EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration




{kind=link}
{kind=link}
Abstract
1. Introduction
2. Definitions of EMT and EndMT
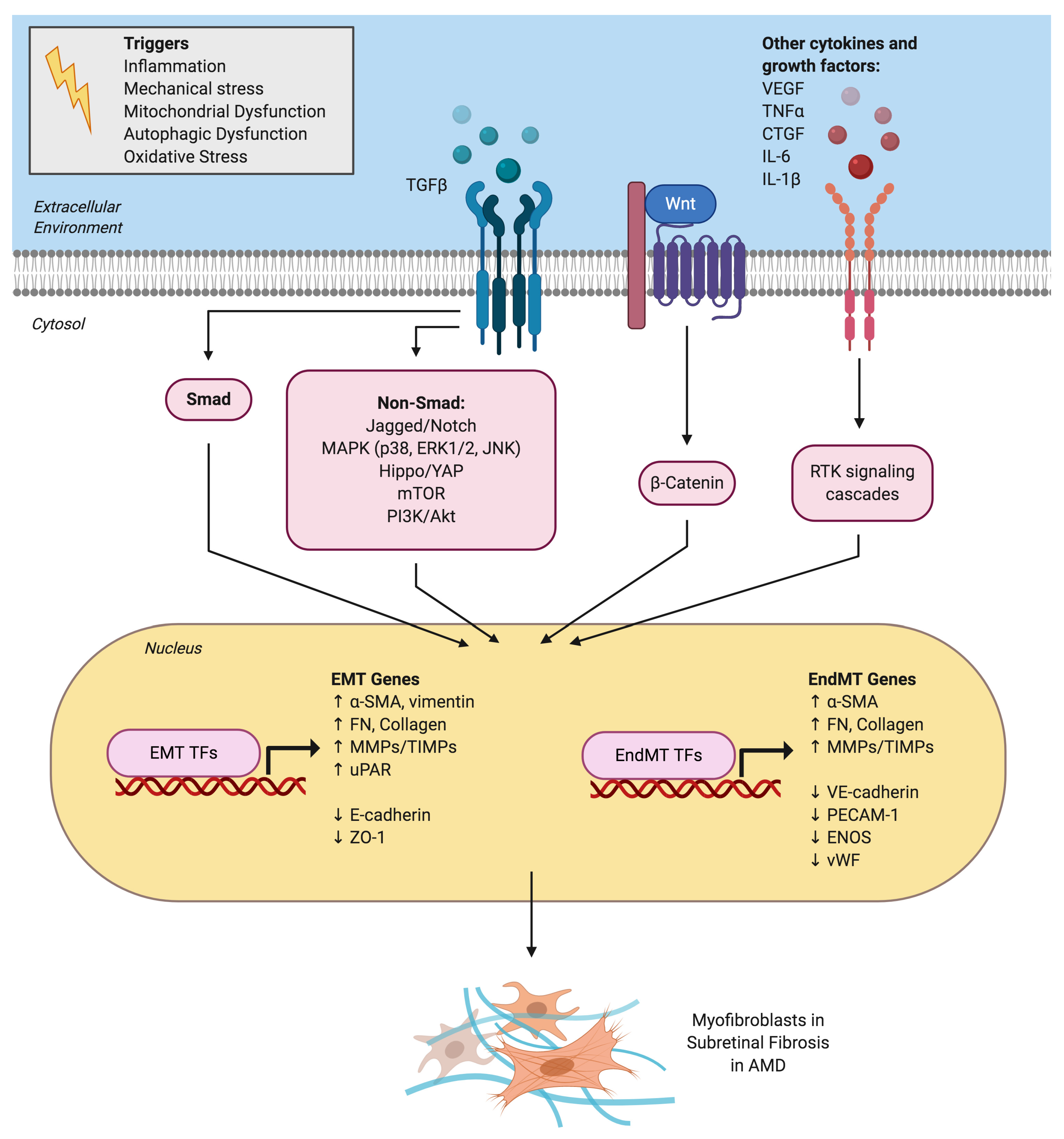
3. TGFβ as the Master Regulator of both EMT and EndMT
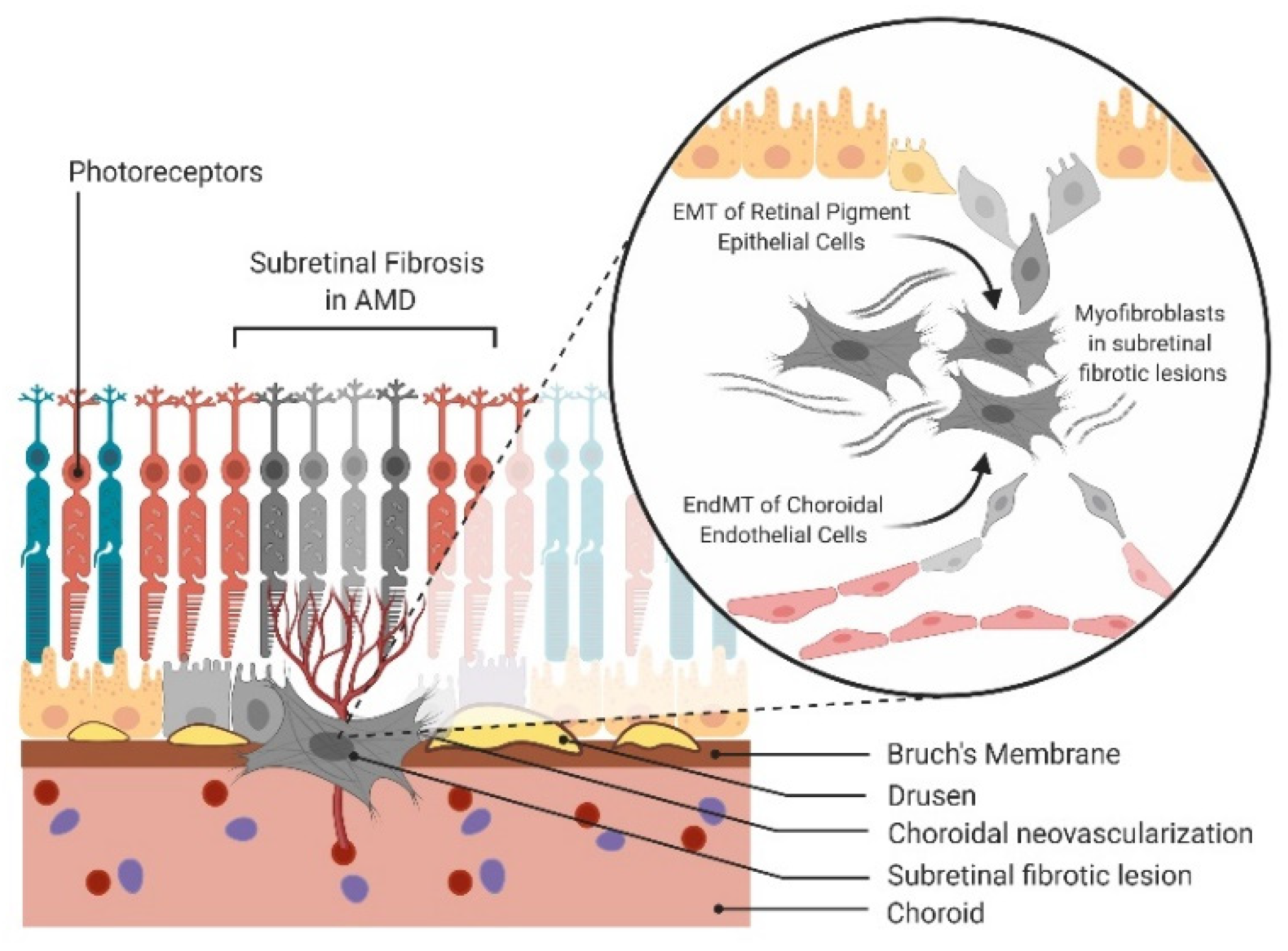
4. Evidence and Molecular Drivers of EMT/EndMT in AMD
4.1. Role of Cytokine-Mediated Signaling Pathways
4.2. Role of Inflammation in EMT/EndMT During AMD
4.3. Metabolic Dysfunction and Autophagy in AMD
5. Role of the Extracellular Matrix in AMD-Associated EMT/EndMT
5.1. ECM Remodelling During AMD Progression
5.2. TGFβ and ECM Changes in AMD
5.3. uPA and ECM Changes in AMD
5.4. Mechanotransduction in AMD
6. Therapeutic Considerations: Modulating Microenvironmental Signals
6.1. Synergistic Interaction between RPE and CECs
6.2. Reversal of EMT and EndMT as a Potential Therapeutic Approach for AMD
6.3. EMT and EndMT Inhibition
7. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMD | age-related macular degeneration |
| AMPK | adenosine monophosphate-activated protein kinase |
| α-SMA | alpha-smooth muscle actin |
| CEC | choroidal endothelial cell |
| COL1A1 | collagen type I alpha 1 chain |
| CNV | choroidal neovascularization |
| CTGF | connective tissue growth factor |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EMT | epithelial–mesenchymal transition |
| EndMT | endothelial–mesenchymal transition |
| ENOS | endothelial nitric oxide synthase |
| ERK | extracellular-signal-regulated kinases |
| ERM | epiretinal membrane |
| FAK | focal adhesion kinase |
| FDG-PET | fluorodeoxyglucose- positron emission tomography |
| FGF | fibroblast growth factor |
| FN | fibronectin |
| GA | geographic atrophy |
| GFP | green fluorescent protein |
| HGF | hepatocyte growth factor |
| HUVECs | human umbilical vein endothelial cells |
| ICAM | intercellular adhesion molecule |
| IGF | insulin-like growth factor |
| IL | interleukin |
| LAP | latency associated peptide |
| LTBP | latent TGFβ-binding protein |
| MAPK | mitogen-activated protein kinase |
| MMPs | matrix metalloproteinase |
| MMT | macrophage-myofibroblast transition |
| mtHsp70 | mitochondrial heat shock protein |
| mTOR | mammalian target of rapamycin |
| nAMD | neovascular age-related macular degeneration |
| NRLP3 | NLR Family Pyrin Domain Containing 3 |
| PAI-1 | plasminogen activator inhibitor type-1 |
| PECAM-1 | platelet-endothelial cell adhesion molecule-1 |
| PET-MRI | positron emission tomography-magnetic resonance imaging |
| PDGF | platelet-derived growth factor |
| PDR | proliferative diabetic retinopathy |
| PGC1α | proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | phosphatidylinositol-3 kinase |
