Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword



Abstract
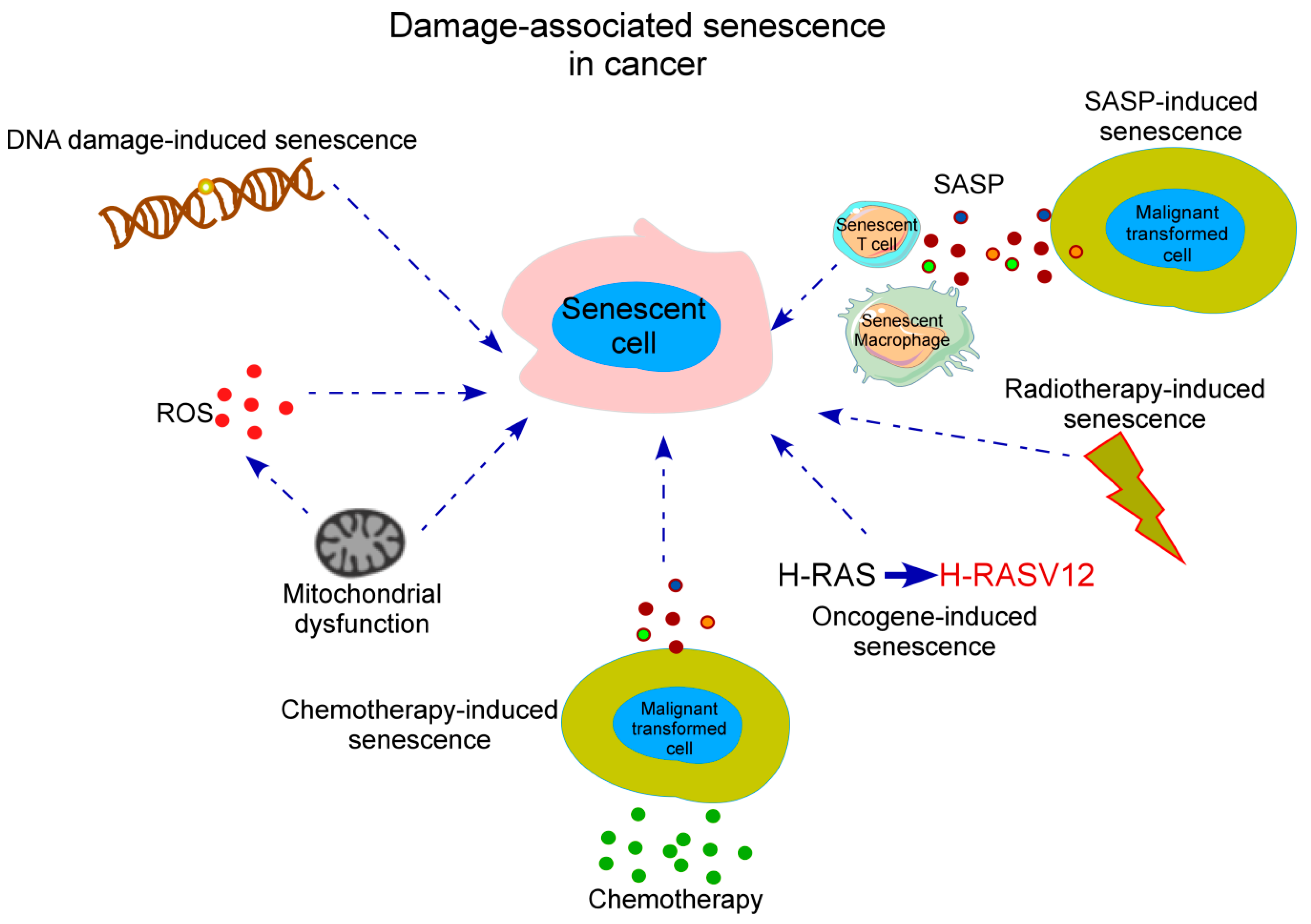
1. Defining Senescence
1.1. From Old to New and Different Concepts of Senescence
1.2. Senescence Biomarkers and the SASP
1.3. Epigenetics in Senescence
1.4. Signaling Pathways that Contribute to Senescence
1.5. Contribution of Inflammaging to Senescence Surveillance
2. Senescence in Cancer
2.1. Before Cancer Treatment: Age-Related Senescence and Cancer
2.2. After Cancer Treatment with Chemotherapy
2.3. After Cancer Treatment with Immunotherapy
3. Senescence in the Immune Response to Fight Cancer
3.1. T Cells
3.2. B Cells
3.3. NK Cells
3.4. Macrophages
3.5. Other Immune Cells
4. Clinical Relevance of Senescence in Immunotherapy: A Balance between Induction and Reversal of Senescence
4.1. Induction or Reversal of Senescence
4.2. Clearance of Senescent Cancer Cells by NK Cells
4.3. Reversal of T Cell Senescence
4.4. Potential Use of B Cells in Immunotherapy and Reversal of B Cell Senescence
4.5. Retuning the Tumor Microenvironment to Promote Macrophage-Mediated Cancer Cell Clearance

{kind=link}
{kind=link}
| Senescent Immune Cell | SASP Factors | References |
|---|---|---|
| CD4+ T cells | IL-6, IL-10, TNFα, IFNγ, TGF-β1 | [102] |
| CD8+ T cells | IL-6, IL-8, TNFα, IL-18, IFNγ, TGF-β1, CCL16, ADAM28 | [102,111,117,118] |
| B cells | IL-6, IL-8, TNFα | [229] |
| NK cells | MMPs, cathepsins | [88] |
| Macrophages | IL-6, TNFα, PDGF-BB, TGFβ | Reviewed in [230] |
| SASP Factor | Senescent Cells That Secrete Factor | Protumorigenic Mechanisms | Antitumorigenic Mechanisms | Reference |
|---|---|---|---|---|
| IL-6 | CD8+ T cells, B cells, macrophages | Recruit MDSCs, impair DC differentiation, inhibit antitumor T cell responses | Recruit macrophages and NKT cells | [231,232,233] |
| IL-8 | Tumor cells in solid tumors and hematological malignancies | Enhancement of angiogenesis, attraction of neutrophils and MDSCs | Reviewed in [234] | |
| IL-1α | Senescent fibroblasts. Breast cancer cells. Colon cancer cells | Regulation of IL-6 and IL-8 protumorigenic effects. Induction of production of tumor survival factors. Oncogene Ras-induced cell senescence, doxorubicin-induced cancer cell senescence and replicative senescence. | Macrophage immune surveillance | [235,236,237,238] |
| IL-1β | Fibroblasts | Inflammaging, induction of ROS-mediated DDR | [58] | |
| IL-10 | Macrophages | Immunosuppression | [233] | |
| CXCL2/CXCR2 | Prostate cancer cells | T cell suppression through macrophage polarization to an anti-inflammatory phenotype. Recruit iMCs that hinder tumor cell senescence. | [239,240] | |
| PGE2 | Hepatic stellate cells | Inhibit antitumor responses through PTGER4 receptor in hepatocellular carcinoma | [241] | |
| CCL2 | Senescent hepatocytes | Promotes accumulation of immunosuppressive iMCs promoting hepatocellular carcinoma through NK cell inhibition | Recruits myeloid cells that differentiate into macrophages to clear senescent precancerous cells | [39] |
| CCL3 (MIP-1 α) | Senescent hepatocytes | Recruit immune NK cells for clearance of senescent cells in hepatocellular carcinoma | [242] | |
| TGF-β1 | Fibroblasts | Inflammaging, induction of ROS-mediated DDR | [58] | |
| TGF-β3 | Senescent adipose-derived mesenchymal stem cells | Decreased angiogenic potential | [243] | |
| CCL5 | Melanoma cells | Recruit TILs to eliminate cancer cells | [244] | |
| TNFα | Induce T cell senescence | [109] | ||
| IFNγ | Bone marrow-derived macrophages | Induce M1 macrophage differentiation | [245] | |
| IFNα | Induce CD8+ T cell senescence, accelerates loss of CD27 and CD28 | [135] |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, E. Senescent Human Fibroblasts Resist Programmed Cell Death, and Failure to Suppress bell Is Involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Perez-Mancera, P.A.; Young, A.R.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Suarez, E.; Samper, E.; Flores, J.M.; Blasco, M.A. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat. Genet. 2000, 26, 114–117. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany N. Y.) 2013, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Marcotte, R.; Lacelle, C.; Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 2004, 125, 777–783. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef]
- Sati, S.; Bonev, B.; Szabo, Q.; Jost, D.; Bensadoun, P.; Serra, F.; Loubiere, V.; Papadopoulos, G.L.; Rivera-Mulia, J.-C.; Fritsch, L.; et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol. Cell 2020, 78, 1–17. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, inflammation and cancer. Semin. Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Gong, W.; Hoffmann, J.M.; Stock, S.; Wang, L.; Liu, Y.; Schubert, M.L.; Neuber, B.; Huckelhoven-Krauss, A.; Gern, U.; Schmitt, A.; et al. Comparison of IL-2 vs IL-7/IL-15 for the generation of NY-ESO-1-specific T cells. Cancer Immunol. Immunother. 2019, 68, 1195–1209. [Google Scholar] [CrossRef]
