Kallikrein-Related Peptidase 14 Activates Zymogens of Membrane Type Matrix Metalloproteinases (MT-MMPs)—A CleavEx Based Analysis

 , , , , , , , , , add
Show full author list
, , , , , , , , , add
Show full author list
Abstract
1. Introduction
2. Results
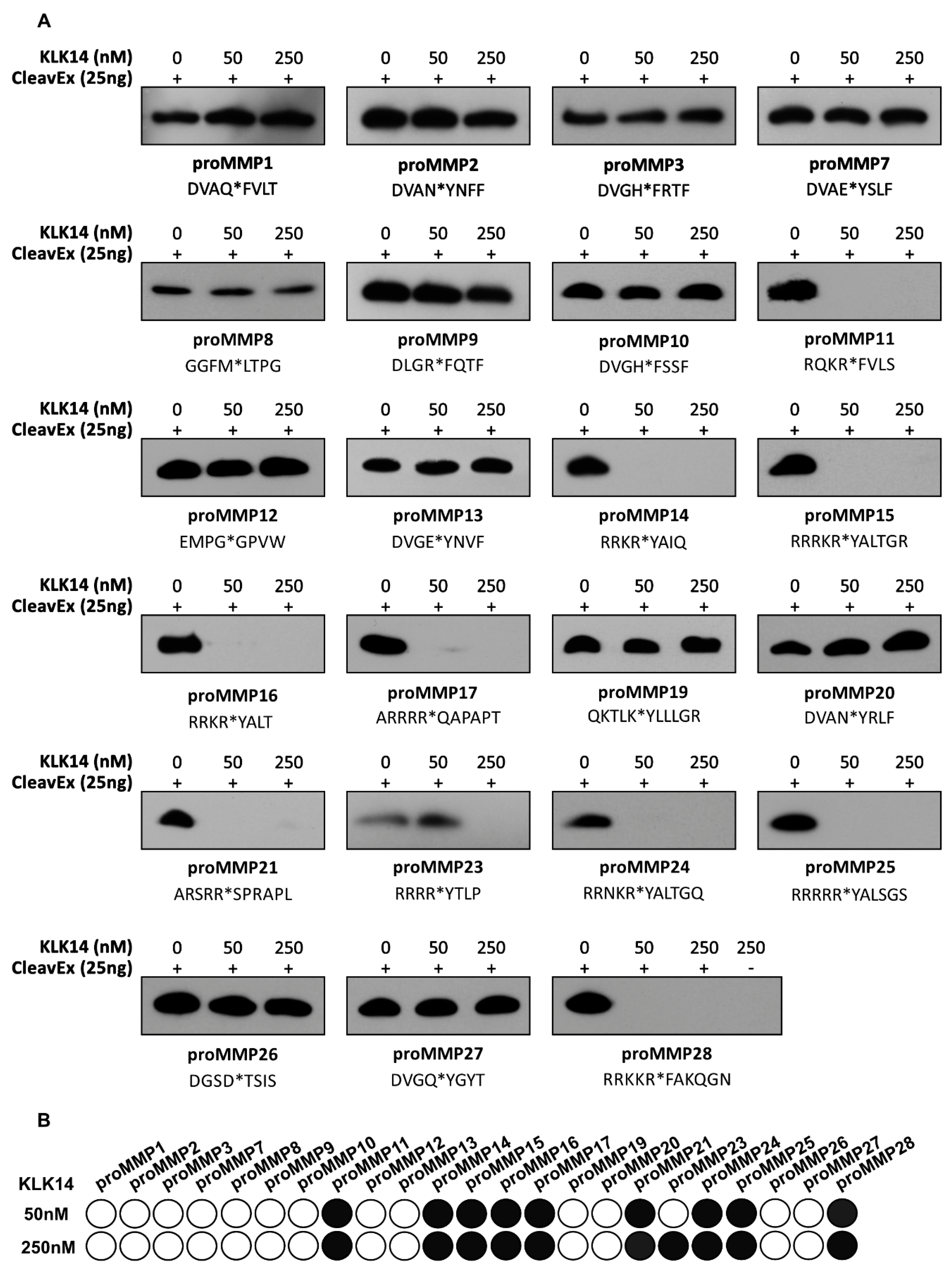
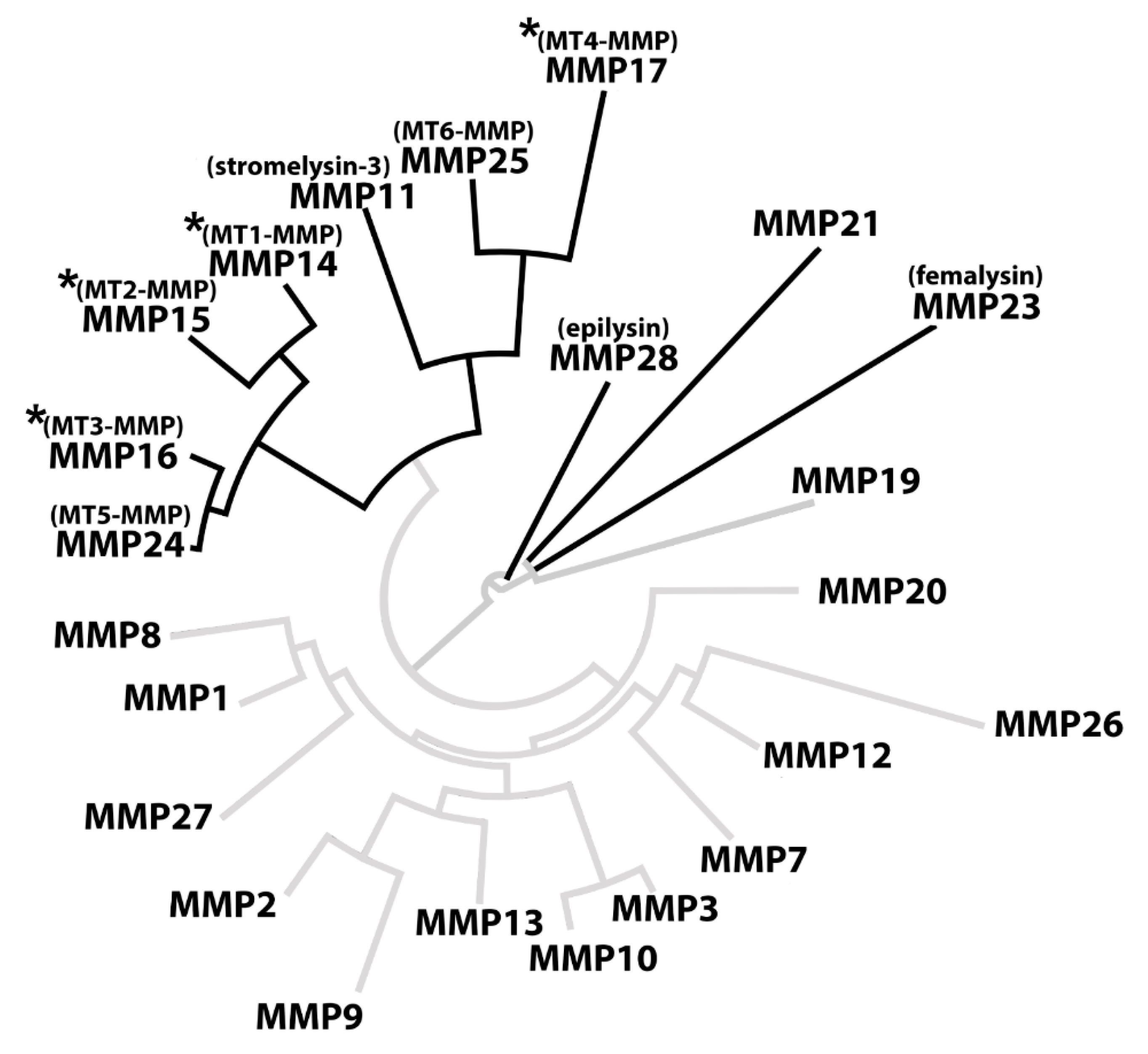
2.1. CleavExproMMP Fusion Protein Screening
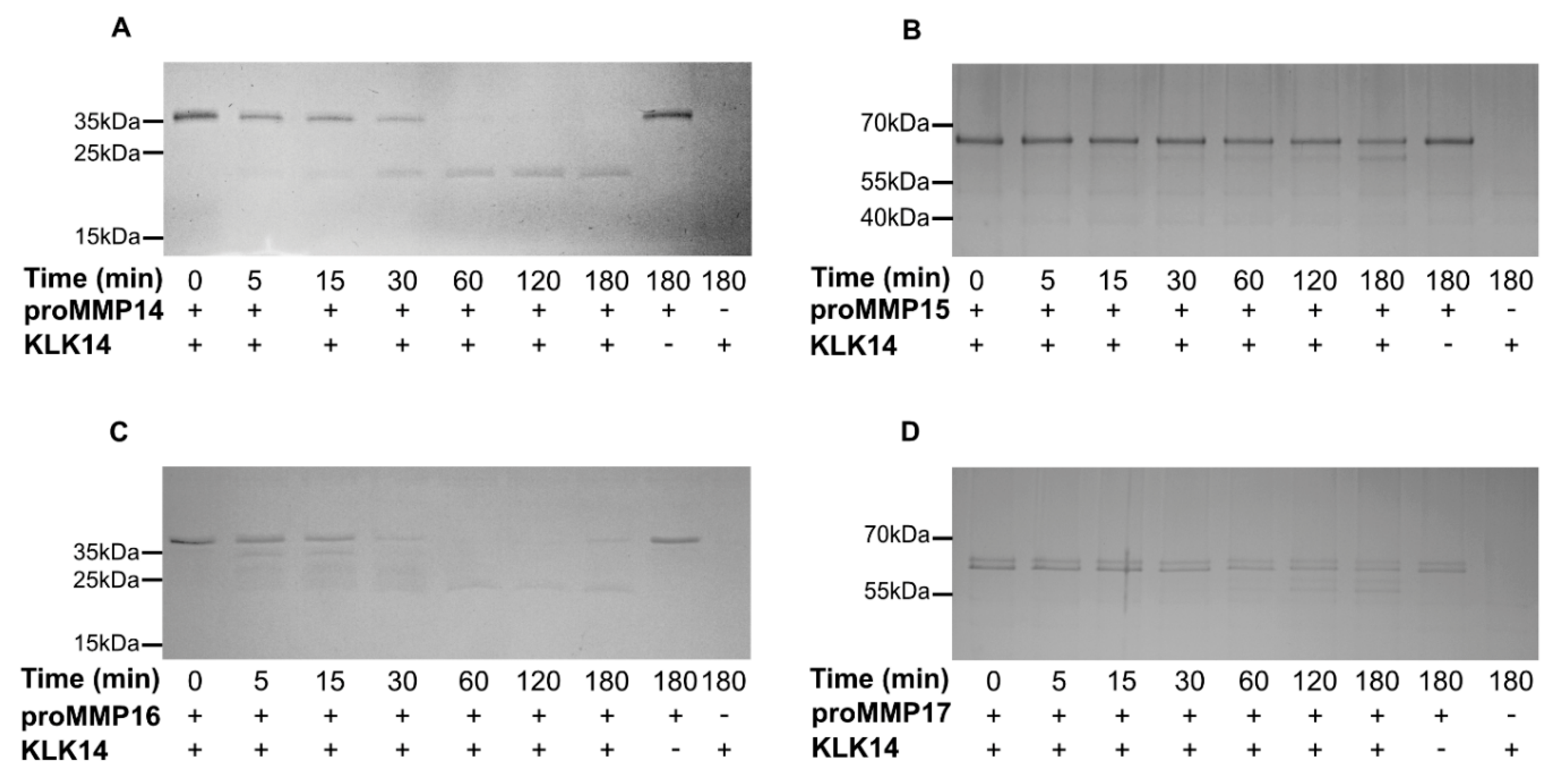
2.2. Processing of Recombinant proMMPs by KLK14
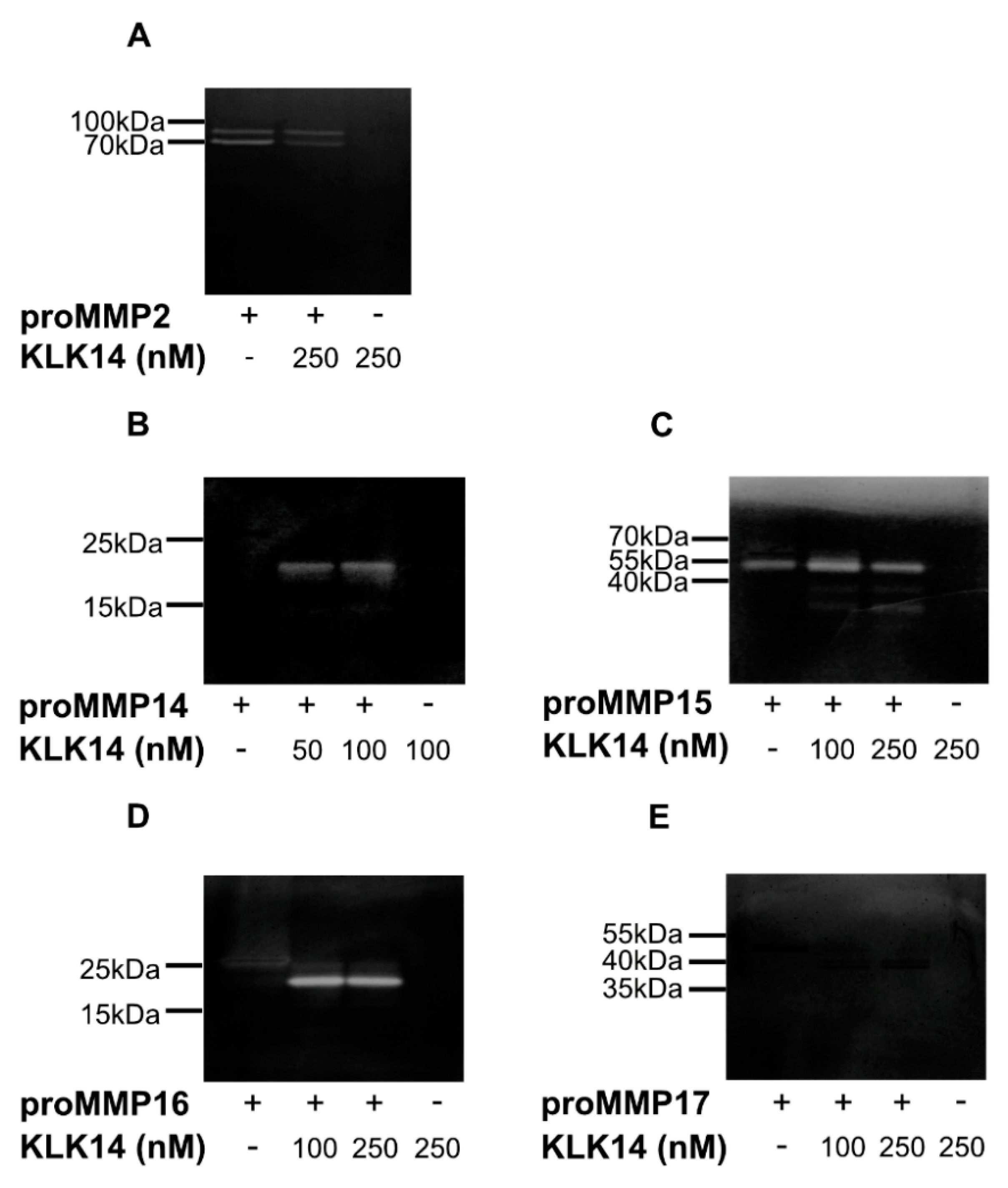
2.3. Functional Activation of proMMPs by KLK14
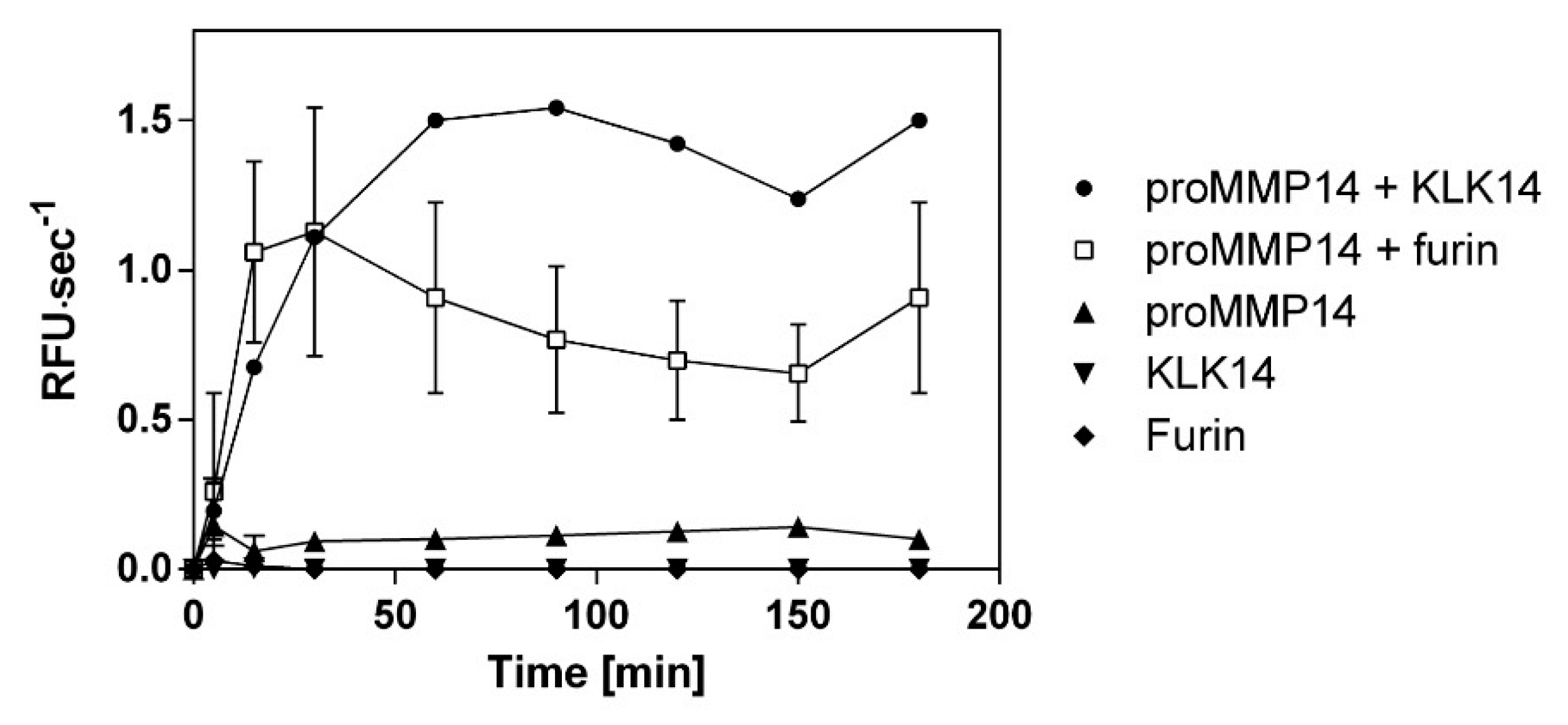
2.4. Comparison of Functional Activation of proMMP14 by KLK14 and Furin
2.5. Cell Surface Processing of proMMP14 by KLK14
3. Discussion
4. Materials and Methods
4.1. Cloning of HmuY-Based CleavEx Fusion Proteins
4.2. Expression and Purification of CleavEx Fusion Proteins
4.3. Expression and Production of KLK14
4.4. Screening the CleavExproMMP Fusion Proteins with KLK14 and N-Terminal Sequencing of KLK14-Released Fragments
4.5. SDS-PAGE Analysis of KLK14-Mediated Recombinant proMMP Processing
4.6. N-Terminal Sequencing of KLK14 Processed Recombinant proMMPs
4.7. Zymogram Analysis of Recombinant proMMP by KLK14
4.8. Functional Activation of Recombinant proMMP14 Using A Synthetic Substrate
4.9. Cell Surface Processing of proMMP14 (MT1-MMP) by KLK14
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM | A disintegrin and metalloproteinase |
| CleavEx | Cleavage of exposed amino acid sequences |
| ECM | Extracellular matrix |
| HmuY | Heme-binding protein |
| KLK | Kallikrein-related peptidase |
| MMP | Matrix metalloproteinase |
| MT-MMP | Membrane-type matrix metalloproteinase |
| MT1-MMP | Membrane-type 1 matrix metalloproteinase, MMP14 |
| TMPRSS2 | Transmembrane serine protease 2 |
References
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Hansen, N.U.B.; Genovese, F.; Leeming, D.; Karsdal, M. The importance of extracellular matrix for cell function and in vivo likeness. Exp. Mol. Pathol. 2015, 98, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulou, G.; Pampalakis, G. Kallikrein-related peptidases: Bridges between immune functions and extracellular matrix degradation. Boil. Chem. 2010, 391, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Löffek, S.; Schilling, O.; Franzke, C.W. Series ‘matrix metalloproteinases in lung health and disease’ edited by J. Müller-Quernheim and O. Eickelberg number 1 in this series: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef]
- Seiki, M. The cell surface: The stage for matrix metalloproteinase regulation of migration. Curr. Opin. Cell Boil. 2002, 14, 624–632. [Google Scholar] [CrossRef]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-Type 1 Matrix Metalloproteinase Cleaves Cd44 and Promotes Cell Migration. J. Cell Boil. 2001, 153, 893–904. [Google Scholar] [CrossRef]
