The Novel gem-Dihydroperoxide 12AC3O Suppresses High Phosphate-Induced Calcification via Antioxidant Effects in p53LMAco1 Smooth Muscle Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
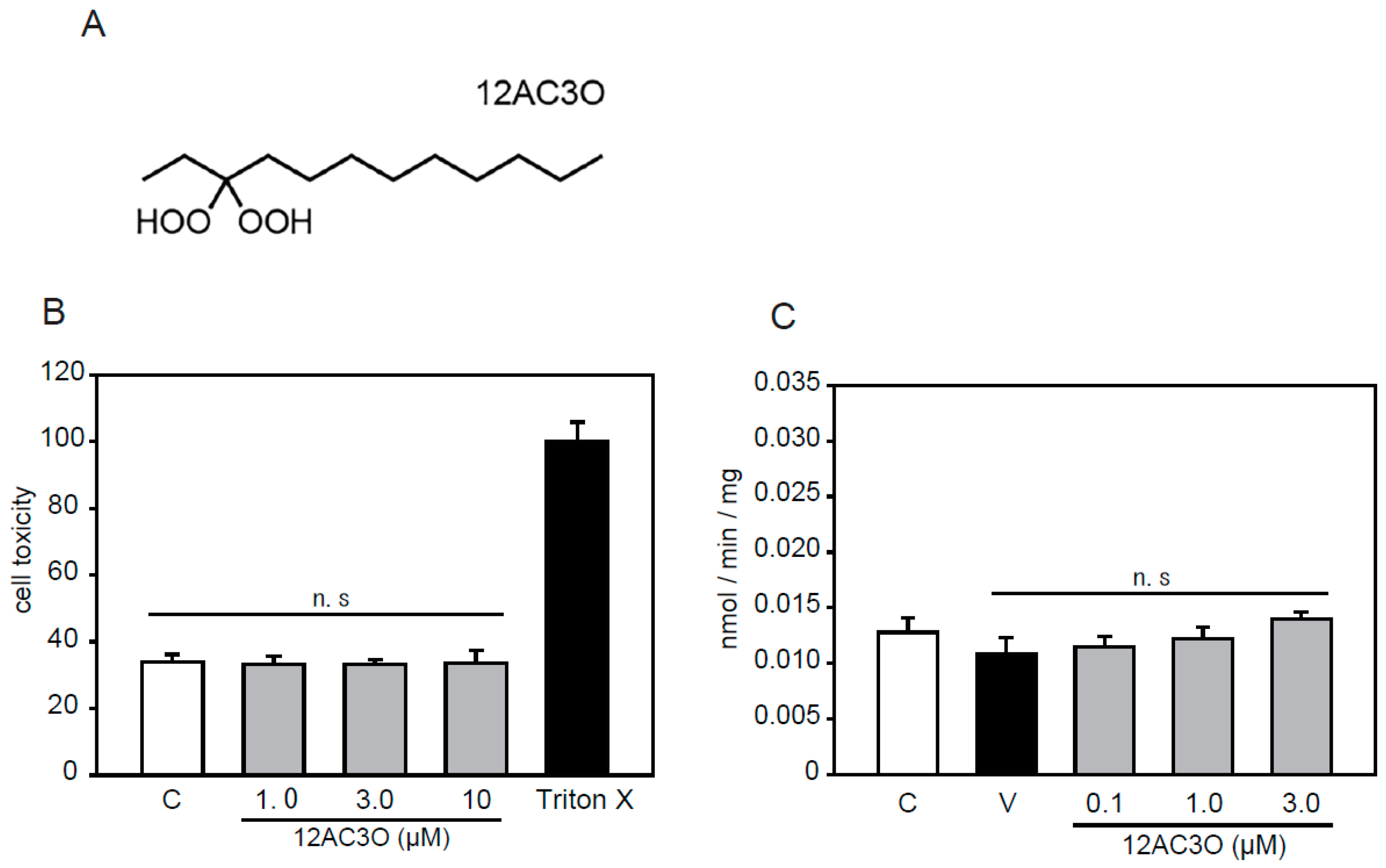
2.1. Selection of DHPs and Cell Toxicity Assay
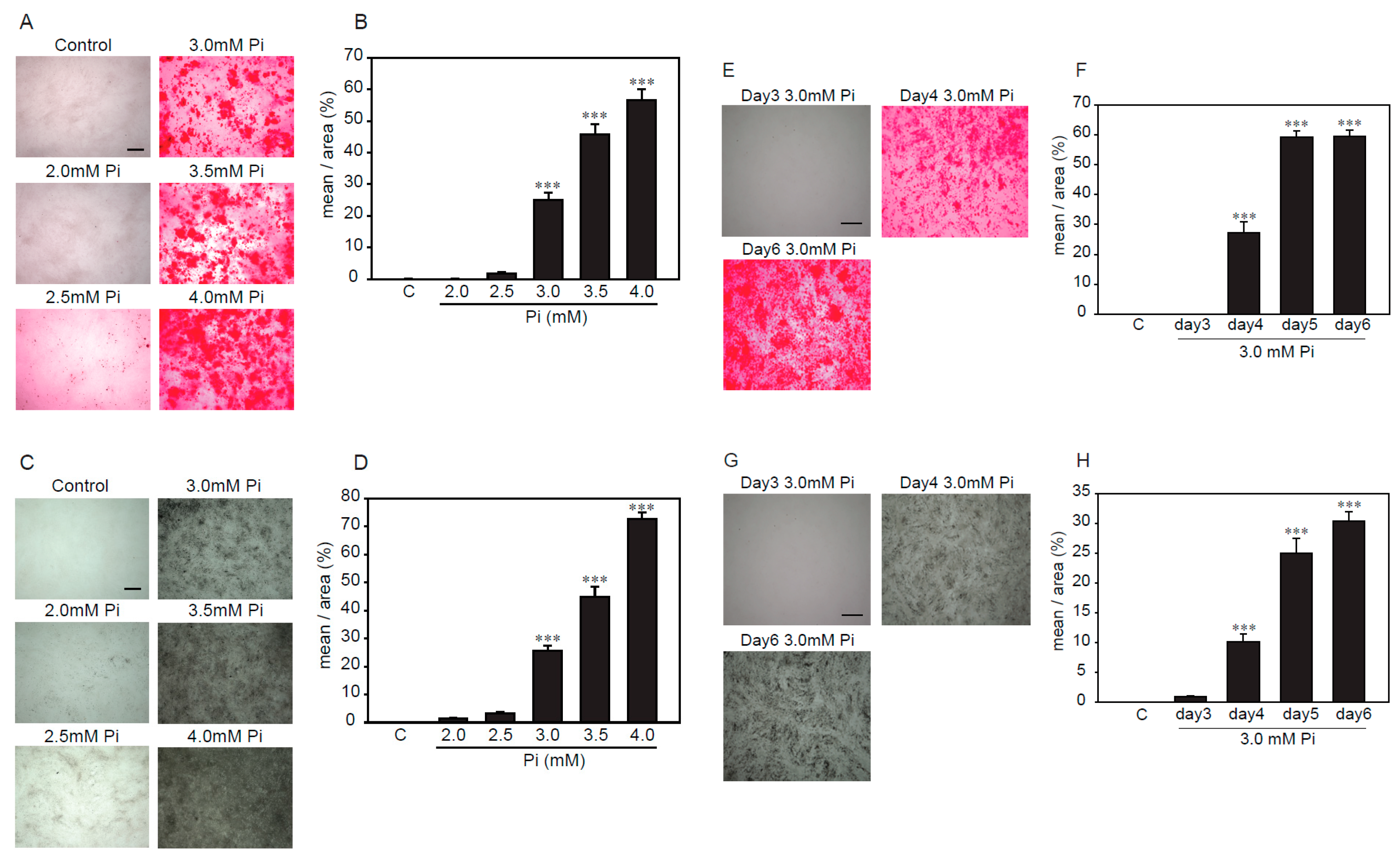
2.2. Establishment of an Experimental Model of High Concentrations of Pi-Induced Calcification in p53LMAco1 Cells
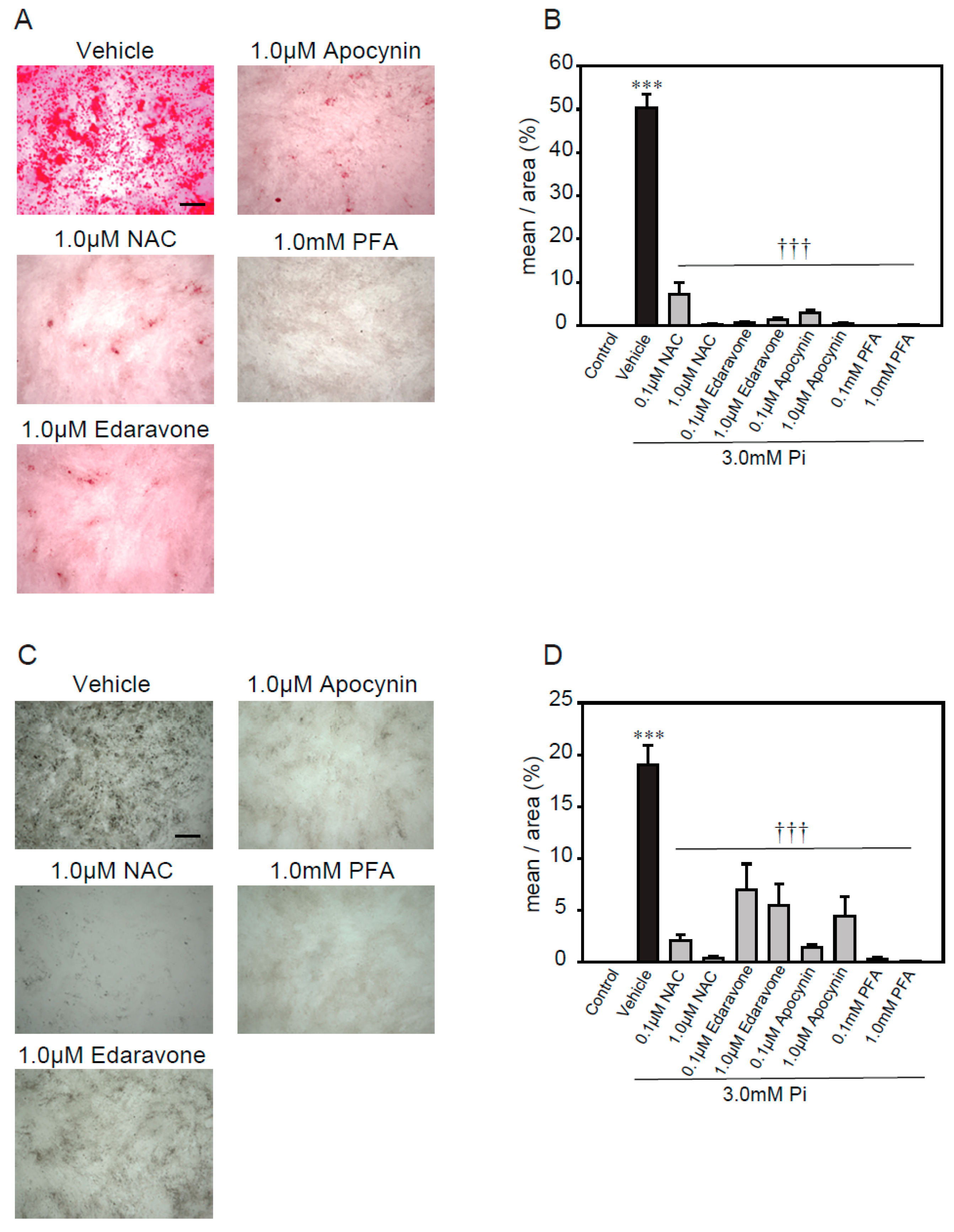
2.3. Antioxidants Suppress High Concentrations of Pi-Induced Calcification
2.4. 12AC3O Suppresses High Concentrations of Pi-Induced Calcification
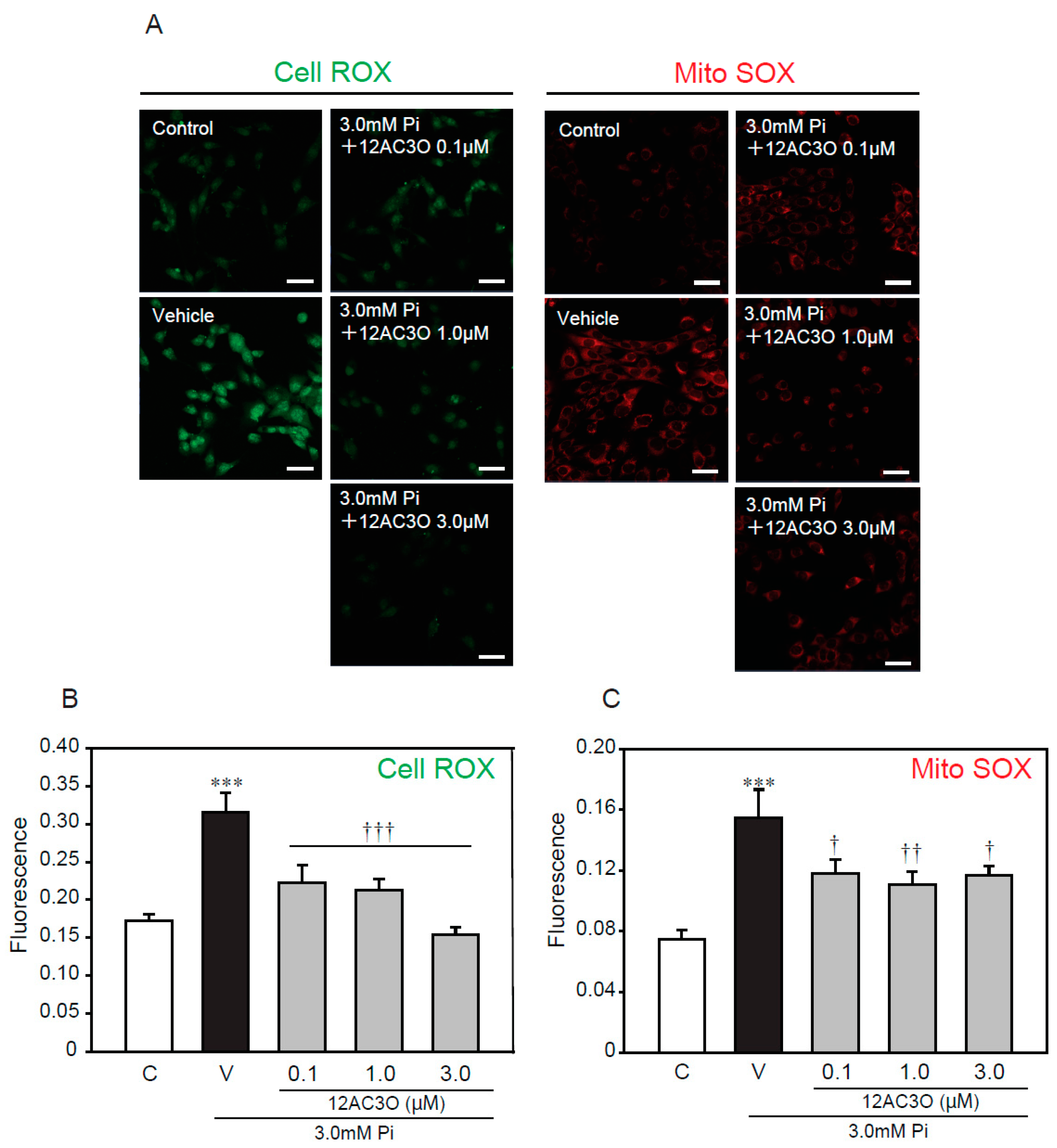
2.5. 12AC3O Suppresses High Concentrations of Pi-Induced Oxidative Stress
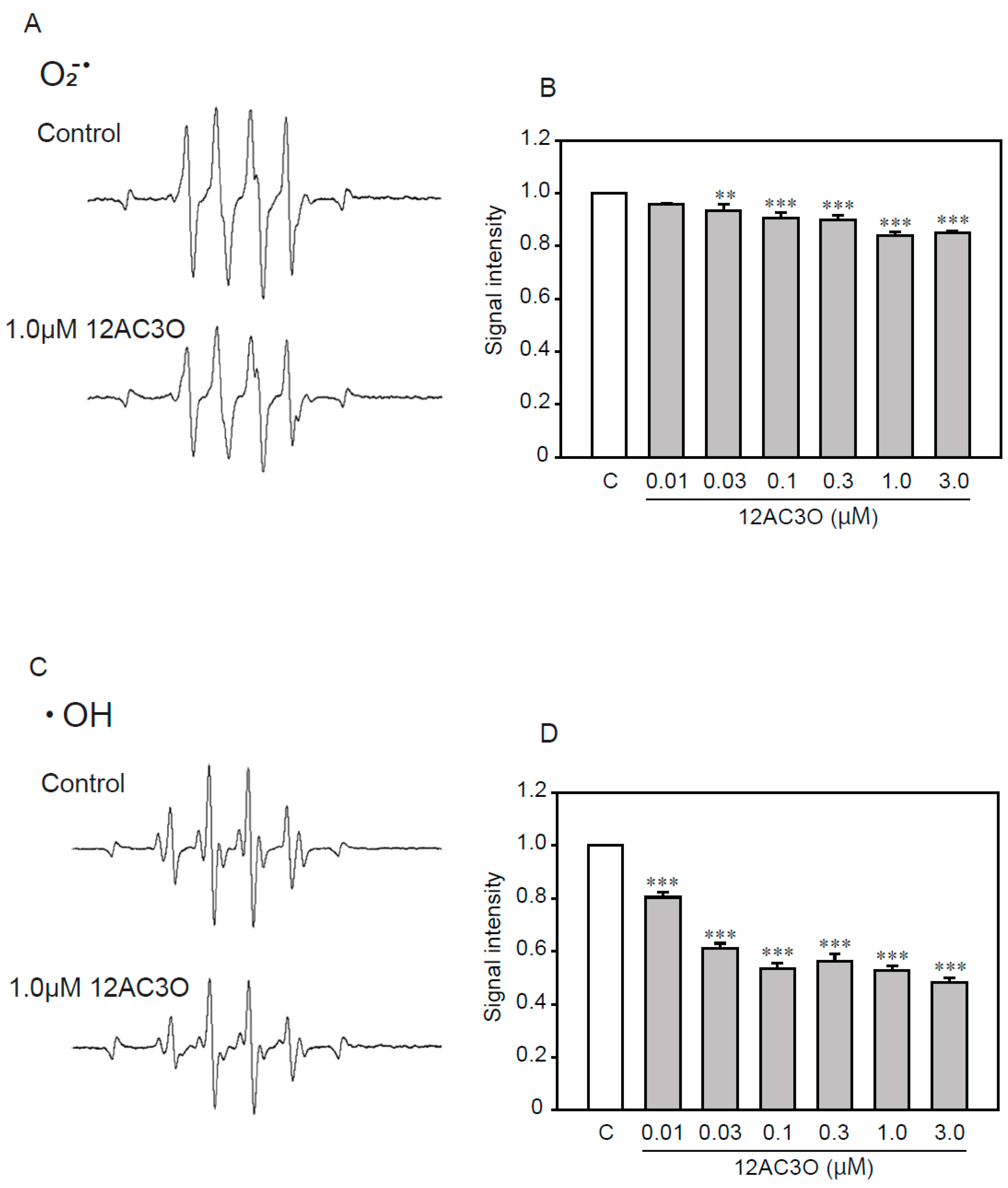
2.6. 12AC3O Directly Traps Superoxide Anion and Hydroxyl Radical
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Neurotoxicity Assays
4.3. 32Pi Transport Assays
4.4. qRT-PCR
4.5. Alizarin Red Staining
4.6. Von Kossa Staining
4.7. ROS Detection
4.8. ESR Analysis
4.9. Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Pi | Phosphate |
| CKD | Chronic kidney disease |
| ROS | Reactive oxygen species |
| DHPs | Gem-dihydroperoxides |
| ESR | Electron spin resonance |
| TNF-α | Tumor necrosis factor-α |
| IBGC | Idiopathic basal ganglia calcification |
| CSF | Cerebral spinal fluid |
| VSMC | Vascular smooth muscle cells |
| BMP | Bone morphogenetic protein |
| NO | Nitric oxide |
| PMBCs | Peripheral blood monocytes |
| NADP | Nicotinamide adenine dinucleotide phosphate |
| NAC | N-acetylcysteine |
| PFA | Phosphonoformic acid |
| NF-kB | Nuclear factor-kappa-B |
| MAPK | Mitogen-activated protein kinase |
| Runx2 | Runt-related transcription factor 2 |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| Msx2 | Muscle segment homeobox 2 |
References
- Keith, D.S.; Nichols, G.A.; Gullion, C.M.; Brown, J.B.; Smith, D.H. Longitudinal Follow-up and Outcomes among a Population with Chronic Kidney Disease in a Large Managed Care Organization. Arch. Intern. Med. 2004, 164, 659–663. [Google Scholar] [CrossRef]