- Okoye, I.S.; Xu, L.; Walker, J.; Elahi, S. The glucocorticoids prednisone and dexamethasone differentially modulate T cell function in response to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef]
- Yi, F.; Frazzette, N.; Cruz, A.C.; Klebanoff, C.A.; Siegel, R.M. Beyond Cell Death: New Functions for TNF Family Cytokines in Autoimmunity and Tumor Immunotherapy. Trends Mol. Med. 2018, 24, 642–653. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Xu, B.W.; Chen, Y.J.; Fu, X.Q.; Zhu, P.L.; Bai, J.X.; Chou, J.Y.; Yin, C.L.; Li, J.K.; Wang, Y.P.; et al. Inhibiting the Src/STAT3 signaling pathway contributes to the anti-melanoma mechanisms of dioscin. Oncol. Lett. 2020, 19, 2508–2514. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; So, J.Y.; Skrypek, N.; Yang, H.H.; Merchant, A.S.; Nelson, G.W.; Chen, W.D.; Ishii, H.; Chen, J.M.; Hu, G.; et al. Induction of DNMT3B by PGE2 and IL6 at distant metastatic sites promotes epigenetic modification and breast cancer colonization. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Patel, H.J.; Patel, B.M. TNF-alpha and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef]
- Maynard, J.P.; Ertunc, O.; Kulac, I.; Baena-Del Valle, J.A.; De Marzo, A.M.; Sfanos, K.S. IL8 Expression Is Associated with Prostate Cancer Aggressiveness and Androgen Receptor Loss in Primary and Metastatic Prostate Cancer. Mol. Cancer Res. 2020, 18, 153–165. [Google Scholar] [CrossRef]
- Orosz, P.; Kruger, A.; Hubbe, M.; Ruschoff, J.; Von Hoegen, P.; Mannel, D.N. Promotion of experimental liver metastasis by tumor necrosis factor. Int. J. Cancer 1995, 60, 867–871. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Eyman, D.; Damodarasamy, M.; Plymate, S.R.; Reed, M.J. CCL5 secreted by senescent aged fibroblasts induces proliferation of prostate epithelial cells and expression of genes that modulate angiogenesis. J. Cell Physiol. 2009, 220, 376–381. [Google Scholar] [CrossRef]
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-beta-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664.e8. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. The Histone Code of Senescence. Cells 2020, 9, 466. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Peters, A.H.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schofer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; van Rooijen, E.; Werner-Klein, M.; et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell 2018, 33, 322–336.e8. [Google Scholar] [CrossRef]
- Duarte, L.F.; Young, A.R.J.; Wang, Z.; Wu, H.A.; Panda, T.; Kou, Y.; Kapoor, A.; Hasson, D.; Mills, N.R.; Ma’ayan, A.; et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014, 5, 5210. [Google Scholar] [CrossRef]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef]
- Zirkel, A.; Nikolic, M.; Sofiadis, K.; Mallm, J.P.; Brackley, C.A.; Gothe, H.; Drechsel, O.; Becker, C.; Altmuller, J.; Josipovic, N.; et al. HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Mol. Cell 2018, 70, 730–744.e6. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- D’Adda Di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Monch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1-and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘Bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Zwerschke, W.; Mazurek, S.; Stöckl, P.; Hütter, E.; Eigenbrodt, E.; Jansen-Dürr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411. [Google Scholar] [CrossRef] [PubMed]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; De Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Dho, S.H.; Ju, S.K.; Maeng, J.S.; Kim, J.Y.; Kwon, K.S. Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology 2012, 13, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschenes-Simard, X.; Ferbeyre, G. Mitochondrial Dysfunction Contributes to Oncogene-Induced Senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; MacKay, G.; Van Der Burg, S.H.; Verdegaal, E.M.E.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- Takebayashi, S.I.; Tanaka, H.; Hino, S.; Nakatsu, Y.; Igata, T.; Sakamoto, A.; Narita, M.; Nakao, M. Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene-induced senescent cells. Aging Cell 2015, 14, 689–697. [Google Scholar] [CrossRef]
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, II; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 2012, 11, 1383–1392. [Google Scholar] [CrossRef]