- Roncucci, L.; Sena, P.; Mariani, F.; Marzona, L.; Benincasa, M.; De Leon, M.P.; Palumbo, C. Matrix metalloproteinases 15 and 19 are stromal regulators of colorectal cancer development from the early stages. Int. J. Oncol. 2012, 41, 260–266. [Google Scholar]
- Tatti, O.; Gucciardo, E.; Pekkonen, P.; Holopainen, T.; Louhimo, R.; Repo, P.; Maliniemi, P.; Lohi, J.; Rantanen, V.; Hautaniemi, S.; et al. MMP16 Mediates a Proteolytic Switch to Promote Cell-Cell Adhesion, Collagen Alignment, and Lymphatic Invasion in Melanoma. Cancer Res. 2015, 75, 2083–2094. [Google Scholar] [CrossRef]
- Gonzalo, P.; Moreno, V.; Gálvez, B.G.; Arroyo, A.G. MT1-MMP and integrins: Hand-to-hand in cell communication. BioFactors 2010, 36, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H. Cell surface activation of progelatinase A (proMMP-2) and cell migration. Cell Res. 1998, 8, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Yana, I.; Weiss, S.J. Regulation of Membrane Type-1 Matrix Metalloproteinase Activation by Proprotein Convertases. Mol. Boil. Cell 2000, 11, 2387–2401. [Google Scholar] [CrossRef] [PubMed]
- Vilen, S.-T.; Suojanen, J.; Salas, F.; Risteli, J.; Ylipalosaari, M.; Itkonen, O.; Koistinen, H.; Baumann, M.; Stenman, U.-H.; Sorsa, T.; et al. Trypsin-2 Enhances Carcinoma Invasion by Processing Tight Junctions and Activating ProMT1-MMP. Cancer Investig. 2012, 30, 583–592. [Google Scholar] [CrossRef]
- Ra, H.-J.; Parks, W.C. Control of matrix metalloproteinase catalytic activity. Matrix Boil. 2007, 26, 587–596. [Google Scholar] [CrossRef]
- Carmeliet, P.; Moons, L.; Lijnen, R.; Baes, M.; Lemaître, V.; Tipping, P.; Drew, A.; Eeckhout, Y.; Shapiro, S.; Lupu, F.; et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat. Genet. 1997, 17, 439–444. [Google Scholar] [CrossRef]
- Reid, J.C.; Matsika, A.; Davies, C.M.; He, Y.; Broomfield, A.; Bennett, N.C.; Magdolen, V.; Srinivasan, B.; Clements, J.A.; Hooper, J. Pericellular regulation of prostate cancer expressed kallikrein-related peptidases and matrix metalloproteinases by cell surface serine proteases. Am. J. Cancer Res. 2017, 7, 2257–2274. [Google Scholar]
- Kalińska, M.; Meyer-Hoffert, U.; Kantyka, T.; Potempa, J. Kallikreins - The melting pot of activity and function. Biochim. 2015, 122, 270–282. [Google Scholar] [CrossRef]
- Shaw, J.L.; Diamandis, E. Distribution of 15 Human Kallikreins in Tissues and Biological Fluids. Clin. Chem. 2007, 53, 1423–1432. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Boil. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Boil. 2015, 44, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.J.; Liu, Y. Matrix metalloproteinases in kidney homeostasis and diseases. Am. J. Physiol. Physiol. 2012, 302, F1351–F1361. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, H.; Guevara, T.; Tallant, C.; Olczak, M.; Sroka, A.; Potempa, J.; Solà, M.; Olczak, T.; Gomis-Rüth, F.X. Unique Structure and Stability of HmuY, a Novel Heme-Binding Protein of Porphyromonas gingivalis. PLoS Pathog. 2009, 5, e1000419. [Google Scholar] [CrossRef]