- Ninomiya, T.; Kiyohara, Y.; Kubo, M.; Tanizaki, Y.; Doi, Y.; Okubo, K.; Wakugawa, Y.; Hata, J.; Oishi, Y.; Shikata, K.; et al. Chronic kidney disease and cardiovascular disease in a general Japanese population: The Hisayama Study. Kidney Int. 2005, 68, 228–236. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Guérin, A.P.; Marchais, S.J.; Métivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transpl. 2003, 18, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, M.; Bungay, P.; Taal, M.W.; McIntyre, C.W. Vascular calcification and cardiovascular function in chronic kidney disease. Nephrol. Dial. Transpl. 2006, 21, 707–714. [Google Scholar] [CrossRef]
- Foley, R.N. Clinical epidemiology of cardiovascular disease in chronic kidney disease. J. Ren. Care 1998, 36, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Goodman, W.G.; Goldin, J.G.; Kuizon, B.D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.E.; et al. Coronary-Artery Calcification in Young Adults with End-Stage Renal Disease Who Are Undergoing Dialysis. N. Engl. J. Med. 2000, 342, 1478–1483. [Google Scholar] [CrossRef]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 520–528. [Google Scholar] [CrossRef]
- Manyam, B.V. What is and what is not “Fahr’s disease”. Park. Relat. Disord. 2005, 11, 73–80. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Shi, L.; Ren, J.; Patti, M.; Wang, T.; De Oliveira, J.R.M.; Sobrido, M.J.; Quintáns, B.; Baquero, M.; et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 2012, 44, 254–256. [Google Scholar] [CrossRef]
- Yamada, M.; Tanaka, M.; Takagi, M.; Kobayashi, S.; Taguchi, Y.; Takashima, S.; Tanaka, K.; Touge, T.; Hatsuta, H.; Murayama, S.; et al. Evaluation of SLC20A2 mutations that cause idiopathic basal ganglia calcification in Japan. Neurology 2014, 82, 705–712. [Google Scholar] [CrossRef]