- Fafián-Labora, J.; Carpintero-Fernández, P.; Jordan, S.J.D.; Shikh-Bahaei, T.; Abdullah, S.M.; Mahenthiran, M.; Rodríguez-Navarro, J.A.; Niklison-Chirou, M.V.; O’Loghlen, A. FASN activity is important for the initial stages of the induction of senescence. Cell Death Dis. 2019, 10, 318. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Drabkin, M.J.; Meyer, J.I.; Kanth, N.; Lobel, S.; Fogel, J.; Grossman, J.; Krumenacker, J.H. Age-stratified Patterns of Thymic Involution on Multidetector CT. J. Thorac. Imaging 2018, 33, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Coder, B.D.; Wang, H.; Ruan, L.; Su, D.M. Thymic involution perturbs negative selection leading to autoreactive T cells that induce chronic inflammation. J. Immunol. 2015, 194, 5825–5837. [Google Scholar] [CrossRef]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Pluquet, O.; Abbadie, C.; Coqueret, O. Connecting cancer relapse with senescence. Cancer Lett. 2019, 463, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Perez-Amill, L.; Marzal, B.; Urbano-Ispizua, A.; Juan, M.; Martin-Antonio, B. CAR-T Cell Therapy: A Door Is Open to Find Innumerable Possibilities of Treatments for Cancer Patients. Turk. J. Haematol. 2018, 35, 217–228. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Braumuller, H.; Wieder, T.; Brenner, E.; Assmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef]
- Martin-Antonio, B.; Sune, G.; Perez-Amill, L.; Castella, M.; Urbano-Ispizua, A. Natural Killer Cells: Angels and Devils for Immunotherapy. Int. J. Mol. Sci. 2017, 18, 1868. [Google Scholar] [CrossRef]
- Martin-Antonio, B.; Sune, G.; Najjar, A.; Perez-Amill, L.; Antonana-Vildosola, A.; Castella, M.; Leon, S.; Velasco-de Andres, M.; Lozano, F.; Lozano, E.; et al. Extracellular NK histones promote immune cell antitumor activity by inducing cell clusters through binding to CD138 receptor. J. Immunother. Cancer 2019, 7, 259. [Google Scholar] [CrossRef]
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.; Lowe, S.W.; Krizhanovsky, V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 2013, 32, 1971–1977. [Google Scholar] [CrossRef]
- Egashira, M.; Hirota, Y.; Shimizu-Hirota, R.; Saito-Fujita, T.; Haraguchi, H.; Matsumoto, L.; Matsuo, M.; Hiraoka, T.; Tanaka, T.; Akaeda, S.; et al. F4/80+ Macrophages Contribute to Clearance of Senescent Cells in the Mouse Postpartum Uterus. Endocrinology 2017, 158, 2344–2353. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and its hallmarks: How to oppose aging strategically? A review of potential options for therapeutic intervention. Front. Immunol. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, F.J.; Franzese, O.; Belaramani, L.L.; Fletcher, J.M.; Gilmour, K.C.; Sharifi, R.; Khan, N.; Hislop, A.D.; Cara, A.; Salmon, M.; et al. The impact of telomere erosion on memory CD8+ T cells in patients with X-linked lymphoproliferative syndrome. Mech. Ageing Dev. 2005, 126, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Boursalian, T.E.; Turk, G.L.; Fink, P.J. Thymic output in aged mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8447–8452. [Google Scholar] [CrossRef]
- Hun, M.L.; Wong, K.; Gunawan, J.R.; Alsharif, A.; Quinn, K.; Chidgey, A.P. Gender Disparity Impacts on Thymus Aging and LHRH Receptor Antagonist-Induced Thymic Reconstitution Following Chemotherapeutic Damage. Front. Immunol. 2020, 11, 302. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Huston, G.E.; Dibble, J.; Duso, D.K.; Swain, S.L. Bim dictates naive CD4 T cell lifespan and the development of age-associated functional defects. J. Immunol. 2010, 185, 4535–4544. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Lee, W.W.; Weyand, C.M. Aging and T-cell diversity. Exp. Gerontol. 2007, 42, 400–406. [Google Scholar] [CrossRef]
- van de Berg, P.J.E.J.; Griffiths, S.J.; Yong, S.-L.; Macaulay, R.; Bemelman, F.J.; Jackson, S.; Henson, S.M.; ten Berge, I.J.M.; Akbar, A.N.; van Lier, R.A.W. Cytomegalovirus Infection Reduces Telomere Length of the Circulating T Cell Pool. J. Immunol. 2010, 184, 3417–3423. [Google Scholar] [CrossRef]
- Henson, S.M.; Lanna, A.; Riddel, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. P38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-Derived γδ Regulatory T Cells Suppress Innate and Adaptive Immunity through the Induction of Immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8+CD28- Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Sanders, K.L.; Edwards, J.L.; Ye, J.; Si, F.; Gao, A.; Huang, L.; Hsueh, E.C.; Ford, D.A.; et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab. 2019, 29, 103–123.e5. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Monteiro, J.; Batliwalla, F.; Ostrer, H.; Gregersen, P.K. Shortened telomeres in clonally expanded CD28-CD8+ T cells imply a replicative history that is distinct from their CD28+CD8+ counterparts. J. Immunol. 1996, 156, 3587–3590. [Google Scholar]