| PVR | proliferative vitreoretinopathy |
| OIR | oxygen-induced retinopathy |
| OxLDL | oxidized low density lipoprotein |
| Rho GTPase | ras-homologous GTPase |
| ROP | retinopathy of prematurity |
| ROS | reactive oxygen species |
| RPE | retinal pigment epithelium |
| RTK | receptor tyrosine kinase |
| RUNX1 | runt-related transcription factor 1 |
| siRNA | small interfering RNA |
| S1P | sphingosine-1-phosphate |
| Taz | transcriptional co-activator with PDZ-binding motif |
| TCF/LEF | t-cell factor/lymphoid enhancer-binding factor |
| Tead | transcriptional enhanced associate domains |
| TFs | transcription factors |
| TGFβ | transforming growth factor-β |
| TGFBR1 | transforming growth factor-β receptor 1 |
| TIE | tyrosine kinase with immunoglobulin-like and EGF-like domains 1 |
| TIMPs | tissue inhibitors of matrix metalloproteinase |
| TNF-α | tumor necrosis factor-α |
| TRPV4 | transient receptor potential vanilloid 4 |
| TSP1 | thrombospondin 1 |
| uPA | urokinase-type plasminogen activator |
| uPAR | urokinase-type plasminogen activator receptor |
| VCAM | vascular cell adhesion molecule |
| VE-Cadherin | vascular endothelial-cadherin |
| VEGF | vascular endothelial growth factor |
| vWF | von Willebrand Factor |
| Wnt | Wingless/Integrated |
| YAP | Yes-associated protein |
| Zeb | zinc finger E-box-binding homeobox |
| ZO-1 | zonula occludens-1 |
References
- Ishikawa, K.; Kannan, R.; Hinton, D.R. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp. Eye Res. 2016, 142, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Radeke, M.J.; Radeke, C.M.; Shih, Y.H.; Hu, J.; Bok, D.; Johnson, L.V.; Coffey, P.J. Restoration of mesenchymal retinal pigmented epithelial cells by TGFbeta pathway inhibitors: Implications for age-related macular degeneration. Genome Med. 2015, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; Chang, T.F.; Li, M.H.; Sun, L.J.; Yan, X.C.; Yang, Z.Y.; Liu, Y.; Xu, W.Q.; Lv, Y.; Su, J.B.; et al. SNAI1, an endothelial-mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis 2018, 21, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Siemerink, M.J.; Augustin, A.J.; Schlingemann, R.O. Mechanisms of ocular angiogenesis and its molecular mediators. Dev. Ophthalmol. 2010, 46, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, S.; Kaplan, H.J. Role of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Exp. Eye Res. 2016, 142, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Boles, N.C.; Fernandes, M.; Swigut, T.; Srinivasan, R.; Schiff, L.; Rada-Iglesias, A.; Wang, Q.; Saini, J.S.; Kiehl, T.; Stern, J.H.; et al. Epigenomic and Transcriptomic Changes During Human RPE EMT in a Stem Cell Model of Epiretinal Membrane Pathogenesis and Prevention by Nicotinamide. Stem Cell Rep. 2020, 14, 631–647. [Google Scholar] [CrossRef]
