Hydrogen Sulfide Treatment Improves Post-Infarct Remodeling and Long-Term Cardiac Function in CSE Knockout and Wild-Type Mice

 ,
,  ,
, 
Abstract
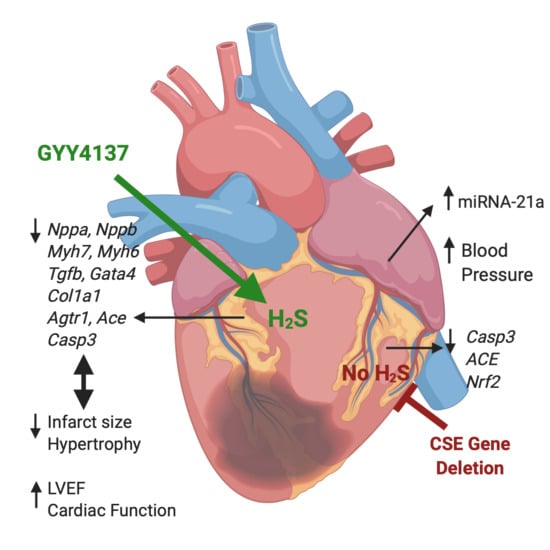
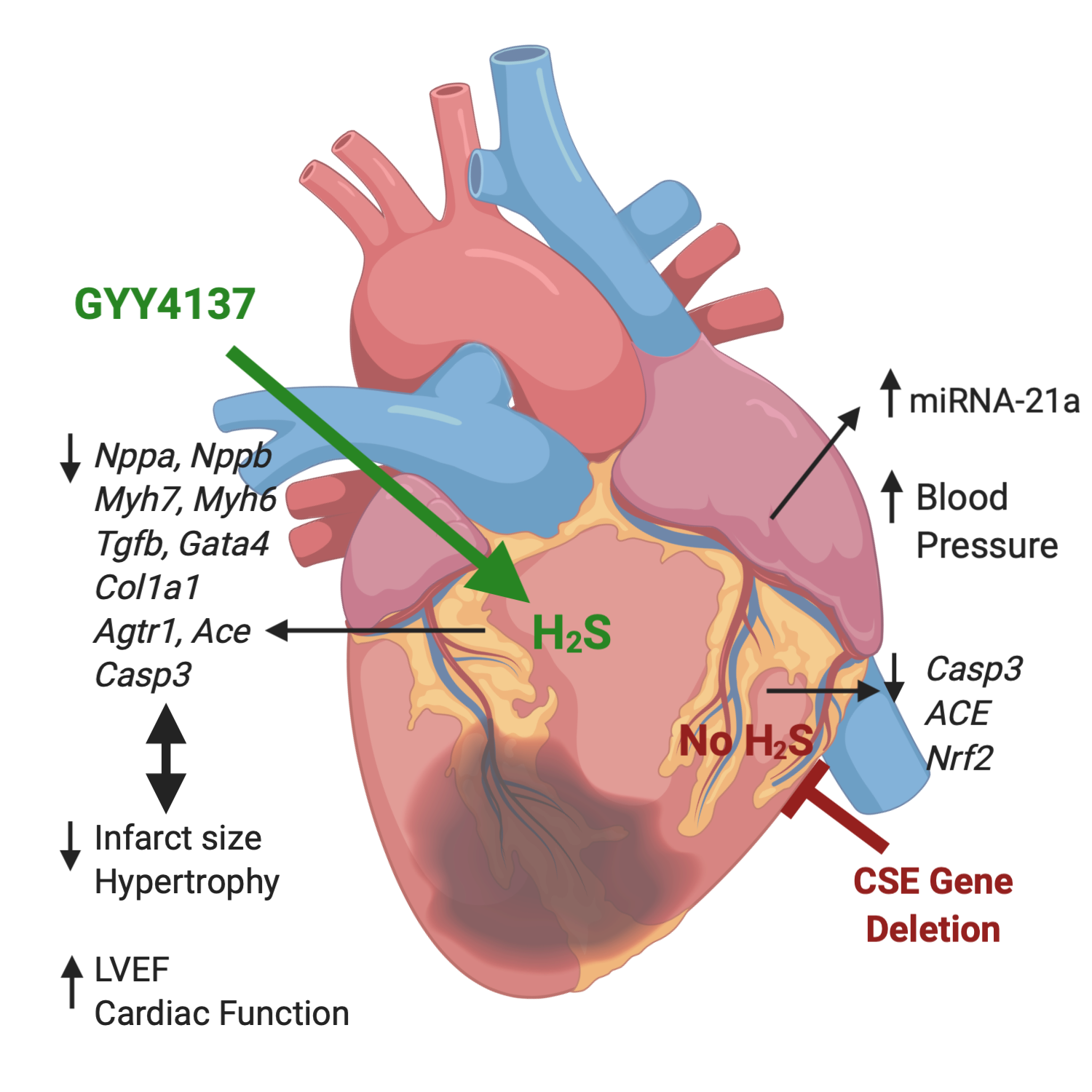
1. Introduction
2. Results
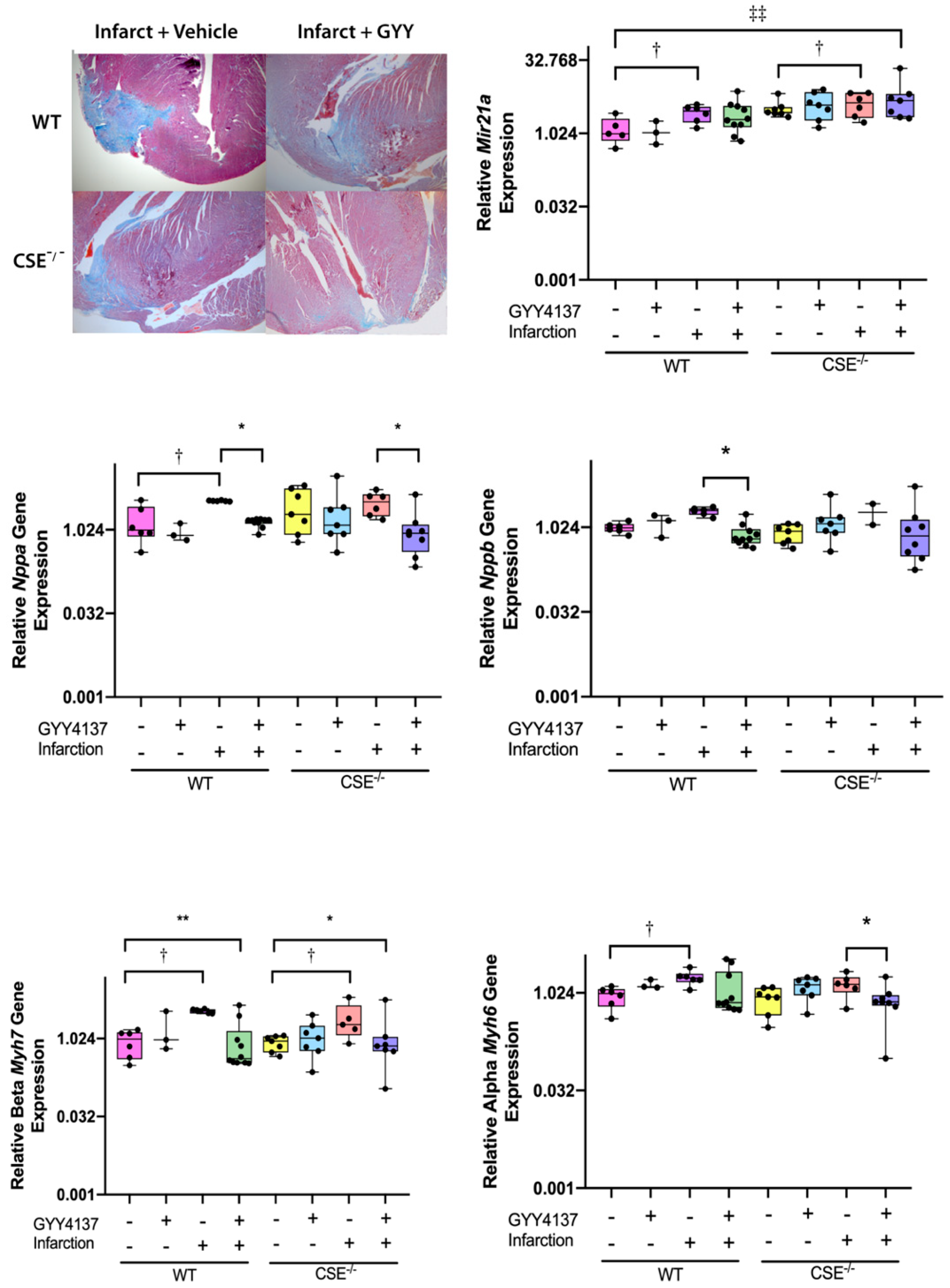
2.1. Infarct Size, Blood Pressure, Heart Weight/Body Weight, Cardiac Function Post-MI
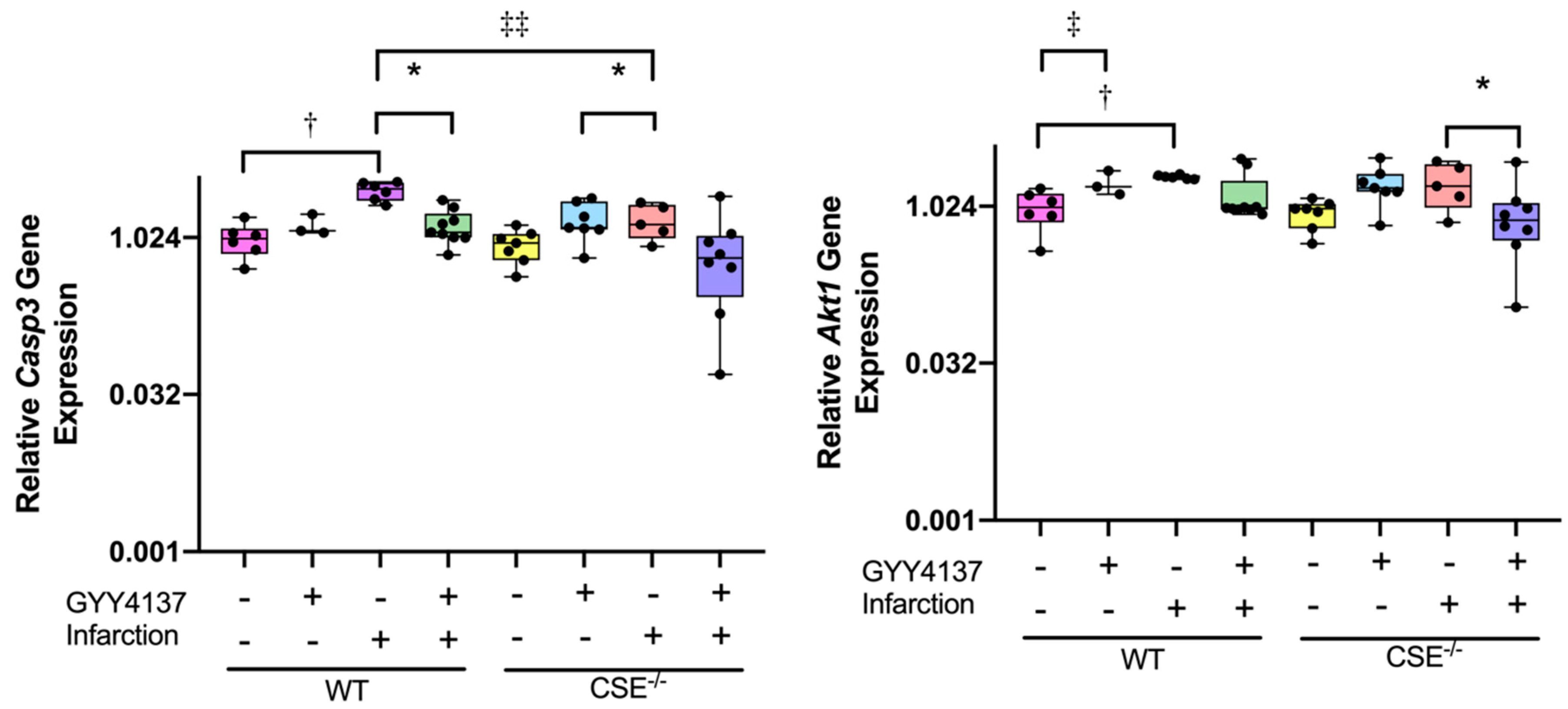
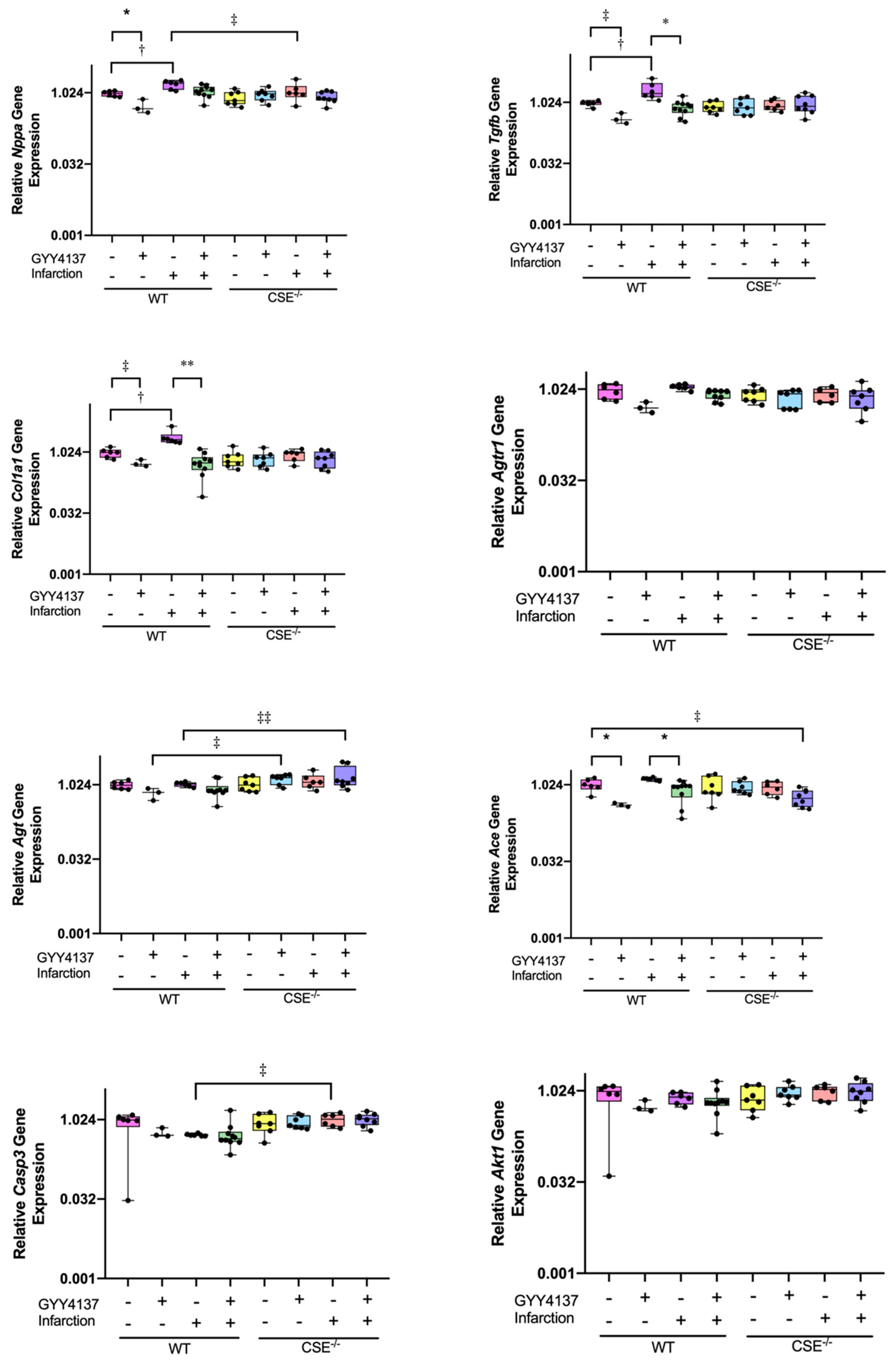
2.2. Expression of Atrial MiR21a and Ventricular Hypertrophy, Fibrosis, and Apoptosis Genes
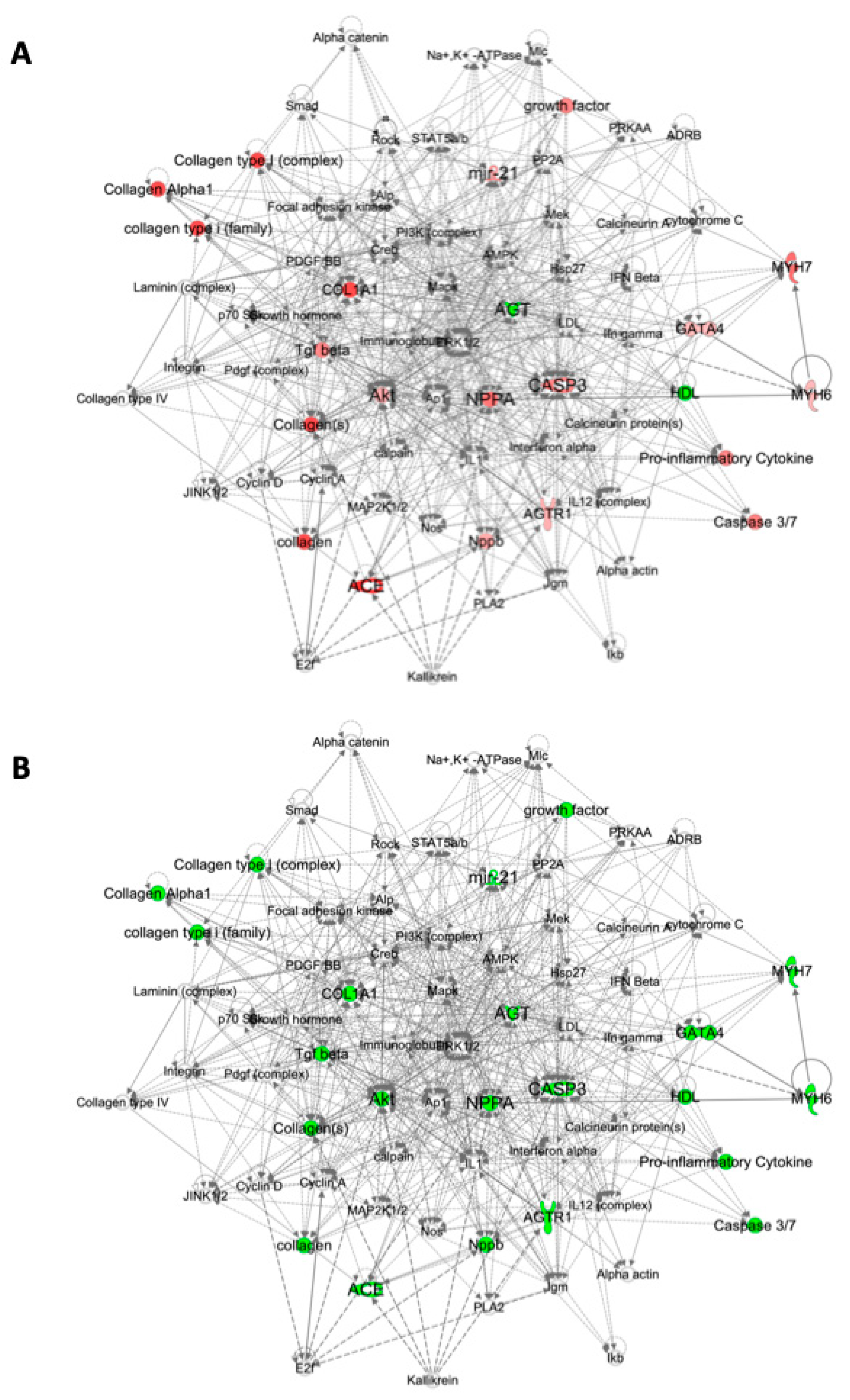
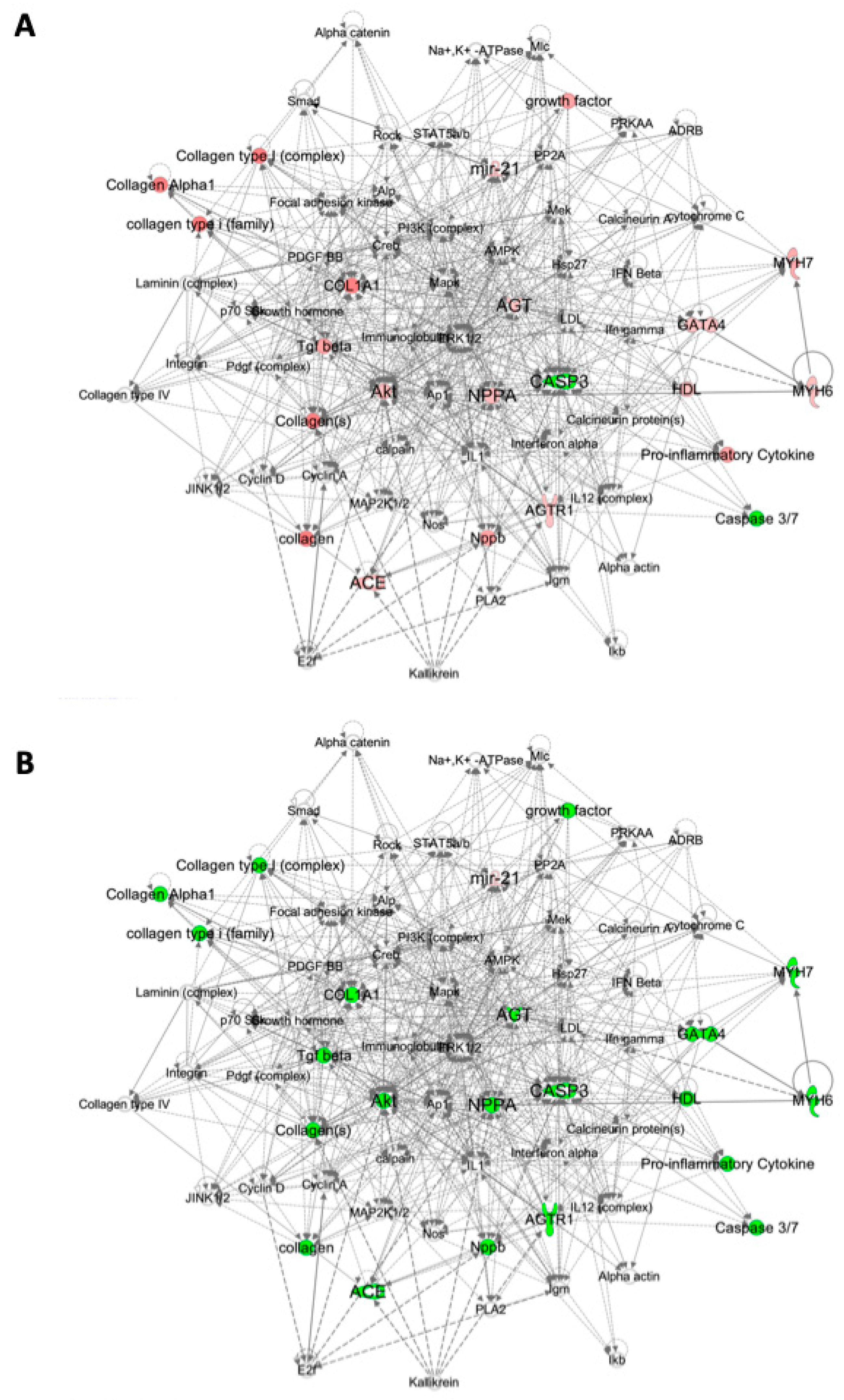
2.3. Pathway Analysis
2.4. Kidney Gene Expression
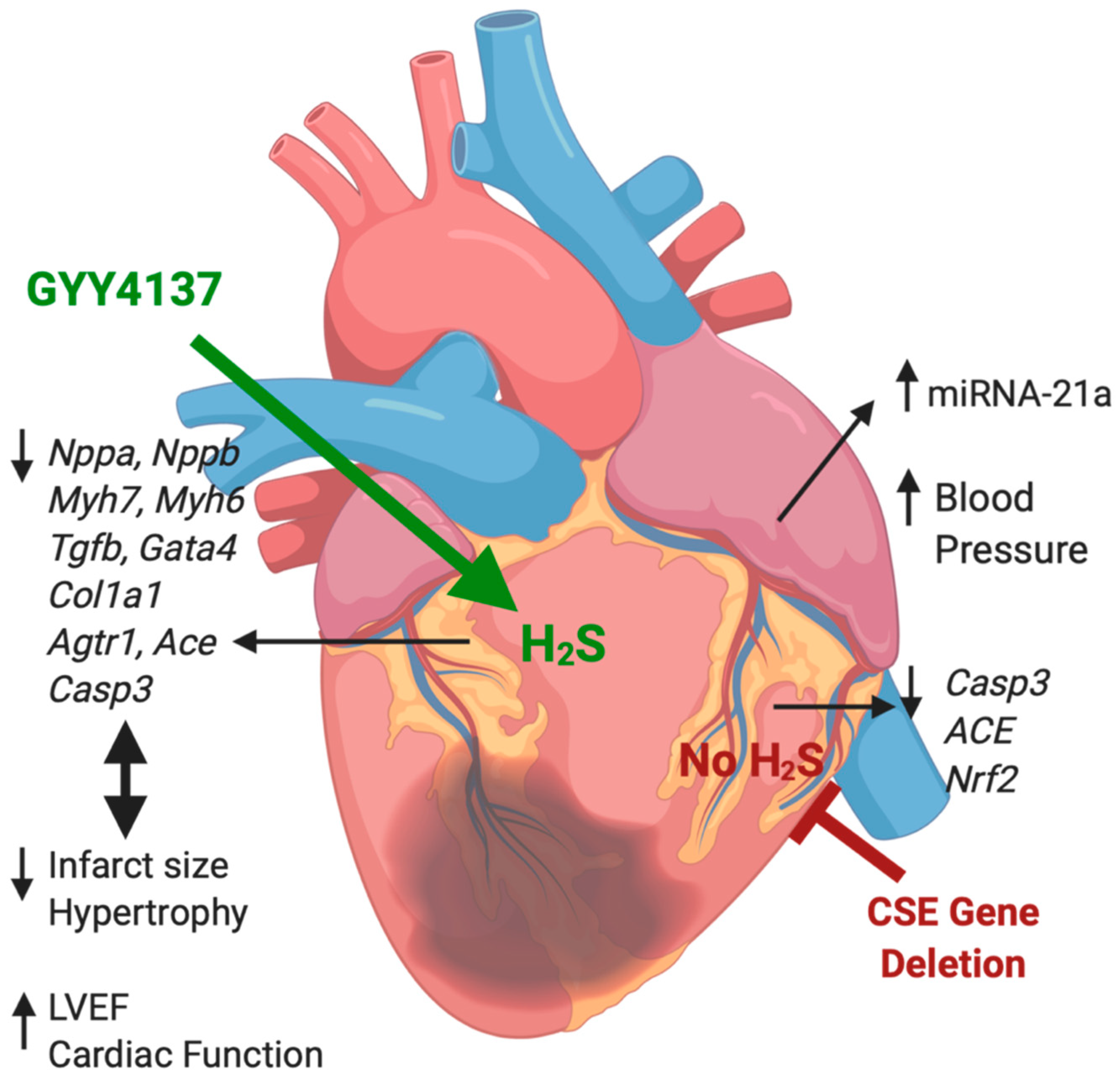
3. Discussion
4. Materials and Methods
4.1. Myocardial Infarction
4.2. Treatment Protocol
4.3. Echocardiography
4.4. Tissue Collection for Histology and RNA Isolation
4.5. Quantitative Real-Time PCR Analysis
4.6. Pathway Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | 2-dimensional |
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| Ace | angiotensin-converting enzyme gene |
| Agt | angiotensinogen gene |
| Agtr | angiotensin type 1 receptor gene |
| Akt1 | thymoma viral proto-oncogene 1gene |
| Bax/Bcl-2 | bcl-2-like protein 4/B-cell lymphoma 2 |
| KATP | ATP-sensitive K channels |
| Casp3 | caspase 3 gene |
| CBS | cystathionine γ-syntase |
| CSE | cystathionine γ-lyase |
| CSE−/− | cystathionine γ-lyase-knockout mice |
| Col1a1 | collagen 1 gene |
| ERK1/2 | extracellular regulated kinase 1/2 |
| FS | fractional shortening |
| Gata4 | GATA Binding Protein 4 gene |
| HPRT | hypoxanthine phosphoribosyltransferase 1 |