- Di Mitri, D.; Azevedo, R.I.; Henson, S.M.; Libri, V.; Riddell, N.E.; Macaulay, R.; Kipling, D.; Soares, M.V.D.; Battistini, L.; Akbar, A.N. Reversible Senescence in Human CD4 + CD45RA + CD27 − Memory T Cells. J. Immunol. 2011, 187, 2093–2100. [Google Scholar] [CrossRef]
- Romero, P.; Zippelius, A.; Kurth, I.; Pittet, M.J.; Touvrey, C.; Iancu, E.M.; Corthesy, P.; Devevre, E.; Speiser, D.E.; Rufer, N. Four Functionally Distinct Populations of Human Effector-Memory CD8 + T Lymphocytes. J. Immunol. 2007, 178, 4112–4119. [Google Scholar] [CrossRef]
- Henson, S.M.; Macaulay, R.; Riddell, N.E.; Nunn, C.J.; Akbar, A.N. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8+ T-cell proliferation by distinct pathways. Eur. J. Immunol. 2015, 45, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, F.J.; Franzese, O.; Finney, H.M.; Fletcher, J.M.; Belaramani, L.L.; Salmon, M.; Dokal, I.; Webster, D.; Lawson, A.D.G.; Akbar, A.N. The Loss of Telomerase Activity in Highly Differentiated CD8 + CD28 − CD27 − T Cells Is Associated with Decreased Akt (Ser 473) Phosphorylation. J. Immunol. 2007, 178, 7710–7719. [Google Scholar] [CrossRef] [PubMed]
- Riddell, N.E.; Griffiths, S.J.; Rivino, L.; King, D.C.B.; Teo, G.H.; Henson, S.M.; Cantisan, S.; Solana, R.; Kemeny, D.M.; Macary, P.A.; et al. Multifunctional cytomegalovirus (CMV)-specific CD8+ T cells are not restricted by telomere-related senescence in young or old adults. Immunology 2015, 144, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Azevedo, R.I.; Jackson, S.E.; Di Mitri, D.; Lachmann, R.; Fuhrmann, S.; Vukmanovic-Stejic, M.; Yong, K.; Battistini, L.; Kern, F.; et al. Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+CD45RA+CD27- T cells: The potential involvement of interleukin-7 in this process. Immunology 2011, 132, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, C.; Guo, W.; Effros, R.B. Resistance to apoptosis in human CD8+ T cells that reach replicative senescence after multiple rounds of antigen-specific proliferation. Exp. Gerontol. 1999, 34, 633–644. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.a.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef]
- Callender, L.A.; Carroll, E.C.; Beal, R.W.J.; Chambers, E.S.; Nourshargh, S.; Akbar, A.N.; Henson, S.M. Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 2018, 17, e12675. [Google Scholar] [CrossRef]
- Callender, L.A.; Carroll, E.C.; Bober, E.A.; Akbar, A.N.; Solito, E.; Henson, S.M. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 2020, 19, e13067. [Google Scholar] [CrossRef]
- Liu, X.; Hoft, D.F.; Peng, G. Senescent T cells within suppressive tumor microenvironments: Emerging target for tumor immunotherapy. J. Clin. Investig. 2020, 130, 1073–1083. [Google Scholar] [CrossRef]
- Kubuki, Y.; Suzuki, M.; Sasaki, H.; Toyama, T.; Yamashita, K.; Maeda, K.; Ido, A.; Matsuoka, H.; Okayama, A.; Nakanishi, T.; et al. Telomerase activity and telomere length as prognostic factors of adult T-cell leukemia. Leuk. Lymphoma 2005, 46, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.; Wong, R.; Mason, M.; Fegan, C.; Man, S.; Pepper, C. Expansion of a CD8 +PD-1 + replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin. Cancer Res. 2012, 18, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhou, J.; Hathcock, K.S.; Robbins, P.; Powell, D.J.; Rosenberg, S.A.; Hodes, R.J. Persistence of tumor infiltrating lymphocytes in adoptive immunotherapy correlates with telomere length. J. Immunother. 2007, 30, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-induced senescent T cells with suppressor function: A potential form of tumor immune evasion. Cancer Res. 2008, 68, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR 8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.W.; Wang, Y.; Valmori, D.; Ayyoub, M.; Han, Y.; Xu, X.L.; Zhao, A.L.; Qu, L.; Gnjatic, S.; Ritter, G.; et al. Ex-vivo analysis of CD8+ T cells infiltrating colorectal tumors identifies a major effector-memory subset with low perforin content. J. Clin. Immunol. 2006, 26, 447–456. [Google Scholar] [CrossRef]
- Urbaniak-Kujda, D.; Kapelko-Słowik, K.; Wołowiec, D.; Dybko, J.; Hałoń, A.; Jaźwiec, B.; Maj, J.; Jankowska-Konsur, A.; Kuliczkowski, K. Increased percentage of CD8+CD28- suppressor lymphocytes in peripheral blood and skin infiltrates correlates with advanced disease in patients with cutaneous T-cell lymphomas. Postepy Hig. Med. Dosw. 2009, 63, 355–359. [Google Scholar]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Johrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, 1–20. [Google Scholar] [CrossRef]