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; De Hertogh, G.; van den Eynde, K.; Alam, K.; Van Raemdonck, K.; Opdenakker, G.; Van Damme, J.; Geboes, K.; Struyf, S. Myofibroblasts in proliferative diabetic retinopathy can originate from infiltrating fibrocytes and through endothelial-to-mesenchymal transition (EndoMT). Exp. Eye Res. 2015, 132, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Saito, A. EMT and EndMT: Regulated in similar ways? J. Biochem. 2013, 153, 493–495. [Google Scholar] [CrossRef]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed. Res. Int. 2014, 2014, 756078. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Welch-Reardon, K.M.; Wu, N.; Hughes, C.C. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Kim, J.H.; Bi, D.; Mitchel, J.A.; Qazvini, N.T.; Tantisira, K.; Park, C.Y.; McGill, M.; Kim, S.H.; Gweon, B.; et al. Unjamming and cell shape in the asthmatic airway epithelium. Nat. Mater. 2015, 14, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Atia, L.; Mitchel, J.A.; Fredberg, J.J.; Butler, J.P. Collective migration and cell jamming in asthma, cancer and development. J. Cell Sci. 2016, 129, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.A.; Echeverría, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Saika, S. TGFbeta pathobiology in the eye. Lab. Investig. 2006, 86, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Ota, M.; Rifkin, D.B. Matrix control of transforming growth factor-beta function. J. Biochem. 2012, 152, 321–329. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed]
- Connor, T.B., Jr.; Roberts, A.B.; Sporn, M.B.; Danielpour, D.; Dart, L.L.; Michels, R.G.; de Bustros, S.; Enger, C.; Kato, H.; Lansing, M.; et al. Correlation of fibrosis and transforming growth factor-beta type 2 levels in the eye. J. Clin Investig. 1989, 83, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.M.; Orlandini, M.; Galvagni, F. The Controversial Role of TGF-β in Neovascular Age-Related Macular Degeneration Pathogenesis. Int. J. Mol. Sci. 2018, 19, 3363. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.M.; Neri, G.; Caldi, E.; Fusco, F.; Bacci, T.; Tarantello, A.; Nuti, E.; Marigliani, D.; Baiocchi, S.; Traversi, C.; et al. TGF-beta concentrations and activity are down-regulated in the aqueous humor of patients with neovascular age-related macular degeneration. Sci. Rep. 2018, 8, 8053. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Saika, S.; Liu, C.Y.; Azhar, M.; Sanford, L.P.; Doetschman, T.; Gendron, R.L.; Kao, C.W.; Kao, W.W. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev. Biol. 2001, 240, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Pfeffer, B.A.; Flanders, K.C.; Guérin, C.J.; Danielpour, D.; Anderson, D.H. Transforming growth factor beta 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp. Eye Res. 1994, 59, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Tanihara, H.; Yoshida, M.; Matsumoto, M.; Yoshimura, N. Identification of transforming growth factor-beta expressed in cultured human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1993, 34, 413–419. [Google Scholar]
- Kvanta, A. Expression and secretion of transforming growth factor-beta in transformed and nontransformed retinal pigment epithelial cells. Ophthalmic Res. 1994, 26, 361–367. [Google Scholar] [CrossRef]
- Hirsch, L.; Nazari, H.; Sreekumar, P.G.; Kannan, R.; Dustin, L.; Zhu, D.; Barron, E.; Hinton, D.R. TGF-beta2 secretion from RPE decreases with polarization and becomes apically oriented. Cytokine 2015, 71, 394–396. [Google Scholar] [CrossRef]
- Mochizuki, M.; Sugita, S.; Kamoi, K. Immunological homeostasis of the eye. Prog. Retin. Eye Res. 2013, 33, 10–27. [Google Scholar] [CrossRef]
- Grossniklaus, H.E.; Green, W.R. Histopathologic and ultrastructural findings of surgically excised choroidal neovascularization. Submacular Surgery Trials Research Group. Arch. Ophthalmol. 1998, 116, 745–749. [Google Scholar] [CrossRef]
- Lopez, P.F.; Sippy, B.D.; Lambert, H.M.; Thach, A.B.; Hinton, D.R. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1996, 37, 855–868. [Google Scholar]
- Grossniklaus, H.E.; Hutchinson, A.K.; Capone, A., Jr.; Woolfson, J.; Lambert, H.M. Clinicopathologic features of surgically excised choroidal neovascular membranes. Ophthalmology 1994, 101, 1099–1111. [Google Scholar] [CrossRef]
- Sarks, J.P.; Sarks, S.H.; Killingsworth, M.C. Evolution of geographic atrophy of the retinal pigment epithelium. Eye 1988, 2, 552–577. [Google Scholar] [CrossRef]
- Guidry, C.; Medeiros, N.E.; Curcio, C.A. Phenotypic variation of retinal pigment epithelium in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2002, 43, 267–273. [Google Scholar]
- Zanzottera, E.C.; Messinger, J.D.; Ach, T.; Smith, R.T.; Curcio, C.A. Subducted and melanotic cells in advanced age-related macular degeneration are derived from retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3269–3278. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Niu, G.; Huber, P.; Carter, W.B. Tumor-induced upregulation of Twist, Snail, and Slug represses the activity of the human VE-cadherin promoter. Arch. Biochem. Biophys. 2009, 482, 77–82. [Google Scholar] [CrossRef]