- Kang, T.; Nagase, H.; Pei, D. Activation of membrane-type matrix metalloproteinase 3 zymogen by the proprotein convertase furin in the trans-Golgi network. Cancer Res. 2002, 62, 675–681. [Google Scholar] [PubMed]
- Kukreja, M.; Shiryaev, S.A.; Cieplak, P.; Muranaka, N.; Routenberg, D.A.; Chernov, A.V.; Kumar, S.; Remacle, A.G.; Smith, J.W.; Kozlov, I.A.; et al. High-Throughput Multiplexed Peptide-Centric Profiling Illustrates Both Substrate Cleavage Redundancy and Specificity in the MMP Family. Chem. Boil. 2015, 22, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Kolkenbrock, H.; Essers, L.; Ulbrich, N.; Will, H. Biochemical Characterization of the Catalytic Domain of Membrane-Type 4 Matrix Metalloproteinase. Boil. Chem. 1999, 380, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Golubkov, V.S.; Chernov, A.V.; Strongin, A.Y. Intradomain Cleavage of Inhibitory Prodomain Is Essential to Protumorigenic Function of Membrane Type-1 Matrix Metalloproteinase (MT1-MMP) in Vivo*. J. Boil. Chem. 2011, 286, 34215–34223. [Google Scholar] [CrossRef]
- Chellaiah, M.A.; Ma, T. Membrane Localization of Membrane Type 1 Matrix Metalloproteinase by CD44 Regulates the Activation of Pro-Matrix Metalloproteinase 9 in Osteoclasts. BioMed Res. Int. 2013, 2013, 302392. [Google Scholar] [CrossRef]
- Sternlicht, M.D.; Werb, Z. How Matrix Metalloproteinases Regulate Cell Behavior. Annu. Rev. Cell Dev. Boil. 2001, 17, 463–516. [Google Scholar] [CrossRef]
- Guo, Y.; Nguyen, K.-A.; Potempa, J. Dichotomy of gingipains action as virulence factors: From cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontology 2010, 54, 15–44. [Google Scholar] [CrossRef]
- Yoon, H.; Laxmikanthan, G.; Lee, J.; Blaber, S.I.; Rodríguez, A.; Kogot, J.M.; Scarisbrick, I.A.; Blaber, M. Activation Profiles and Regulatory Cascades of the Human Kallikrein-related Peptidases. J. Boil. Chem. 2007, 282, 31852–31864. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Blaber, S.I.; Evans, D.M.; Trim, J.; Juliano, M.A.; Scarisbrick, I.A.; Blaber, M. Activation profiles of human kallikrein-related peptidases by proteases of the thrombostasis axis. Protein Sci. 2008, 17, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Blaber, S.I.; Li, W.; Scarisbrick, I.A.; Blaber, M. Activation profiles of human kallikrein-related peptidases by matrix metalloproteinases. Boil. Chem. 2013, 394, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Golubkov, V.S.; Cieplak, P.; Chekanov, A.V.; Ratnikov, B.I.; Aleshin, A.E.; Golubkova, N.V.; Postnova, T.I.; Radichev, I.A.; Rozanov, D.V.; Zhu, W.; et al. Internal Cleavages of the Autoinhibitory Prodomain Are Required for Membrane Type 1 Matrix Metalloproteinase Activation, although Furin Cleavage Alone Generates Inactive Proteinase*. J. Boil. Chem. 2010, 285, 27726–27736. [Google Scholar] [CrossRef]