| hsa-miR-93-5p | human microRNA-93-5p |
| HSP | heat shock protein |
| H2S | hydrogen sulfide |
| HW/BW | heart weight/body weight ratio |
| IP | intraperitoneal |
| LVEF | left ventricular (LV) ejection fraction |
| MAP | mean arterial blood pressure |
| MI | myocardial infarction |
| Mir21a | microRNA-21a |
| mRNA | messenger ribonucleic acid |
| Myh7 | β-myosin heavy chain |
| Myh6 | α-myosin heavy chain |
| NaHS | sodium hydrosulfide |
| Na2S | sodium sulfide |
| NIH | United States National Institutes of Health |
| NO | nitric oxide |
| Nppa | atrial natriuretic peptide gene |
| Nppb | B-type natriuretic peptide gene |
| Nrf2 | nuclear factor, erythroid 2 Like 2 gene |
| p38 MAPK | p38 mitogen activated protein kinases |
| PI3K | phosphoinositide 3-kinases |
| PKC | protein kinase C |
| RNA | ribonucleic acid |
| rRNA | ribosomal ribonucleic acid |
| RNU1A1 | small nuclear RNA U1a1 |
| SNORD68 | small nucleolar RNA C/D box 68 |
| SHR | spontaneously hypertensive rats |
| STAT-3 | signal transducers and activators of transcription-3 |
| SC | subcutaneous |
| Tgfb | transforming growth factor β1 |
| WT | wild-type mice |
References
- Calvert, J.; Coetzee, W.; Lefer, D. Novel Insights into Hydrogen Sulfide–Mediated Cytoprotection. Antioxid. Redox Signal. 2010, 12, 1203–1207. [Google Scholar] [CrossRef]
- Li, Z.; Polhemus, D.; Lefer, D. Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ. Res. 2018, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Lilyanna, S.; Peh, M.; Liewa, O.; Wang, P.; Moore, P.; Richards, A.; Martinez, E. GYY4137 attenuates remodeling, preserves cardiac function and modulates the natriuretic peptide response to ischemia. J. Mol. Cell Cardiol. 2015, 87, 27–37. [Google Scholar] [CrossRef]