- Ghosh, S.; Shang, P.; Terasaki, H.; Stepicheva, N.; Hose, S.; Yazdankhah, M.; Weiss, J.; Sakamoto, T.; Bhutto, I.A.; Xia, S.; et al. A Role for betaA3/A1-Crystallin in Type 2 EMT of RPE Cells Occurring in Dry Age-Related Macular Degeneration. Investig. Ophthalmol Vis. Sci. 2018, 59, AMD104–AMD113. [Google Scholar] [CrossRef]
- Hirasawa, M.; Noda, K.; Noda, S.; Suzuki, M.; Ozawa, Y.; Shinoda, K.; Inoue, M.; Ogawa, Y.; Tsubota, K.; Ishida, S. Transcriptional factors associated with epithelial-mesenchymal transition in choroidal neovascularization. Mol. Vis. 2011, 17, 1222–1230. [Google Scholar]
- Feng, Z.; Li, R.; Shi, H.; Bi, W.; Hou, W.; Zhang, X. Combined silencing of TGF-beta2 and Snail genes inhibit epithelial-mesenchymal transition of retinal pigment epithelial cells under hypoxia. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 875–884. [Google Scholar] [CrossRef]
- Saika, S.; Kono-Saika, S.; Tanaka, T.; Yamanaka, O.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Yoo, J.; Flanders, K.C.; et al. Smad3 is required for dedifferentiation of retinal pigment epithelium following retinal detachment in mice. Lab. Investig. 2004, 84, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Niessen, C.M. Tight junctions/adherens junctions: Basic structure and function. J. Investig. Dermatol 2007, 127, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G. Endothelial cell-to-cell junctions: Adhesion and signaling in physiology and pathology. Cold Spring Harb. Perspect. Med. 2012, 2, a006528. [Google Scholar] [CrossRef]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef]
- Liebner, S.; Cattelino, A.; Gallini, R.; Rudini, N.; Iurlaro, M.; Piccolo, S.; Dejana, E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004, 166, 359–367. [Google Scholar] [CrossRef]
- Mylavarapu, S.; Kumar, H.; Kumari, S.; Sravanthi, L.S.; Jain, M.; Basu, A.; Biswas, M.; Mylavarapu, S.V.S.; Das, A.; Roy, M. Activation of Epithelial-Mesenchymal Transition and Altered beta-Catenin Signaling in a Novel Indian Colorectal Carcinoma Cell Line. Front. Oncol. 2019, 9, 54. [Google Scholar] [CrossRef]
- Pratt, C.H.; Vadigepalli, R.; Chakravarthula, P.; Gonye, G.E.; Philp, N.J.; Grunwald, G.B. Transcriptional regulatory network analysis during epithelial-mesenchymal transformation of retinal pigment epithelium. Mol. Vis. 2008, 14, 1414–1428. [Google Scholar]
- Chen, H.C.; Zhu, Y.T.; Chen, S.Y.; Tseng, S.C. Wnt signaling induces epithelial-mesenchymal transition with proliferation in ARPE-19 cells upon loss of contact inhibition. Lab. Investig. 2012, 92, 676–687. [Google Scholar] [CrossRef]