- Chung, D.J.; Pronschinske, K.B.; Shyer, J.A.; Sharma, S.; Leung, S.; Curran, S.A.; Lesokhin, A.M.; Devlin, S.M.; Giralt, S.A.; Young, J.W. T-cell exhaustion in Multiple myeloma relapse after autotransplant: Optimal timing of immunotherapy. Cancer Immunol. Res. 2016, 4, 61–71. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Parish, S.T.; Kim, S.; Sekhon, R.K.; Wu, J.E.; Kawakatsu, Y.; Effros, R.B. Adenosine deaminase modulation of telomerase activity and replicative senescence in human CD8 T lymphocytes. J. Immunol. 2010, 184, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Maybruck, B.T.; Pfannenstiel, L.W.; Diaz-Montero, M.; Gastman, B.R. Tumor-derived exosomes induce CD8+ T cell suppressors. J. Immunother. Cancer 2017, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; Coutavas, E.; Levati, L.; Seidel, J.; Rustin, M.H.A.; Henson, S.M.; Akbar, A.N.; Franzese, O. IFN-α Inhibits Telomerase in Human CD8 + T Cells by Both hTERT Downregulation and Induction of p38 MAPK Signaling. J. Immunol. 2013, 191, 3744–3752. [Google Scholar] [CrossRef]
- Dodeller, F.; Schulze-Koops, H. The p38 mitogen-activated protein kinase signaling cascade in CD4 T cells. Arthritis Res. Ther. 2006, 8, 205. [Google Scholar] [CrossRef]
- Salvador, J.M.; Mittelstadt, P.R.; Guszczynski, T.; Copeland, T.D.; Yamaguchi, H.; Appella, E.; Fornace, A.J., Jr.; Ashwell, J.D. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 2005, 6, 390–395. [Google Scholar] [CrossRef]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef]
- Lanna, A.; Gomes, D.C.O.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.W.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Andreu, P.; Johansson, M.; Affara, N.I.; Pucci, F.; Tan, T.; Junankar, S.; Korets, L.; Lam, J.; Tawfik, D.; DeNardo, D.G.; et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 2010, 17, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Nayar, S.; Dasgupta, P.; Galustian, C. Extending the lifespan and efficacies of immune cells used in adoptive transfer for cancer immunotherapies—A review. Oncoimmunology 2015, 4, e1002720. [Google Scholar] [CrossRef]
- Wu, C.; Espinoza, D.A.; Koelle, S.J.; Yang, D.; Truitt, L.; Schlums, H.; Lafont, B.A.; Davidson-Moncada, J.K.; Lu, R.; Kaur, A.; et al. Clonal expansion and compartmentalized maintenance of rhesus macaque NK cell subsets. Sci. Immunol. 2018, 3, eaat9781. [Google Scholar] [CrossRef]
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef]
- Gounder, S.S.; Abdullah, B.J.J.; Radzuanb, N.; Zain, F.; Sait, N.B.M.; Chua, C.; Subramani, B. Effect of Aging on NK Cell Population and Their Proliferation at Ex Vivo Culture Condition. Anal. Cell Pathol. (Amst.) 2018, 2018, 7871814. [Google Scholar] [CrossRef]
- Manser, A.R.; Uhrberg, M. Age-related changes in natural killer cell repertoires: Impact on NK cell function and immune surveillance. Cancer Immunol. Immunother. 2016, 65, 417–426. [Google Scholar] [CrossRef]
- Przemska-Kosicka, A.; Childs, C.E.; Maidens, C.; Dong, H.; Todd, S.; Gosney, M.A.; Tuohy, K.M.; Yaqoob, P. Age-Related Changes in the Natural Killer Cell Response to Seasonal Influenza Vaccination Are Not Influenced by a Synbiotic: A Randomised Controlled Trial. Front. Immunol. 2018, 9, 591. [Google Scholar] [CrossRef]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Li, L.; McCarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.Z.; Cooper, L.; et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br. J. Haematol. 2017, 177, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Streltsova, M.A.; Erokhina, S.A.; Kanevskiy, L.M.; Lee, D.A.; Telford, W.G.; Sapozhnikov, A.M.; Kovalenko, E.I. Analysis of NK cell clones obtained using interleukin-2 and gene-modified K562 cells revealed the ability of “senescent” NK cells to lose CD57 expression and start expressing NKG2A. PLoS ONE 2018, 13, e0208469. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Verges, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 2010, 116, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Dace, D.S.; Apte, R.S. Effect of senescence on macrophage polarization and angiogenesis. Rejuvenation Res. 2008, 11, 177–185. [Google Scholar] [CrossRef]
- Linehan, E.; Fitzgerald, D.C. Ageing and the immune system: Focus on macrophages. Eur. J. Microbiol. Immunol. (Bp) 2015, 5, 14–24. [Google Scholar] [CrossRef]
- Jackaman, C.; Tomay, F.; Duong, L.; Abdol Razak, N.B.; Pixley, F.J.; Metharom, P.; Nelson, D.J. Aging and cancer: The role of macrophages and neutrophils. Ageing Res. Rev. 2017, 36, 105–116. [Google Scholar] [CrossRef]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef]
- Reimann, M.; Lee, S.; Loddenkemper, C.; Dorr, J.R.; Tabor, V.; Aichele, P.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Tumor stroma-derived TGF-beta limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell 2010, 17, 262–272. [Google Scholar] [CrossRef]