- Nicholson, C.; Calvert, J. Hydrogen sulfide and ischemia–reperfusion injury. Pharmacol. Res. 2010, 62, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.; Calvert, J.; Butler, J.; Lefer, D. The Cardioprotective Actions of Hydrogen Sulfide in Acute Myocardial Infarction and Heart Failure. Scientifica 2014, 2014, 8. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Z.; Ho, P.; Loke, Y.; Zhu, Y.; Huang, S.; Tan, C.; Whiteman, M.; Lu, J.; Moore, P. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J. Appl. Physiol. 2007, 102, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Geng, B.; Chang, L.; Pan, C.; Qi, Y.; Zhao, J.; Pang, Y.; Du, J.; Tang, C. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. BBRC 2004, 318, 756–763. [Google Scholar] [CrossRef]
- Givvimani, S.; Munjal, C.; Gargoum, R.; Sen, U.; Tyagi, N.; Vacek, J.; Tyagi, S. Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J. Appl. Physiol. 2011, 110, 1093–1100. [Google Scholar] [CrossRef]
- Polhemus, D.; Kondo, K.; Bhushan, S.; Bir, S.; Kevil, C.; Murohara, T.; Lefer, D.; Calvert, J. Hydrogen Sulfide Attenuates Cardiac Dysfunction After Heart Failure Via Induction of Angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, X.; Pan, T.-T.; Neo, K.; Lee, S.; Khin, E.; Moore, P.; Bian, J.-S. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflug. Arch. 2008, 455, 607–616. [Google Scholar] [CrossRef]
- Sivarajah, A.; Collino, M.; Yasin, M.; Benetti, E.; Gallicchio, M.; Mazzon, E.; Cuzzocrea, S.; Fantozzi, R.; Thiemermann, C. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 2009, 31, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Calvert, J.; Duranski, M.; Ramachandran, A.; Lefer, D. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H801–H806. [Google Scholar] [CrossRef] [PubMed]