- Osenkowski, P.; Meroueh, S.O.; Pavel, D.; Mobashery, S.; Fridman, R. Mutational and Structural Analyses of the Hinge Region of Membrane Type 1-Matrix Metalloproteinase and Enzyme Processing. J. Boil. Chem. 2005, 280, 26160–26168. [Google Scholar] [CrossRef]
- Hotary, K.; Allen, E.; Punturieri, A.; Yana, I.; Weiss, S.J. Regulation of Cell Invasion and Morphogenesis in a Three-Dimensional Type I Collagen Matrix by Membrane-Type Matrix Metalloproteinases 1, 2, and 3. J. Cell Boil. 2000, 149, 1309–1323. [Google Scholar] [CrossRef]
- Ota, I.; Li, X.-Y.; Hu, Y.; Weiss, S.J. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc. Natl. Acad. Sci. USA 2009, 106, 20318–20323. [Google Scholar] [CrossRef]
- Pahwa, S.; Bhowmick, M.; Amar, S.; Cao, J.; Strongin, A.Y.; Fridman, R.; Weiss, S.J.; Fields, G.B. Characterization and regulation of MT1-MMP cell surface-associated activity. Chem. Boil. Drug Des. 2018, 93, 1251–1264. [Google Scholar] [CrossRef]
- Osenkowski, P.; Toth, M.; Fridman, R. Processing, shedding, and endocytosis of membrane type 1-matrix metalloproteinase (MT1-MMP). J. Cell. Physiol. 2004, 200, 2–10. [Google Scholar] [CrossRef]
- Lohi, J.; Lehti, K.; Westermarck, J.; Kähäri, V.-M.; Keski-Oja, J. Regulation of Membrane-Type Matrix Metalloproteinase-1 Expression by Growth Factors and Phorbol 12-Myristate 13-Acetate. JBIC J. Boil. Inorg. Chem. 1996, 239, 239–247. [Google Scholar] [CrossRef]
- Cao, J.; Rehemtulla, A.; Bahou, W.; Zucker, S. Membrane Type Matrix Metalloproteinase 1 Activates Pro-gelatinase A without Furin Cleavage of the N-terminal Domain. J. Boil. Chem. 1996, 271, 30174–30180. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kondo, T.; Seiki, M.; Ito, A. Cell type-specific involvement of furin in membrane type 1 matrix metalloproteinase-mediated progelatinase A activation. Ann. N. Y. Acad. Sci. 1999, 878, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kondo, T.; Fujisawa, T.; Seiki, M.; Ito, A. Furin-independent Pathway of Membrane Type 1-Matrix Metalloproteinase Activation in Rabbit Dermal Fibroblasts. J. Boil. Chem. 1999, 274, 37280–37284. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.G.; Chekanov, A.V.; Golubkov, V.S.; Savinov, A.Y.; Rozanov, D.; Strongin, A.Y. O-Glycosylation Regulates Autolysis of Cellular Membrane Type-1 Matrix Metalloproteinase (MT1-MMP). J. Boil. Chem. 2006, 281, 16897–16905. [Google Scholar] [CrossRef]
- Golubkov, V.S.; Chekanov, A.V.; Shiryaev, S.A.; Aleshin, A.E.; Ratnikov, B.I.; Gawlik, K.; Radichev, I.; Motamedchaboki, K.; Smith, J.W.; Strongin, A.Y. Proteolysis of the membrane type-1 matrix metalloproteinase prodomain: Implications for a two-step proteolytic processing and activation. J. Biol. Chem. 2007, 282, 36283–36291. [Google Scholar] [CrossRef]
- Beaufort, N.; Plaza, K.; Utzschneider, D.T.; Schwarz, A.; Burkhart, J.M.; Creutzburg, S.; Debela, M.; Schmitt, M.; Ries, C.; Magdolen, V. Interdependence of kallikrein-related peptidases in proteolytic networks. Boil. Chem. 2010, 391, 581–587. [Google Scholar] [CrossRef]