- Hozumi, I.; Kurita, H.; Ozawa, K.; Furuta, N.; Inden, M.; Sekine, S.I.; Yamada, M.; Hayashi, Y.; Kimura, A.; Inuzuka, T.; et al. Inorganic phosphorus (Pi) in CSF is a biomarker for SLC20A2-associated idiopathic basal ganglia calcification (IBGC1). J. Neurol. Sci. 2018, 388, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Jane, A.; Leopold, M. Vascular Calcification: Mechanisms of Vascular Smooth Muscle Cell Calcification. Physiol. Behav. 2015, 176, 139–148. [Google Scholar]
- Lau, W.L.; Pai, A.; Moe, S.M.; Giachelli, C.M. Direct Effects of Phosphate on Vascular Cell Function. Adv. Chronic Kidney Dis. 2011, 18, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- Giachelli, C.M. The emerging role of phosphate in vascular calcification. Kidney Int. 2009, 75, 890–897. [Google Scholar] [CrossRef]
- Lau, W.L.; Festing, M.H.; Giachelli, C.M. Phosphate and vascular calcification: Emerging role of the sodium-dependent phosphate co-transporter PiT-1. Thromb. Haemost. 2010, 104, 464–470. [Google Scholar] [CrossRef]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Parhami, F.; Demer, L.L. Tumor necrosis factor-α promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 2000, 102, 2636–2642. [Google Scholar] [CrossRef]
- Dalfino, G.; Simone, S.; Porreca, S.; Cosola, C.; Balestra, C.; Manno, C.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Bone morphogenetic protein-2 may represent the molecular link between oxidative stress and vascular stiffness in chronic kidney disease. Atherosclerosis 2010, 211, 418–423. [Google Scholar] [CrossRef]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.V.; Mokas, S.; Larivière, R.; Richard, D.E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Sun, Y.; Chen, J.; Yuan, K.; Mao, X.; Heath, J.M.; Anderson, P.G.; Tintut, Y.; Demer, L.L.; Wang, D.; et al. Runx2-upregulated receptor activator of nuclear factor κb ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Deuell, K.A.; Callegari, A.; Giachelli, C.M.; Rosenfeld, M.E.; Scatena, M. RANKL enhances macrophage paracrine pro-calcific activity in high phosphate-treated smooth muscle cells: Dependence on IL-6 and TNF-α. J. Vasc. Res. 2012, 49, 510–521. [Google Scholar] [CrossRef]
