2890. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Hu, Q.; Liu, Y.; Wang, Z.; Cui, X.; Li, R.; Yan, W.; You, Y. IDH1/2 mutation status combined with Ki-67 labeling index defines distinct prognostic groups in glioma. Oncotarget 2015, 6, 30232–30238. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef]
- Wang, S.; Li, M.; Xing, L.; Yu, J. High expression level of peptidylprolyl isomerase A is correlated with poor prognosis of liver hepatocellular carcinoma. Oncol. Lett. 2019, 18, 4691–4702. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.S. Current implications of cyclophilins in human cancers. J. Exp. Clin. Cancer Res. CR 2010, 29, 97. [Google Scholar] [CrossRef]
- Herman, A.; Gruden, K.; Blejec, A.; Podpecan, V.; Motaln, H.; Rozman, P.; Hren, M.; Zupancic, K.; Veber, M.; Verbovsek, U.; et al. Analysis of Glioblastoma Patients’ Plasma Revealed the Presence of MicroRNAs with a Prognostic Impact on Survival and Those of Viral Origin. PLoS ONE 2015, 10, e0125791. [Google Scholar] [CrossRef]
- Bouwens, T.A.; Trouw, L.A.; Veerhuis, R.; Dirven, C.M.F.; Lamfers, M.L.M.; Al-Khawaja, H. Complement activation in Glioblastoma Multiforme pathophysiology: Evidence from serum levels and presence of complement activation products in tumor tissue. J. Neuroimmunol. 2015, 278, 271–276. [Google Scholar] [CrossRef]
- Koochekpour, S.; Zhuang, Y.; Beroukhim, R.; Hsieh, C.-L.; Hofer, M.D.; Zhau, H.E.; Hiraiwa, M.; Pattan, D.Y.; Ware, J.L.; Luftig, R.B.; et al. Amplification and overexpression of prosaposin in prostate cancer. Genes Chromosomes Cancer 2005, 44, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhou, J.; Luo, P.; Gao, H.; Ma, Y.; Chen, Y.-S.; Li, L.; Zou, D.; Zhang, Y.; Jing, Z. Prosaposin promotes the proliferation and tumorigenesis of glioma through toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway. EBioMedicine 2018, 37, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhou, J.; Hou, D.; Luo, P.; Gao, H.; Ma, Y.; Chen, Y.S.; Li, L.; Zou, D.; Zhang, H.; et al. Prosaposin is a biomarker of mesenchymal glioblastoma and regulates mesenchymal transition through the TGF-beta1/Smad signaling pathway. J. Pathol. 2019, 249, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Halvorsen, O.J.; Gravdal, K.; Bhattacharya, N.; Lee, J.M.; Liu, N.W.; Johnston, B.T.; Johnston, A.B.; Haukaas, S.A.; Aamodt, K.; et al. Prosaposin inhibits tumor metastasis via paracrine and endocrine stimulation of stromal p53 and Tsp-1. Proc. Natl. Acad. Sci. USA 2009, 106, 12115–12120. [Google Scholar] [CrossRef]
- Koochekpour, S.; Hu, S.; Vellasco-Gonzalez, C.; Bernardo, R.; Azabdaftari, G.; Zhu, G.; Zhau, H.E.; Chung, L.W.K.; Vessella, R.L. Serum prosaposin levels are increased in patients with advanced prostate cancer. Prostate 2012, 72, 253–269. [Google Scholar] [CrossRef]
- Hamilton, J.D.; Rapp, M.; Schneiderhan, T.; Sabel, M.; Hayman, A.; Scherer, A.; Kropil, P.; Budach, W.; Gerber, P.; Kretschmar, U.; et al. Glioblastoma multiforme metastasis outside the CNS: Three case reports and possible mechanisms of escape. J. Clin. Oncol. 2014, 32, e80–e84. [Google Scholar] [CrossRef]
- Dowling, P.; Meleady, P.; Dowd, A.; Henry, M.; Glynn, S.; Clynes, M. Proteomic analysis of isolated membrane fractions from superinvasive cancer cells. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2007, 1774, 93–101. [Google Scholar] [CrossRef]
- Walker, L.C.; Waddell, N.; Ten Haaf, A.; kConFab Investigators; Grimmond, S.; Spurdle, A.B. Use of expression data and the CGEMS genome-wide breast cancer association study to identify genes that may modify risk in BRCA1/2 mutation carriers. Breast Cancer Res. Treat. 2008, 112, 229–236. [Google Scholar] [CrossRef]