- Filippou, P.S.; Karagiannis, G.S.; Musrap, N.; Diamandis, E. Kallikrein-related peptidases (KLKs) and the hallmarks of cancer. Crit. Rev. Clin. Lab. Sci. 2016, 53, 277–291. [Google Scholar] [CrossRef]
- Tokuhara, C.K.; Santesso, M.R.; De Oliveira, G.S.N.; Ventura, T.M.O.; Doyama, J.T.; Zambuzzi, W.; Oliveira, R.C. Updating the role of matrix metalloproteinases in mineralized tissue and related diseases. J. Appl. Oral Sci. 2019, 27, e20180596. [Google Scholar] [CrossRef]
- Caley, M.; Martins, V.L.; O’Toole, E. Metalloproteinases and Wound Healing. Adv. Wound Care 2015, 4, 225–234. [Google Scholar] [CrossRef]
- Turunen, S.P.; Tatti-Bugaeva, O.; Lehti, K. Membrane-type matrix metalloproteases as diverse effectors of cancer progression. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 1974–1988. [Google Scholar] [CrossRef]
- Sotiropoulou, G.; Pampalakis, G.; Diamandis, E. Functional Roles of Human Kallikrein-related Peptidases. J. Boil. Chem. 2009, 284, 32989–32994. [Google Scholar] [CrossRef] [PubMed]
- Borgoño, C.A.; Diamandis, E. The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer 2004, 4, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Lin, Z.; Jiang, J.; Lu, S.; Chen, M.; Que, H.; He, X.; Que, G.; Mao, J.; Xiao, J.; et al. MMP14 regulates cell migration and invasion through epithelial-mesenchymal transition in nasopharyngeal carcinoma. Am. J. Transl. Res. 2015, 7, 950–958. [Google Scholar] [PubMed]
- Von Nandelstadh, P.; Gucciardo, E.; Lohi, J.; Li, R.; Sugiyama, N.; Carpen, O.; Lehti, K. Actin-associated protein palladin promotes tumor cell invasion by linking extracellular matrix degradation to cell cytoskeleton. Mol. Boil. Cell 2014, 25, 2556–2570. [Google Scholar] [CrossRef]
- Lowy, A.M.; Clements, W.M.; Bishop, J.; Kong, L.; Bonney, T.; Sisco, K.; Aronow, B.; Fenoglio-Preiser, C.; Groden, J.L. β-Catenin/Wnt Signaling Regulates Expression of the Membrane Type 3 Matrix Metalloproteinase in Gastric Cancer. Cancer Res. 2006, 66, 4734–4741. [Google Scholar] [CrossRef]
- Hotary, K.B.; Yana, I.; Sabeh, F.; Li, X.-Y.; Holmbeck, K.; Birkedal-Hansen, H.; Allen, E.D.; Hiraoka, N.; Weiss, S.J. Matrix Metalloproteinases (MMPs) Regulate Fibrin-invasive Activity via MT1-MMP–dependent and –independent Processes. J. Exp. Med. 2002, 195, 295–308. [Google Scholar] [CrossRef]
- Chabottaux, V.; Sounni, N.E.; Pennington, C.J.; English, W.R.; Brûle, F.V.D.; Blacher, S.; Gilles, C.; Munaut, C.; Maquoi, E.; Lopez-Otin, C.; et al. Membrane-Type 4 Matrix Metalloproteinase Promotes Breast Cancer Growth and Metastases. Cancer Res. 2006, 66, 5165–5172. [Google Scholar] [CrossRef]
- Yousef, G.M.; Stephan, C.; Scorilas, A.; Ellatif, M.A.; Jung, K.; Kristiansen, G.; Jung, M.; Polymeris, M.-E.; Diamandis, E. Differential expression of the human kallikrein gene 14 (KLK14) in normal and cancerous prostatic tissues. Prostate 2003, 56, 287–292. [Google Scholar] [CrossRef]