- Nagpure, B.; Bian, J.-S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell Longev. 2016, 2006, 6904327. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Bhushan, S.; King, A.; Prabhu, S.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.; Gojon, G.; Sr Gojon, G.; et al. H2S Protects Against Pressure Overload–Induced Heart Failure via Upregulation of Endothelial Nitric Oxide Synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Das, A.; Mezzaroma, A.; Chau, V.; Marchetti, C.; Durrant, D.; Samidurai, A.; Van Tassell, B.; Yin, C.; Ockaili, R.; et al. Induction of MicroRNA-21 With Exogenous Hydrogen Sulfide Attenuates Myocardial Ischemic and Inflammatory Injury in Mice. Circ. Cardiovasc. Genet. 2014, 7, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Cardin, S.; Guasch, E.; Luo, X.; Naud, P.; Quang, K.; Shi, Y.; Tardif, J.-C.; Comtois, P.; Nattel, S. Role for MicroRNA-21 in Atrial Profibrillatory Fibrotic Remodeling Associated With Experimental Postinfarction Heart Failure. Circ. Arrythmia Electrophysiol. 2012, 5, 1027–1035. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signal. 2009, 11, 7013–7714. [Google Scholar] [CrossRef]
- Wang, R. The Gasotransmitter Role of Hydrogen Sulfide. Antioxid. Redox Signal. 2003, 5, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, A.; McDonald, M.; Thiemermann, C. The production of hydrogen sulfide limits myocardial ischemia and reperfusion injury and contributes to the cardioprotective effects of preconditioning with endotoxin, but not ischemia in the rat. Shock 2006, 26, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine γ-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Shen, X.; Whiteman, M.; Xin, H.; Shen, Y.; Xin, X.; Moore, P.; Zhu, Y.