- Llanos, S.; Serrano, M. Senescence and Cancer: In the Name of Immunosuppression. Cancer Cell 2016, 30, 507–508. [Google Scholar] [CrossRef][Green Version]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Harbort, C.J.; Soeiro-Pereira, P.V.; von Bernuth, H.; Kaindl, A.M.; Costa-Carvalho, B.T.; Condino-Neto, A.; Reichenbach, J.; Roesler, J.; Zychlinsky, A.; Amulic, B. Neutrophil oxidative burst activates ATM to regulate cytokine production and apoptosis. Blood 2015, 126, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Johnson, C.A.; Prasad, N.; Yang, J.; Chen, S.C.; Ayers, G.D.; Pawlikowski, J.S.; Raman, D.; Sosman, J.A.; Kelley, M.; et al. Connecting the Dots: Therapy-Induced Senescence and a Tumor-Suppressive Immune Microenvironment. J. Natl. Cancer Inst. 2016, 108, djv406. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- von Kobbe, C. Targeting senescent cells: Approaches, opportunities, challenges. Aging (Albany N. Y.) 2019, 11, 12844–12861. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2019, 9, 1576. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Stokes, K.L.; Cortez-Retamozo, V.; Acosta, J.; Lauderback, B.; Robles-Oteiza, C.; Cicchini, M.; Pittet, M.J.; Feldser, D.M. Natural killer cells limit the clearance of senescent lung adenocarcinoma cells. Oncogenesis 2019, 8, 24. [Google Scholar] [CrossRef]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent upregulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [PubMed]
- Ciaglia, E.; Laezza, C.; Abate, M.; Pisanti, S.; Ranieri, R.; D’Alessandro, A.; Picardi, P.; Gazzerro, P.; Bifulco, M. Recognition by natural killer cells of N6-isopentenyladenosine-treated human glioma cell lines. Int. J. Cancer 2018, 142, 176–190. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Waldhauer, I.; Goehlsdorf, D.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.G.; Steinle, A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008, 68, 6368–6376. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Atteridge, C.L.; Wang, X.; Lundgren, A.D.; Wu, J.D. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J. Immunol. 2010, 184, 3346–3350. [Google Scholar] [CrossRef] [PubMed]
- Munoz, D.P.; Yannone, S.M.; Daemen, A.; Sun, Y.; Vakar-Lopez, F.; Kawahara, M.; Freund, A.M.; Rodier, F.; Wu, J.D.; Desprez, P.Y.; et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 2019, 5, e124716. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Cecere, F.; Vulpis, E.; Fionda, C.; Molfetta, R.; Soriani, A.; Petrucci, M.T.; Ricciardi, M.R.; Fuerst, D.; Amendola, M.G.; et al. Genotoxic Stress Induces Senescence-Associated ADAM10-Dependent Release of NKG2D MIC Ligands in Multiple Myeloma Cells. J. Immunol. 2015, 195, 736–748. [Google Scholar] [CrossRef]
- Sepult, C.; Bellefroid, M.; Rocks, N.; Donati, K.; Gerard, C.; Gilles, C.; Ludwig, A.; Duysinx, B.; Noel, A.; Cataldo, D. ADAM10 mediates malignant pleural mesothelioma invasiveness. Oncogene 2019, 38, 3521–3534. [Google Scholar] [CrossRef]
- Ko, S.Y.; Lin, S.C.; Wong, Y.K.; Liu, C.J.; Chang, K.W.; Liu, T.Y. Increase of disintergin metalloprotease 10 (ADAM10) expression in oral squamous cell carcinoma. Cancer Lett. 2007, 245, 33–43. [Google Scholar] [CrossRef]
- Carl-McGrath, S.; Lendeckel, U.; Ebert, M.; Roessner, A.; Rocken, C. The disintegrin-metalloproteinases ADAM9, ADAM12, and ADAM15 are upregulated in gastric cancer. Int. J. Oncol. 2005, 26, 17–24. [Google Scholar] [CrossRef]
- Wolpert, F.; Tritschler, I.; Steinle, A.; Weller, M.; Eisele, G. A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. Neuro Oncol. 2014, 16, 382–391. [Google Scholar] [CrossRef]
- Sheppard, S.; Guedes, J.; Mroz, A.; Zavitsanou, A.M.; Kudo, H.; Rothery, S.M.; Angelopoulos, P.; Goldin, R.; Guerra, N. The immunoreceptor NKG2D promotes tumour growth in a model of hepatocellular carcinoma. Nat. Commun. 2017, 8, 13930. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.G.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany N. Y.) 2016, 8, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, T.; Kotani, H.; Iida, Y.; Koyanagi, A.; Tanino, R.; Tsubata, Y.; Isobe, T.; Harada, M. Pemetrexed sensitizes human lung cancer cells to cytotoxic immune cells. Cancer Sci. 2020, 111, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, C.; Ricci, B.; Vulpis, E.; Fionda, C.; Ricciardi, M.R.; Petrucci, M.T.; Masuelli, L.; Peri, A.; Cippitelli, M.; Zingoni, A.; et al. Drug-Induced Senescent Multiple Myeloma Cells Elicit NK Cell Proliferation by Direct or Exosome-Mediated IL15 Trans-Presentation. Cancer Immunol. Res. 2018, 6, 860–869. [Google Scholar] [CrossRef]