- Borczuk, A.C.; Gorenstein, L.; Walter, K.L.; Assaad, A.A.; Wang, L.; Powell, C.A. Non-Small-Cell Lung Cancer Molecular Signatures Recapitulate Lung Developmental Pathways. Am. J. Pathol. 2003, 163, 1949–1960. [Google Scholar] [CrossRef]
- Roh, S.H.; Kasembeli, M.; Bakthavatsalam, D.; Chiu, W.; Tweardy, D.J. Contribution of the Type II Chaperonin, TRiC/CCT, to Oncogenesis. Int. J. Mol. Sci. 2015, 16, 26706–26720. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Joachimiak, L.; Bracher, A.; Monkemeyer, L.; Walzthoeni, T.; Chen, B.; Pechmann, S.; Holmes, S.; Cong, Y.; Ma, B.; et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 2012, 20, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Yam, A.Y.; Xia, Y.F.; Lin, H.-T.J.; Burlingame, A.; Gerstein, M.; Frydman, J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 2008, 15, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Vallin, J.; Grantham, J. The role of the molecular chaperone CCT in protein folding and mediation of cytoskeleton-associated processes: Implications for cancer cell biology. Cell Stress Chaperones 2019, 24, 17–27. [Google Scholar] [CrossRef]
- Kasembeli, M.; Lau, W.C.; Roh, S.H.; Eckols, T.K.; Frydman, J.; Chiu, W.; Tweardy, D.J. Modulation of STAT3 folding and function by TRiC/CCT chaperonin. PLoS Biol. 2014, 12, e1001844. [Google Scholar] [CrossRef]
- Trinidad, A.G.; Muller, P.; Cuellar, J.; Klejnot, M.; Nobis, M.; Valpuesta, J.M.; Vousden, K.H. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol. Cell 2013, 50, 805–817. [Google Scholar] [CrossRef]
- Behrends, C.; Langer, C.; Boteva, R.; Bottcher, U.M.; Stemp, M.J.; Schaffar, G.; Rao, B.V.; Giese, A.; Kretzschmar, H.; Siegers, K.; et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol. Cell. 2006, 23, 887–897. [Google Scholar] [CrossRef]
- Posokhova, E.; Song, H.; Belcastro, M.; Higgins, L.; Bigley, L.R.; Michaud, N.A.; Martemyanov, K.A.; Sokolov, M. Disruption of the chaperonin containing TCP-1 function affects protein networks essential for rod outer segment morphogenesis and survival. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef]
- Bouhouche, A.; Benomar, A.; Bouslam, N.; BChkili, T.; Yahyaoui, M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 2006, 43, 441–443. [Google Scholar] [CrossRef]
- Lee, M.J.; Stephenson, D.; Groves, M.J.; Sweeney, M.G.; Davis, M.B.; An, S.-F.; Houlden, H.; Salih, M.A.M.; Timmerman, V.; de Jonghe, P.; et al. Hereditary sensory neuropathy is caused by a mutation in the delta subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct4) gene. Hum. Mol. Genet. 2003, 12, 1917–1925. [Google Scholar] [CrossRef]
- Khaled, A.R.; Showalter, A.; Limaye, A.; Bassiouni, R.; Khaled, A.S.; Oyer, J.; Igarashi, R.; Copik, A. Harnessing the immune response for the treatment of cancer by inducing immunogenic cell death through inhibition of a group II chaperonin. J. Immunol. 2017, 198 (Suppl. 1), 76.6. [Google Scholar]
- Sergeeva, O.A.; Chen, B.; Haase-Pettingell, C.; Ludtke, S.J.; Chiu, W.; King, J.A. Human CCT4 and CCT5 chaperonin subunits expressed in Escherichia coli form biologically active homo-oligomers. J. Biol. Chem. 2013, 288, 17734–17744. [Google Scholar] [CrossRef] [PubMed]