-Z. Hydrogen Sulfide Mitigates Myocardial Infarction via Promotion of Mitochondrial Biogenesis-Dependent M2 Polarization of Macrophages. Antioxid. Redox Signal. 2016, 25, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.; Calvert, J.; Morrison, J.; Doeller, J.; Kraus, D.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.-C.; Chang, C.-H.; Chen, Y.-C. Biomarkers for acute cardiorenal syndrome. Nephrol. Dial. Transplant. 2018, 23, 68–71. [Google Scholar] [CrossRef]
- Zhang, S.; Pan, C.; Zhou, F.; Yuan, Z.; Wang, H.; Cui, W.; Zhang, G. Hydrogen Sulfide as a Potential Therapeutic Target in Fibrosis. Oxid. Med. Cell Longev. 2015, 2015, 593407. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Whiteman, M.; Guan, Y.; Neo, K.; Cheng, Y.; Lee, S.; Zhao, Y.; Baskar, R.; Tan, C.-H.; Moore, P. Characterization of a Novel, Water-Soluble Hydrogen Sulfide–Releasing Molecule (GYY4137): New Insights Into the Biology of Hydrogen Sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine γ-Lyase-deficient Mice Require Dietary Cysteine to Protect against Acute Lethal Myopathy and Oxidative Injury. J. Biol. Chem. 2010, 285, 26358–26368. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Xin, X.; Xin, H.; Shen, X.; Zhu, Y.-Z. Hydrogen Sulfide Recruits Macrophage Migration by Integrin β1-Src-FAK/Pyk2-Rac Pathway in Myocardial Infarction. Sci. Rep. 2016, 6, 22363. [Google Scholar] [CrossRef]