- Fritzsche, F.; Gansukh, T.; Borgoño, C.A.; Burkhardt, M.; Pahl, S.; Mayordomo, E.; Winzer, K.-J.; Weichert, W.; Denkert, C.; Jung, K.; et al. Expression of human Kallikrein 14 (KLK14) in breast cancer is associated with higher tumour grades and positive nodal status. Br. J. Cancer 2006, 94, 540–547. [Google Scholar] [CrossRef][Green Version]
- Papachristopoulou, G.; Avgeris, M.; Charlaftis, A.; Scorilas, A. Quantitative expression analysis and study of the novel human kallikrein-related peptidase 14 gene (KLK14) in malignant and benign breast tissues. Thromb. Haemost. 2010, 105, 131–137. [Google Scholar] [CrossRef]
- Trudel, D.; Fradet, Y.; Meyer, F.; Harel, F.; Têtu, B. Membrane-type-1 matrix metalloproteinase, matrix metalloproteinase 2, and tissue inhibitor of matrix proteinase 2 in prostate cancer: Identification of patients with poor prognosis by immunohistochemistry. Hum. Pathol. 2008, 39, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Kong, C.Z.; Zhang, Z.; Zhu, Y.Y. Loss of P53 facilitates invasion and metastasis of prostate cancer cells. Mol. Cell. Biochem. 2013, 384, 121–127. [Google Scholar] [CrossRef]
- Li, Y.; Cai, G.; Yuan, S.; Jun, Y.; Li, N.; Wang, L.; Chen, F.; Ling, R.; Yun, J. The overexpression membrane type 1 matrix metalloproteinase is associated with the progression and prognosis in breast cancer. Am. J. Transl. Res. 2015, 7, 120–127. [Google Scholar] [PubMed]
- Yao, G.; He, P.; Chen, L.; Hu, X.; Gu, F.; Ye, C. MT1-MMP in breast cancer: Induction of VEGF-C correlates with metastasis and poor prognosis. Cancer Cell Int. 2013, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Li, Q.; Li, S.; He, J.; Cao, W.; Lan, J.; Sun, B.; Zou, H.; Wang, C.; Liu, R.; et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Sci. Rep. 2016, 6, 22179. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Zhu, L.-F.; Xu, X.-H.; Ning, T.-Y.; Ye, J.-H.; Liu, L.-K. Membrane Type 1 Matrix Metalloproteinase induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. BMC Cancer 2013, 13, 171. [Google Scholar] [CrossRef]
- Cao, J.; Chiarelli, C.; Richman, O.; Zarrabi, K.; Kozarekar, P.; Zucker, S. Membrane Type 1 Matrix Metalloproteinase Induces Epithelial-to-Mesenchymal Transition in Prostate Cancer. J. Boil. Chem. 2008, 283, 6232–6240. [Google Scholar] [CrossRef]
- Kantyka, T.; Fischer, J.; Wu, Z.; Declercq, W.; Reiss, K.; Schröder, J.-M.; Meyer-Hoffert, U. Inhibition of kallikrein-related peptidases by the serine protease inhibitor of Kazal-type 6. Peptides 2011, 32, 1187–1192. [Google Scholar] [CrossRef]
- Matsudaira, P. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J. Boil. Chem. 1987, 262, 10035–10038. [Google Scholar]