- Ong, D.S.T.; Hu, B.; Ho, Y.W.; Sauve, C.G.; Bristow, C.A.; Wang, Q.; Multani, A.S.; Chen, P.; Nezi, L.; Jiang, S.; et al. PAF promotes stemness and radioresistance of glioma stem cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9086–E9095. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Feng, L.; Liu, D.; Zhang, L.; Wu, B.; Jiang, W.; Han, Z.; Cheng, S. Quantitative proteomics reveals the novel co-expression signatures in early brain development for prognosis of glioblastoma multiforme. Oncotarget 2016, 7, 14161–14171. [Google Scholar] [CrossRef]
- Guest, S.T.; Kratche, Z.R.; Bollig-Fischer, A.; Haddad, R.; Ethier, S.P. Two members of the TRiC chaperonin complex, CCT2 and TCP1 are essential for survival of breast cancer cells and are linked to driving oncogenes. Exp. Cell Res. 2015, 332, 223–235. [Google Scholar] [CrossRef]
- Jabs, V.; Edlund, K.; König, H.; Grinberg, M.; Madjar, K.; Rahnenführer, J.; Ekman, S.; Bergkvist, M.; Holmberg, L.; Ickstadt, K.; et al. Integrative analysis of genome-wide gene copy number changes and gene expression in non-small cell lung cancer. PLoS ONE 2017, 12, e0187246. [Google Scholar] [CrossRef]
- Deng, L.; Xiong, P.; Luo, Y.; Bu, X.; Qian, S.; Zhong, W. Bioinformatics analysis of the molecular mechanism of diffuse intrinsic pontine glioma. Oncol. Lett. 2016, 12, 2524–2530. [Google Scholar] [CrossRef]
- Pucci-Minafra, I.; Di Cara, G.; Musso, R.; Cancemi, P.; Albanese, N.N.; Roz, E.; Minafra, S. Retrospective Proteomic Screening of 100 Breast Cancer Tissues. Proteomes 2017, 5, 15. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. DJ-1(PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef]
- Hinkle, D.A.; Mullett, S.J.; Gabris, B.E.; Hamilton, R.L. DJ-1 expression in glioblastomas shows positive correlation with p53 expression and negative correlation with epidermal growth factor receptor amplification. Neuropathology 2011, 31, 29–37. [Google Scholar] [CrossRef]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Shivaji, S. The Role of DJ-1 in the Pathogenesis of Endometriosis. PLoS ONE 2011, 6, e18074. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, H.; Zhang, L.; Liu, X.; Zhang, C.; Wang, Y.; He, Q.; Zhang, Y.; Li, Y.; Chen, Q.; et al. DJ-1 promotes colorectal cancer progression through activating PLAGL2/Wnt/BMP4 axis. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Sharma, S. An appraisal of RECQ1 expression in cancer progression. Front. Genet. 2014, 5, 426. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Brosh, R.M., Jr. Human RECQ1 Is a DNA Damage Responsive Protein Required for Genotoxic Stress Resistance and Suppression of Sister Chromatid Exchanges. PLoS ONE 2007, 2, e1297. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Maldonado, R.; Faoro, V.; Bajpai, S.; Berti, M.; Odreman, F.; Vindigni, M.; Ius, T.; Ghasemian, A.; Bonin, S.; Skrap, M.; et al. The human RECQ1 helicase is highly expressed in glioblastoma and plays an important role in tumor cell proliferation. Mol. Cancer 2011, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nasiri Kenari, A.; Kastaniegaard, K.; Greening, D.W.; Shambrook, M.; Stensballe, A.; Cheng, L.; Hill, A.F. Proteomic and Post-Translational Modification Profiling of Exosome-Mimetic Nanovesicles Compared to Exosomes. Proteomics 2019, 19, 1800161. [Google Scholar] [CrossRef]
- Dragovic, R.A.; Dobson, P.J.; Harrison, P.; Sargent, I.L.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.P.; Hole, P.; Carr, B.; et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 780–788. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2018, 56, 4566–4581. [Google Scholar] [CrossRef]
- Mallawaaratchy, D.M.; Buckland, M.E.; McDonald, K.L.; Li, C.C.; Ly, L.; Sykes, E.K.; Christopherson, R.I.; Kaufman, K.L. Membrane proteome analysis of glioblastoma cell invasion. J. Neuropathol. Exp. Neurol. 2015, 74, 425–441. [Google Scholar] [CrossRef]