- Ang, A.; Rivers-Auty, J.; Hegde, A.; Ishii, I.; Bhatia, M. The effect of CSE gene deletion in caerulein-induced acute pancreatitis in the mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G712–G721. [Google Scholar] [CrossRef] [PubMed]
- Adam, O.; Löhfelm, B.; Thum, T.; Gupta, S.; Puhl, S.-L.; Schäfers, H.-J.; Böhm, M.; Laufs, U. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res. Cardiol. 2012, 107, 278. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.; Jupiter, R.; Pankey, E.; Reddy, I.; Edward, J.; Swan, K.; Peak, T.; Mostany, R.; Kadowitz, P. Analysis of cardiovascular responses to the H S donors Na S and NaHS in the rat. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H605–H614. [Google Scholar] [CrossRef]
- Qipshidze, N.; Metreveli, N.; Mishra, P.; Lominadze, D.; Tyagi, S. Hydrogen Sulfide Mitigates Cardiac Remodeling During Myocardial Infarction via Improvement of Angiogenesis. Int. J. Biol. Sci. 2012, 8, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Zhu, J.; Xiao, Y.; Huang, Z.; Zhang, Y.; Tang, X.; Xie, L.; Chen, Y.; Shao, Y.; Ferro, A.; et al. Hydrogen Sulfide Donor GYY4137 Protects against Myocardial Fibrosis. Oxid. Med. Cell Longev. 2015, 2015, 691070. [Google Scholar] [CrossRef] [PubMed]
- Ellmers, L.; Scott, N.; Medicherla, S.; Pilbrow, A.; Bridgman, P.; Yandle, T.; Richards, A.; Protter, A.; Cameron, V. Transforming Growth Factor-β Blockade Down-Regulates the Renin-Angiotensin System and Modifies Cardiac Remodeling after Myocardial Infarction. Endocrinology 2008, 149, 5828–5834. [Google Scholar] [CrossRef]
- Ellmers, L.; Rademaker, M.; Charles, C.; Yandle, T.; Richards, A. (Pro)renin Receptor Blockade Ameliorates Cardiac Injury and Remodeling and Improves Function After Myocardial Infarction. J. Card. Failure 2016, 22, 64–71. [Google Scholar] [CrossRef]