- Plaza, K.; Kalinska, M.; Bochenska, O.; Meyer-Hoffert, U.; Wu, Z.; Fischer, J.; Falkowski, K.; Sasiadek, L.; Bielecka, E.; Potempa, B.; et al. Gingipains of Porphyromonas gingivalis Affect the Stability and Function of Serine Protease Inhibitor of Kazal-type 6 (SPINK6), a Tissue Inhibitor of Human Kallikreins*. J. Boil. Chem. 2016, 291, 18753–18764. [Google Scholar] [CrossRef]
- Morrison, C.J.; Butler, G.S.; Bigg, H.F.; Roberts, C.R.; Soloway, P.D.; Overall, C.M. Cellular Activation of MMP-2 (Gelatinase A) by MT2-MMP Occurs via a TIMP-2-independent Pathway. J. Biol. Chem. 2001, 276, 47402–47410. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CleavExproMMP | Identified Sequence |
|---|---|
| proMMP11 | RQKR27FVLSDEP |
| proMMP14 | RRKR27YAIQDE |
| proMMP15 | RRRKR28YALTGRD |
| proMMP16 | RRKR27YALTDEP |
| proMMP17 | ARRRR28QAPAPTDE |
| proMMP21 | ARSRRSPR31APLDEPN |
| proMMP23 | RRRR27YTLPDEPN |
| proMMP24 | RRNKR28YALTGQDE |
| proMMP25 | RRRRR28YALSGSDE |
| proMMP28 | RRKKR28FAKQGNDE |
| Protease | Band | N-Terminal Sequence | Annotation |
|---|---|---|---|
| proMMP14 | 1 | NH2-ALAS24LGSAQSSSFS | Proform |
| 2 | RRKR111YAIQGLKWQH | Active form | |
| proMMP15 | 1 | NH2-E47DAEVHAENWLLY | Proform |
| 2 | PGKR131YALTGRKWNH | Active form | |
| 3 | DLRG298IQQLYGTPDGQP | Hinge region | |
| proMMP16 | 1 | NH2-A32TVCGTEQYF | Proform |
| 2 | ALAA75MQQFYGINMT | Truncated proform | |
| 3 | KKPR100CGVPDQTRGS | Truncated proform | |
| 4 | RRKR119YALTGQKWQ | Active form | |
| proMMP17 | 1 | NH2-A39PAPAPRAEDLSL | Proform |
| 2 | NH2-A39PAPAPRAEDLSL | Proform | |
| 3 | TQAR122RRPQAPAPTKW | Active form | |
| 4 | TQAR122RRPQAPAPTKW | Active form |
| Protein Name | Primer | Primer Sequence (5′-3′) |
|---|---|---|
| reverse * | ATATGCGGCCGCTTATTTAACGGGGTATGTATAAGCGA | |
| proMMP15 | forward 1 | GCCCTCACCGGGAGGGACGAGCCGAACCAACCCT |
| proMMP15 | forward 2 | CGGAAGCGCTACGCCCTCACCGGGAGGGAC |
| proMMP15 | forward 3 | ATATGTCGACCGGCGTCGGAAGCGCTACGCCCTCA |
| proMMP17 | forward 1 | GCTCCAGCCCCCACCGACGAGCCGAACCAACCCT |
| proMMP17 | forward 2 | AGGAGACGCCAGGCTCCAGCCCCCACCGAC |
| proMMP17 | forward 3 | ATATGTCGACGCTCGCAGGAGACGCCAGGCTCCAG |
| proMMP19 | forward 1 | CTGTTGCTGGGCCGCGACGAGCCGAACCAACCCT |
| proMMP19 | forward 2 | ACCCTTAAATACCTGTTGCTGGGCCGCGAC |
| proMMP19 | forward 3 | ATATGTCGACCAGAAGACCCTTAAATACCTGTTGCTGG |
| proMMP21 | forward 1 | CCGCGGGCGCCGCTGGACGAGCCGAACCAACCCT |
| proMMP21 | forward 2 | TCCAGGCGCTCCCCGCGGGCGCCGCTGGAC |
| proMMP21 | forward 3 | ATATGTCGACGCCCGCTCCAGGCGCTCCCCGCGG |
| proMMP24 | forward 1 | GCCCTGACTGGACAGGACGAGCCGAACCAACCCT |
| proMMP24 | forward 2 | GAAACAAGCGCTATGCCCTGACTGGACAGGAC |
| proMMP24 | forward 3 | ATATGTCGACCGGAGAAACAAGCGCTATGCCCT |
| proMMP25 | forward 1 | CTGAGCGGCAGCGACGAGCCGAACCAACCCT |
| proMMP25 | forward 2 | CGCCGGTACGCTCTGAGCGGCAGCGAC |
| proMMP25 | forward 3 | ATATGTCGACAGGCGGCGTCGCCGGTACGCTCTGAGC |
| proMMP28 | forward 1 | GCAAAGCAAGGTAACGACGAGCCGAACCAACCCT |
| proMMP28 | forward 2 | TAAGAAACGCTTTGCAAAGCAAGGTAACGACGAG |
| proMMP28 | forward 3 | ATATGTCGACAGGCGTAAGAAACGCTTTGCAAAGCAAG |