- Boersema, P.J.; Mohammed, S.; Heck, A.J. Hydrophilic interaction liquid chromatography (HILIC) in proteomics. Anal. Bioanal. Chem. 2008, 391, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Azimi, A.; Yang, P.; Ali, M.; Howard, V.; Mann, G.J.; Kaufman, K.L.; Fernandez-Penas, P. Data independent acquisition proteomic analysis can discriminate between actinic keratosis, Bowen’s disease and cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2020, 140, 212–222.e11. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.; Rinner, O.; Picotti, P.; Hüttenhain, R.; Beck, M.; Brusniak, M.-Y.; Hengartner, M.O.; Aebersold, R. mProphet: Automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 2011, 8, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Eldh, M.; Olofsson Bagge, R.; Lasser, C.; Svanvik, J.; Sjostrand, M.; Mattsson, J.; Lindner, P.; Choi, D.S.; Gho, Y.S.; Lotvall, J. MicroRNA in exosomes isolated directly from the liver circulation in patients with metastatic uveal melanoma. BMC Cancer 2014, 14, 962. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic-Histological Subtype | ||||
| Cohort | Sample (n) | Gender | Age at Resection (Mean ± SD) | Histopathological Diagnosis |
| GBM | 24 | 17M/7F | 60.8 ± 14.1 | Primary Glioblastoma, IDH-wildtype, WHO grade IV |
| AST | 13 | 10M/3F | 40.1 ± 13.8 | Astrocytoma, IDH-mutant; WHO grade II (n = 5), grade III (n = 6) and grade IV (n = 2) |
| OLI | 4 | 4M | 36.5 ± 3.7 | Oligodendroglioma, IDH-mutant; WHO grade II (n = 1) and grade III (n = 3) |
| WHO Grade | ||||
| Cohort | Sample (n) | Gender | Age at Resection (Mean ± SD) | Histopathological Diagnosis |
| GIV | 26 | 19M/7F | 59.4 ± 14.6 | WHO grade IV astrocytoma; IDH-wildtype (n = 24) and IDH-mutant Glioblastoma (n = 2) |
| GIII | 9 | 8M/1F | 39.8 ± 13.5 | WHO grade III glioma; IDH-mutant astrocytoma and IDH-mutant oligodendroglioma |
| GII | 6 | 4M/2F | 37.3 ± 11.9 | Grade II glioma; IDH-mutant astrocytoma and IDH-mutant oligodendroglioma |
| Controls | ||||
| Cohort | Sample (n) | Gender | Age at Resection (Mean ± SD) | Histopathological Diagnosis |
| MEN | 5 | 3M/2F | 57.6 ± 11.0 | Meningioma, grade I (non-glioma controls) |
| HC | 6 | 3M/3F | 46.8 ± 7.4 | Healthy controls |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hallal, S.; Azimi, A.; Wei, H.; Ho, N.; Lee, M.Y.T.; Sim, H.-W.; Sy, J.; Shivalingam, B.; Buckland, M.E.; Alexander-Kaufman, K.L. A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance. Int. J. Mol. Sci. 2020, 21, 4754. https://doi.org/10.3390/ijms21134754
Hallal S, Azimi A, Wei H, Ho N, Lee MYT, Sim H-W, Sy J, Shivalingam B, Buckland ME, Alexander-Kaufman KL. A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance. International Journal of Molecular Sciences. 2020; 21(13):4754. https://doi.org/10.3390/ijms21134754
Chicago/Turabian StyleHallal, Susannah, Ali Azimi, Heng Wei, Nicholas Ho, Maggie Yuk Ting Lee, Hao-Wen Sim, Joanne Sy, Brindha Shivalingam, Michael Edward Buckland, and Kimberley Louise Alexander-Kaufman. 2020. "A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance" International Journal of Molecular Sciences 21, no. 13: 4754. https://doi.org/10.3390/ijms21134754
APA StyleHallal, S., Azimi, A., Wei, H., Ho, N., Lee, M. Y. T., Sim, H.-W., Sy, J., Shivalingam, B., Buckland, M. E., & Alexander-Kaufman, K. L. (2020). A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance. International Journal of Molecular Sciences, 21(13), 4754. https://doi.org/10.3390/ijms21134754