- Shioi, A.; Katagi, M.; Okuno, Y.; Mori, K.; Jono, S.; Koyama, H.; Nishizawa, Y. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: Roles of tumor necrosis factor-α and oncostatin M derived from macrophages. Circ. Res. 2002, 91, 9–16. [Google Scholar] [CrossRef]
- Tintut, Y.; Demer, L. Role of osteoprotegerin and its ligands and competing receptors in atherosclerotic calcification. J. Investig. Med. 2006, 54, 395–401. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Shao, J.S.; Lai, C.F.; Huang, E.; Cai, J.; Behrmann, A.; Cheng, S.L.; Towler, D.A. Aortic Msx2-Wnt calcification cascade is regulated by TNF-α-dependent signals in diabetic Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2589–2596. [Google Scholar] [CrossRef]
- Shinohara, H.; Taniguchi, K.; Kumazaki, M.; Yamada, N.; Ito, Y.; Otsuki, Y.; Uno, B.; Hayakawa, F.; Minami, Y.; Naoe, T.; et al. Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronic myeloid leukemia. Cancer Lett. 2015, 360, 28–38. [Google Scholar] [CrossRef]
- Zhang, M.; Harashima, N.; Moritani, T.; Huang, W.; Harada, M. The roles of ROS and caspases in TRAIL-induced apoptosis and necroptosis in human pancreatic cancer cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Chang, H.B.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar]
- Sudo, R.; Sato, F.; Azechi, T.; Wachi, H. 7-Ketocholesterol-induced lysosomal dysfunction exacerbates vascular smooth muscle cell calcification via oxidative stress. Genes Cells 2015, 20, 982–991. [Google Scholar] [CrossRef]
- Luong, T.T.D.; Schelski, N.; Boehme, B.; Makridakis, M.; Vlahou, A.; Lang, F.; Pieske, B.; Alesutan, I.; Voelkl, J. Fibulin-3 Attenuates Phosphate-Induced Vascular Smooth Muscle Cell Calcification by Inhibition of Oxidative Stress. Cell. Physiol. Biochem. 