- Inao, T.; Kotani, H.; Iida, Y.; Kartika, I.D.; Okimoto, T.; Tanino, R.; Shiba, E.; Harada, M. Different sensitivities of senescent breast cancer cells to immune cell-mediated cytotoxicity. Cancer Sci. 2019, 110, 2690–2699. [Google Scholar] [CrossRef]
- Sharma, C.; Wang, H.X.; Li, Q.; Knoblich, K.; Reisenbichler, E.S.; Richardson, A.L.; Hemler, M.E. Protein Acyltransferase DHHC3 Regulates Breast Tumor Growth, Oxidative Stress, and Senescence. Cancer Res. 2017, 77, 6880–6890. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef]
- Marcais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. (Toronto Ont.) 2020, 27 (Suppl. 2), S87–S97. [Google Scholar]
- Das, S.; Johnson, D.B. Immune-related adverse events and antitumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef]
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Gato, M.; Martínez-Aguillo, M.; Morilla, I.; Fernández, G.; Hernández, B.; López, P.; Alberdi, N.; et al. Early detection of hyperprogressive disease in non-small cell lung cancer by monitoring of systemic T cell dynamics. Cancers 2020, 12, 344. [Google Scholar] [CrossRef] [PubMed]
- Sokol, L.; Cripe, L.; Kantarjian, H.; Sekeres, M.A.; Parmar, S.; Greenberg, P.; Goldberg, S.L.; Bhushan, V.; Shammo, J.; Hohl, R.; et al. Randomized, dose-escalation study of the p38α MAPK inhibitor SCIO-469 in patients with myelodysplastic syndrome. Leukemia 2013, 27, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Haluska, P.; Tolcher, A.W.; Erlichman, C.; Papadopoulos, K.P.; Lensing, J.L.; Beeram, M.; Molina, J.R.; Rasco, D.W.; Arcos, R.R.; et al. A first-in-human phase i study of the oral p38 MAPK inhibitor, ralimetinib (LY2228820 Dimesylate), in patients with advanced cancer. Clin. Cancer Res. 2016, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; Heitz, F.; Buderath, P.; Powell, M.; Sehouli, J.; Lee, C.M.; Hamilton, A.; Fiorica, J.; Moore, K.N.; Teneriello, M.; et al. A randomized, double-blind, placebo-controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2020, 156, 23–31. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Dandapani, M.; Russell, F.M.; Grzes, K.M.; Atrih, A.; Foretz, M.; Viollet, B.; Lamont, D.J.; Cantrell, D.A.; Hardie, D.G. Phenformin, But Not Metformin, Delays Development of T Cell Acute Lymphoblastic Leukemia/Lymphoma via Cell-Autonomous AMPK Activation. Cell Rep. 2019, 27, 690–698.e4. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Antitumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Moshofsky, K.B.; Cho, H.J.; Wu, G.; Romine, K.A.; Newman, M.T.; Kosaka, Y.; McWeeney, S.K.; Lind, E.F. Acute myeloid leukemia–induced T-cell suppression can be reversed by inhibition of the MAPK pathway. Blood Adv. 2019, 3, 3038–3051. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Keam, B.; Ahn, Y.O.; Park, H.R.; Kim, M.; Kim, T.M.; Kim, D.W.; Heo, D.S. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating antitumor immunity in head and neck squamous cell carcinoma. Oncoimmunology 2019, 8, e1515057. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Guo, Z.; Kiniwa, Y.; Voo, K.S.; Peng, W.; Fu, T.; Wang, D.Y.; Li, Y.; Wang, H.Y.; Wang, R.F. Immunology: Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 2005, 309, 1380–1384. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Wang, H.Y.; Peng, W.; Kiniwa, Y.; Seo, K.H.; Wang, R.F. Tumor-Infiltrating γδ T Cells Suppress T and Dendritic Cell Function via Mechanisms Controlled by a Unique Toll-like Receptor Signaling Pathway. Immunity 2007, 27, 334–348. [Google Scholar] [CrossRef]
- Ferris, R.L.; Saba, N.F.; Gitlitz, B.J.; Haddad, R.; Sukari, A.; Neupane, P.; Morris, J.C.; Misiukiewicz, K.; Bauman, J.E.; Fenton, M.; et al. Effect of Adding Motolimod to Standard Combination Chemotherapy and Cetuximab Treatment of Patients With Squamous Cell Carcinoma of the Head and Neck. JAMA Oncol. 2018, 4, 1583. [Google Scholar] [CrossRef]
- Dietsch, G.N.; Randall, T.D.; Gottardo, R.; Northfelt, D.W.; Ramanathan, R.K.; Cohen, P.A.; Manjarrez, K.L.; Newkirk, M.; Bryan, J.K.; Hershberg, R.M. Late-Stage Cancer Patients Remain Highly Responsive to Immune Activation by the Selective TLR8 Agonist Motolimod (VTX-2337). Clin. Cancer Res. 2015, 21, 5445–5452. [Google Scholar] [CrossRef]
- Monk, B.J.; Brady, M.F.; Aghajanian, C.; Lankes, H.A.; Rizack, T.; Leach, J.; Fowler, J.M.; Higgins, R.; Hanjani, P.; Morgan, M.; et al. A phase 2, randomized, double-blind, placebo- controlled study of chemo-immunotherapy combination using motolimod with pegylated liposomal doxorubicin in recurrent or persistent ovarian cancer: A Gynecologic Oncology Group partners study. Ann. Oncol. 2017, 28, 996–1004. [Google Scholar] [CrossRef]