- Ellmers, L.; Scott, N.; Cameron, V.; Richards, A.; Rademaker, M. Chronic Urocortin 2 Administration Improves Cardiac Function and Ameliorates Cardiac Remodeling After Experimental Myocardial Infarction. J. Cardiovas. Pharmacol. 2015, 65, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT Sham Vehicle | WT Sham GYY4137 | WT Infarct Vehicle | WT Infarct GYY4137 | CSE KO Sham Vehicle | CSE KO Sham GYY4137 | CSE KO Infarct Vehicle | CSE KO Infarct GYY4137 | |
|---|---|---|---|---|---|---|---|---|
| MAP mmHg | 73.5 ± 3.74 | 68.8 ± 4.8 | 66.3 ± 3.4 | 67.5 ± 2.6 | 94.8 ± 3.2 ‡‡ | 101.4 ± 3.2 ‡‡ | 86.4 ± 3.4 ‡ | 76.12 ± 3.4 *,‡ |
| Infarct mm2 | - | - | 5.80 ± 0.71 | 1.50 ± 0.58 * | - | - | 7.33 ± 2.03 | 1.75 ± 0.37 * |
| HW/BW | 5.39 ± 0.17 | 5.73 ± 0.23 | 6.81 ± 0.17 †† | 5.79 ± 0.13 ** | 5.06 ± 0.15 | 5.71 ± 0.15 | 6.76 ± 0.17 †† | 5.91 ± 0.14 ** |
| LVEF% | 74.7 ± 1.28 | 70.1 ± 1.80 | 46.8 ± 1.28 †† | 64.2 ± 0.99 ** | 72.3 ± 1.18 | 68.9 ± 1.18 | 40.8 ± 1.28 ††,‡ | 62.7 ± 1.18 ** |
| FS% | 37.5 ± 0.89 | 32.0 ± 1.26 | 17.67 ± 0.89 †† | 29.0 ± 0.69 ** | 35.57 ± 0.82 | 39.57 ± 0.82 | 15.83 ± 0.89 †† | 30.13 ± 0.77 ** |
| LV mass g | 0.71 ± 0.01 | 0.68 ± 0.01 | 0.74 ± 0.03 | 0.69 ± 0.03 | 0.69 ± 0.01 | 0.73 ± 0.02 | 0.69 ± 0.02 | 0.69 ± 0.01 |
| LVPWs mm | 1.36 ± 0.08 | 1.18 ± 0.07 | 0.99 ± 0.14 † | 1.28 ± 0.14 | 1.06 ± 0.80 ‡ | 1.49 ± 0.08 | 0.97 ± 0.09 †† | 1.08 ± 0.07 |
| IVS% | 0.96 ± 0.04 | 1.01 ± 0.04 | 1.31 ± 0.08 †† | 0.80 ± 0.08 ** | 0.96 ± 0.04 | 0.81 ± 0.04 | 0.87 ± 0.05 ‡‡ | 0.84 ± 0.04 |