2018, 46, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Ortmann, R.; Jomaa, H.; Schlitzer, M. New Antimalarial Drugs. Angew. Chem. Int. Ed. 2003, 42, 5274–5293. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Dunstan, H.; Sauerbrey, A.; Miyachi, H.; Chitambar, C.R. The anti-malarial artesunate is also active against cancer. Int. J. Oncol. 2001, 18, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, M.; Zhang, R.; Wang, H. Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy. J. Cell. Mol. Med. 2009, 13, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Tada, N.; Cui, L.; Okubo, H.; Miura, T.; Itoh, A. A facile catalyst-free synthesis of gem-dihydroperoxides with aqueous hydrogen peroxide. Chem. Commun. 2010, 46, 1772–1774. [Google Scholar] [CrossRef] [PubMed]
- Žmitek, K.; Zupan, M.; Stavber, S.; Iskra, J. Iodine as a catalyst for efficient conversion of ketones to gem-dihydroperoxides by aqueous hydrogen peroxide. Org. Lett. 2006, 8, 2491–2494. [Google Scholar] [CrossRef]
- Kuranaga, Y.; Yamada, N.; Kashiwaya, M.; Nakamura, M.; Cui, L.; Kumazaki, M.; Shinohara, H.; Sugito, N.; Taniguchi, K.; Ito, Y.; et al. Anti-Oncogenic gem-dihydroperoxides induce apoptosis in cancer cells by trapping reactive oxygen species. Int. J. Mol. Sci. 2016, 17, 177–184. [Google Scholar] [CrossRef]
- Ueda, T.; Inden, M.; Asaka, Y.; Masaki, Y.; Kurita, H.; Tanaka, W.; Yamaguchi, E.; Itoh, A.; Hozumi, I. Effects of gem-dihydroperoxides against mutant copper-zinc superoxide dismutase-mediated neurotoxicity. Mol. Cell. Neurosci. 2018, 92, 177–184. [Google Scholar] [CrossRef]
- Wang, H.; Yan, S.; Chai, H.; Riha, G.M.; Li, M.; Yao, Q.; Chen, C. Shear stress induces endothelial transdifferentiation from mouse smooth muscle cells. Biochem. Biophys. Res. Commun. 2006, 346, 860–865. [Google Scholar] [CrossRef]
- Lu, H.; Enosawa, S.; Ohmi, K.; Suzuki, S. The proliferative response of p53 knock-out mouse-derived vascular smooth muscle cell line, P53LMAC01, to PDGF, when compared with human aortic smooth muscle cells. Transpl. Immunol. 2001, 8, 253–257. [Google Scholar] [CrossRef]