| Protein Name | Primer | Primer Sequence (5P′-3′) |
|---|---|---|
| reverse * | GTCGACCTGCAGGCTCGC | |
| proMMP1 | forward | GAUGUGGCGCAGUUUGUGCUGACCGACGAGCCGAACCAACCC |
| proMMP2 | forward | GAUGUGGCGAACUAUAACUUUUUUGACGAGCCGAACCAACCC |
| proMMP3 | forward | GAUGUGGGUCAUUUUCGUACCUUUGACGAGCCGAACCAACCC |
| proMMP7 | forward | GAUGUGGCGGAAUAUAGCCUGUUUGACGAGCCGAACCAACCC |
| proMMP8 | forward | GGUGGUUUUAUGCUGACCCCGGGUGACGAGCCGAACCAACCC |
| proMMP9 | forward | GAUCUGGGU CGUUUUCAGACCUUUGACGAGCCGAACCAACCC |
| proMMP10 | forward | GAUGUGGGUCAUUUUAGCAGCUUUGACGAGCCGAACCAACCC |
| proMMP11 | forward | CGUCAGAAACGUUUUGUGCUGAGCGACGAGCCGAACCAACCC |
| proMMP12 | forward | GAAAUGCCGGGUGGUCCGGUGUGGGACGAGCCGAACCAACCC |
| proMMP13 | forward | GAUGUGGGUGAAUAUAACGUGUUUGACGAGCCGAACCAACCC |
| proMMP14 | forward | CGUCGUAAACGUUAUGCGAUUCAGGACGAGCCGAACCAACCC |
| proMMP16 | forward | CGUCGUAAACGUUAUGCGCUGACCGACGAGCCGAACCAACCC |
| proMMP20 | forward | GAUGUGGCGAACUAUCGUCUGUUUGACGAGCCGAACCAACCC |
| proMMP23 | forward | CGUCGUCGUCGUUAUACCCUGCCGGACGAGCCGAACCAACCC |
| proMMP26 | forward | GAUGGUAGCGAUACCAGCAUUAGCGACGAGCCGAACCAACCC |
| proMMP27 | forward | GAUGUGGGUCAGUAUGGUUAUACCGACGAGCCGAACCAACCC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falkowski, K.; Bielecka, E.; Thøgersen, I.B.; Bocheńska, O.; Płaza, K.; Kalińska, M.; Sąsiadek, L.; Magoch, M.; Pęcak, A.; Wiśniewska, M.; et al. Kallikrein-Related Peptidase 14 Activates Zymogens of Membrane Type Matrix Metalloproteinases (MT-MMPs)—A CleavEx Based Analysis. Int. J. Mol. Sci. 2020, 21, 4383. https://doi.org/10.3390/ijms21124383
Falkowski K, Bielecka E, Thøgersen IB, Bocheńska O, Płaza K, Kalińska M, Sąsiadek L, Magoch M, Pęcak A, Wiśniewska M, et al. Kallikrein-Related Peptidase 14 Activates Zymogens of Membrane Type Matrix Metalloproteinases (MT-MMPs)—A CleavEx Based Analysis. International Journal of Molecular Sciences. 2020; 21(12):4383. https://doi.org/10.3390/ijms21124383
Chicago/Turabian StyleFalkowski, Katherine, Ewa Bielecka, Ida B. Thøgersen, Oliwia Bocheńska, Karolina Płaza, Magdalena Kalińska, Laura Sąsiadek, Małgorzata Magoch, Aleksandra Pęcak, Magdalena Wiśniewska, and et al. 2020. "Kallikrein-Related Peptidase 14 Activates Zymogens of Membrane Type Matrix Metalloproteinases (MT-MMPs)—A CleavEx Based Analysis" International Journal of Molecular Sciences 21, no. 12: 4383. https://doi.org/10.3390/ijms21124383
APA StyleFalkowski, K., Bielecka, E., Thøgersen, I. B., Bocheńska, O., Płaza, K., Kalińska, M., Sąsiadek, L., Magoch, M., Pęcak, A., Wiśniewska, M., Gruba, N., Wysocka, M., Wojtysiak, A., Brzezińska-Bodal, M., Sychowska, K., Pejkovska, A., Rehders, M., Butler, G., Overall, C. M., ... Kantyka, T. (2020). Kallikrein-Related Peptidase 14 Activates Zymogens of Membrane Type Matrix Metalloproteinases (MT-MMPs)—A CleavEx Based Analysis. International Journal of Molecular Sciences, 21(12), 4383. https://doi.org/10.3390/ijms21124383