| EVS mL | 0.05 ± 0.02 | 0.05 ± 0.02 | 0.05 ± 0.03 | 0.06 ± 0.03 | 0.03 ± 0.02 | 0.07 ± 0.02 | 0.04 ± 0.02 | 0.05 ± 0.02 |
| EDV mL | 0.12 ± 0.08 | 0.07 ± 0.07 | 0.13 ± 0.15 | 0.13 ± 0.15 | 0.22 ± 0.08 | 0.19 ± 0.08 | 0.26 ± 0.09 | 0.20 ± 0.07 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellmers, L.J.; Templeton, E.M.; Pilbrow, A.P.; Frampton, C.; Ishii, I.; Moore, P.K.; Bhatia, M.; Richards, A.M.; Cameron, V.A. Hydrogen Sulfide Treatment Improves Post-Infarct Remodeling and Long-Term Cardiac Function in CSE Knockout and Wild-Type Mice. Int. J. Mol. Sci. 2020, 21, 4284. https://doi.org/10.3390/ijms21124284
Ellmers LJ, Templeton EM, Pilbrow AP, Frampton C, Ishii I, Moore PK, Bhatia M, Richards AM, Cameron VA. Hydrogen Sulfide Treatment Improves Post-Infarct Remodeling and Long-Term Cardiac Function in CSE Knockout and Wild-Type Mice. International Journal of Molecular Sciences. 2020; 21(12):4284. https://doi.org/10.3390/ijms21124284
Chicago/Turabian StyleEllmers, Leigh J., Evelyn M. Templeton, Anna P. Pilbrow, Chris Frampton, Isao Ishii, Philip K. Moore, Madhav Bhatia, A. Mark Richards, and Vicky A. Cameron. 2020. "Hydrogen Sulfide Treatment Improves Post-Infarct Remodeling and Long-Term Cardiac Function in CSE Knockout and Wild-Type Mice" International Journal of Molecular Sciences 21, no. 12: 4284. https://doi.org/10.3390/ijms21124284
APA StyleEllmers, L. J., Templeton, E. M., Pilbrow, A. P., Frampton, C., Ishii, I., Moore, P. K., Bhatia, M., Richards, A. M., & Cameron, V. A. (2020). Hydrogen Sulfide Treatment Improves Post-Infarct Remodeling and Long-Term Cardiac Function in CSE Knockout and Wild-Type Mice. International Journal of Molecular Sciences, 21(12), 4284. https://doi.org/10.3390/ijms21124284