- Saito, T.; Itoh, H.; Yamashita, J.; Doi, K.; Chun, T.H.; Tanaka, T.; Inoue, M.; Masatsugu, K.; Fukunaga, Y.; Sawada, N.; et al. Angiotensin II suppresses growth arrest specific homeobox (Gax) expression via redox-sensitive mitogen-activated protein kinase (MAPK). Regul. Pept. 2005, 127, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Morita, I.; Nishi, H.; Murota, S.I. Preventive effect of MCI-186 on 15-HPETE induced vascular endothelial cell injury in vitro. Prostaglandins Leukot. Essent. Fat. Acids 1988, 33, 81–87. [Google Scholar] [CrossRef]
- Tóth, A.E.; Walter, F.R.; Bocsik, A.; Sántha, P.; Veszelka, S.; Nagy, L.; Puskás, L.G.; Couraud, P.O.; Takata, F.; Dohgu, S.; et al. Edaravone protects against methylglyoxal-induced barrier damage in human brain endothelial cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- De Batista, P.R.; Palacios, R.; Martín, A.; Hernanz, R.; Médici, C.T.; Silva, M.A.S.C.; Rossi, E.M.; Aguado, A.; Vassallo, D.V.; Salaices, M.; et al. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Hernanz, R.; Martínez-Revelles, S.; Palacios, R.; Martín, A.; Cachofeiro, V.; Aguado, A.; García-Redondo, L.; Barrús, M.T.; De Batista, P.R.; Briones, A.M.; et al. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br. J. Pharmacol. 2015, 172, 3159–3176. [Google Scholar] [CrossRef]
- Mune, S.; Shibata, M.; Hatamura, I.; Saji, F.; Okada, T.; Maeda, Y.; Sakaguchi, T.; Negi, S.; Shigematsu, T. Mechanism of phosphate-induced calcification in rat aortic tissue culture: Possible involvement of Pit-1 and apoptosis. Clin. Exp. Nephrol. 2009, 13, 571–577. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Distinctive molecular signature and activated signaling pathways in aortic smooth muscle cells of patients with myocardial infarction. Atherosclerosis 2018, 271, 237–244. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Woo, C.C.; Giannakakis, A.; Lin, X.Y.; Cheow, E.S.H.; Lee, C.N.; Richards, M.; Sze, S.K.; Nookaew, I.; Kuznetsov, V.A.; et al. Transcriptome alterations of vascular smooth muscle cells in aortic wall of myocardial infarction patients. Data Br. 2018, 17, 1112–1135. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Wang, P.; Zhou, P.; Chen, W.; Peng, D.A.N. Combined effects of hyperphosphatemia and hyperglycemia on the calcification of cultured human aortic smooth muscle cells. Exp. Ther. Med. 2019, 17, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.J.; Zhang, Y.M.; Qi, J.P.; Liu, R.; Zhang, H.; He, L.C. Ferulic acid inhibits H2O2-induced oxidative stress and inflammation in rat vascular smooth muscle cells via inhibition of the NADPH oxidase and NF-κB pathway. Int. Immunopharmacol. 2015, 28, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Xu, M.J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef]
- Almajdoob, S.; Hossain, E.; Anand-Srivastava, M.B. Resveratrol attenuates hyperproliferation of vascular smooth muscle cells from spontaneously hypertensive rats: Role of ROS and ROS-mediated cell signaling. Vascul. Pharmacol. 2018, 101, 48–56. [Google Scholar] [CrossRef]
- Sun, Y.; Byon, C.H.; Yuan, K.; Chen, J.; Mao, X.; Heath, J.M.; Javed, A.; Zhang, K.; Anderson, P.G.; Chen, Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 2012, 111, 543–552. [Google Scholar] [CrossRef]
- Okamura, D.M.; Pennathur, S. The balance of powers: Redox regulation of fibrogenic pathways in kidney injury. Redox Biol. 2015, 6, 495–504. [Google Scholar] [CrossRef]
- Carracedo, J.; Alique, M.; Vida, C.; Bodega, G.; Ceprián, N.; Morales, E.; Praga, M.; de Sequera, P.; Ramírez, R. Mechanisms of Cardiovascular Disorders in Patients With Chronic Kidney Disease: A Process Related to Accelerated Senescence. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Takase, N.; Inden, M.; Sekine, S.I.; Ishii, Y.; Yonemitsu, H.; Iwashita, W.; Kurita, H.; Taketani, Y.; Hozumi, I. Neuroprotective effect of 5-Aminolevulinic acid against low inorganic phosphate in neuroblastoma SH-SY5Y cells. Sci. Rep. 2017, 7, 5768. [Google Scholar] [CrossRef]