- Monk, B.J.; Facciabene, A.; Brady, W.E.; Aghajanian, C.A.; Fracasso, P.M.; Walker, J.L.; Lankes, H.A.; Manjarrez, K.L.; Danet-Desnoyers, G.H.; Bell-McGuinn, K.M.; et al. Integrative Development of a TLR8 Agonist for Ovarian Cancer Chemoimmunotherapy. Clin. Cancer Res. 2017, 23, 1955–1966. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Morishima, C.; Eaton, K.D.; Baik, C.S.; Goulart, B.H.; Anderson, L.N.; Manjarrez, K.L.; Dietsch, G.N.; Bryan, J.K.; Hershberg, R.M.; et al. Phase Ib Trial of the Toll-like Receptor 8 Agonist, Motolimod (VTX-2337), Combined with Cetuximab in Patients with Recurrent or Metastatic SCCHN. Clin. Cancer Res. 2017, 23, 2442–2450. [Google Scholar] [CrossRef]
- Shayan, G.; Kansy, B.A.; Gibson, S.P.; Srivastava, R.M.; Bryan, J.K.; Bauman, J.E.; Ohr, J.; Kim, S.; Duvvuri, U.; Clump, D.A.; et al. Phase Ib Study of Immune Biomarker Modulation with Neoadjuvant Cetuximab and TLR8 Stimulation in Head and Neck Cancer to Overcome Suppressive Myeloid Signals. Clin. Cancer Res. 2018, 24, 62–72. [Google Scholar] [CrossRef]
- Roex, G.; Feys, T.; Beguin, Y.; Kerre, T.; Poire, X.; Lewalle, P.; Vandenberghe, P.; Bron, D.; Anguille, S. Chimeric Antigen Receptor-T-Cell Therapy for B-Cell Hematological Malignancies: An Update of the Pivotal Clinical Trial Data. Pharmaceutics 2020, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reynies, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougouin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Zhang, H.; Alsaleh, G.; Feltham, J.; Sun, Y.; Napolitano, G.; Riffelmacher, T.; Charles, P.; Frau, L.; Hublitz, P.; Yu, Z.; et al. Polyamines Control eIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol. Cell 2019, 76, 110–125.e9. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Ageing and obesity similarly impair antibody responses. Clin. Exp. Immunol. 2017, 187, 64–70. [Google Scholar] [CrossRef]
- Prieto, L.I.; Baker, D.J. Cellular Senescence and the Immune System in Cancer. Gerontology 2019, 65, 505–512. [Google Scholar] [CrossRef]
- Kansara, M.; Leong, H.S.; Lin, D.M.; Popkiss, S.; Pang, P.; Garsed, D.W.; Walkley, C.R.; Cullinane, C.; Ellul, J.; Haynes, N.M.; et al. Immune response to RB1-regulated senescence limits radiation-induced osteosarcoma formation. J. Clin. Investig. 2013, 123, 5351–5360. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines (Basel) 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Kuan, E.L.; Ziegler, S.F. A tumor-myeloid cell axis, mediated via the cytokines IL-1alpha and TSLP, promotes the progression of breast cancer. Nat. Immunol. 2018, 19, 366–374. [Google Scholar] [CrossRef]
- Su, Y.; Xu, C.; Sun, Z.; Liang, Y.; Li, G.; Tong, T.; Chen, J. S100A13 promotes senescence-associated secretory phenotype and cellular senescence via modulation of non-classical secretion of IL-1alpha. Aging (Albany N. Y.) 2019, 11, 549–572. [Google Scholar] [CrossRef]
- Wiggins, K.A.; Parry, A.J.; Cassidy, L.D.; Humphry, M.; Webster, S.J.; Goodall, J.C.; Narita, M.; Clarke, M.C.H. IL-1alpha cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell 2019, 18, e12946. [Google Scholar] [CrossRef]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5. [Google Scholar] [CrossRef]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Raulet, D.H. Immunosurveillance of senescent cancer cells by natural killer cells. Oncoimmunology 2014, 3, e27616. [Google Scholar] [CrossRef] [PubMed]
- Ratushnyy, A.; Ezdakova, M.; Buravkova, L. Secretome of Senescent Adipose-Derived Mesenchymal Stem Cells Negatively Regulates Angiogenesis. Int. J. Mol. Sci. 2020, 21, 1802. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Pawlikowski, J.S.; Liu, Y.; Hawkins, O.E.; Davis, T.A.; Smith, J.; Weller, K.P.; Horton, L.W.; McClain, C.M.; Ayers, G.D.; et al. Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res. 2015, 75, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Fu, M.; Xin, H.B. Polarizing Macrophages In Vitro. Methods Mol. Biol. 2018, 1784, 119–126. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2020, 21, 4346. https://doi.org/10.3390/ijms21124346
Battram AM, Bachiller M, Martín-Antonio B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. International Journal of Molecular Sciences. 2020; 21(12):4346. https://doi.org/10.3390/ijms21124346
Chicago/Turabian StyleBattram, Anthony M., Mireia Bachiller, and Beatriz Martín-Antonio. 2020. "Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword" International Journal of Molecular Sciences 21, no. 12: 4346. https://doi.org/10.3390/ijms21124346
APA StyleBattram, A. M., Bachiller, M., & Martín-Antonio, B. (2020). Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. International Journal of Molecular Sciences, 21(12), 4346. https://doi.org/10.3390/ijms21124346