- Lu, Y.; Bian, Y.; Wang, Y.; Bai, R.; Wang, J.; Xiao, C. Globular adiponectin reduces vascular calcification via inhibition of ER-stress-mediated smooth muscle cell apoptosis. Int. J. Clin. Exp. Pathol. 2015, 8, 2545–2554. [Google Scholar]
- Olah, Z.; Lehel, C.; Anderson, W.B.; Eiden, M.V.; Wilson, C.A. The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J. Biol. Chem. 1994, 269, 25426–25431. [Google Scholar] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takase, N.; Inden, M.; Hirai, S.; Yamada, Y.; Kurita, H.; Takeda, M.; Yamaguchi, E.; Itoh, A.; Hozumi, I. The Novel gem-Dihydroperoxide 12AC3O Suppresses High Phosphate-Induced Calcification via Antioxidant Effects in p53LMAco1 Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 4628. https://doi.org/10.3390/ijms21134628
Takase N, Inden M, Hirai S, Yamada Y, Kurita H, Takeda M, Yamaguchi E, Itoh A, Hozumi I. The Novel gem-Dihydroperoxide 12AC3O Suppresses High Phosphate-Induced Calcification via Antioxidant Effects in p53LMAco1 Smooth Muscle Cells. International Journal of Molecular Sciences. 2020; 21(13):4628. https://doi.org/10.3390/ijms21134628
Chicago/Turabian StyleTakase, Naoko, Masatoshi Inden, Shunsuke Hirai, Yumeka Yamada, Hisaka Kurita, Mitsumi Takeda, Eiji Yamaguchi, Akichika Itoh, and Isao Hozumi. 2020. "The Novel gem-Dihydroperoxide 12AC3O Suppresses High Phosphate-Induced Calcification via Antioxidant Effects in p53LMAco1 Smooth Muscle Cells" International Journal of Molecular Sciences 21, no. 13: 4628. https://doi.org/10.3390/ijms21134628
APA StyleTakase, N., Inden, M., Hirai, S., Yamada, Y., Kurita, H., Takeda, M., Yamaguchi, E., Itoh, A., & Hozumi, I. (2020). The Novel gem-Dihydroperoxide 12AC3O Suppresses High Phosphate-Induced Calcification via Antioxidant Effects in p53LMAco1 Smooth Muscle Cells. International Journal of Molecular Sciences, 21(13), 4628. https://doi.org/10.3390/ijms21134628