- Umazume, K.; Tsukahara, R.; Liu, L.; Fernandez de Castro, J.P.; McDonald, K.; Kaplan, H.J.; Tamiya, S. Role of retinal pigment epithelial cell beta-catenin signaling in experimental proliferative vitreoretinopathy. Am. J. Pathol. 2014, 184, 1419–1428. [Google Scholar] [CrossRef]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef]
- Lilien, J.; Balsamo, J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr. Opin. Cell Biol. 2005, 17, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Umazume, K.; Liu, L.; Scott, P.A.; de Castro, J.P.; McDonald, K.; Kaplan, H.J.; Tamiya, S. Inhibition of PVR with a tyrosine kinase inhibitor, dasatinib, in the swine. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xin, Y.; Ye, F.; Wang, W.; Lu, Q.; Kaplan, H.J.; Dean, D.C. Taz-tead1 links cell-cell contact to zeb1 expression, proliferation, and dedifferentiation in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3372–3378. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiao, W.; Liu, X.; Zeng, M.; Luo, L.; Wu, M.; Ye, S.; Liu, Y. Blockade of Jagged/Notch pathway abrogates transforming growth factor beta2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells. Curr. Mol. Med. 2014, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiao, W.; Wang, W.; Luo, L.; Ye, S.; Liu, Y. The complex interplay between ERK1/2, TGFβ/Smad, and Jagged/Notch signaling pathways in the regulation of epithelial-mesenchymal transition in retinal pigment epithelium cells. PLoS ONE 2014, 9, e96365. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, K.; Nakatsuka, K.; Matsuo, N.; Yoshioka, H. p38 MAPK mediates the expression of type I collagen induced by TGF-beta 2 in human retinal pigment epithelial cells ARPE-19. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Schiff, L.; Boles, N.C.; Fernandes, M.; Nachmani, B.; Gentile, R.; Blenkinsop, T.A. P38 inhibition reverses TGFbeta1 and TNFalpha-induced contraction in a model of proliferative vitreoretinopathy. Commun. Biol. 2019, 2, 162. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef]
- Zarranz-Ventura, J.; Fernandez-Robredo, P.; Recalde, S.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-beta inhibition reduces progression of early choroidal neovascularization lesions in rats: P17 and P144 peptides. PLoS ONE 2013, 8, e65434. [Google Scholar] [CrossRef] [PubMed]
- Recalde, S.; Zarranz-Ventura, J.; Fernandez-Robredo, P.; Garcia-Gomez, P.J.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-beta inhibition decreases diode laser-induced choroidal neovascularization development in rats: P17 and P144 peptides. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7090–7097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Z.L. Transforming growth factor-beta neutralizing antibodies inhibit subretinal fibrosis in a mouse model. Int. J. Ophthalmol. 2012, 5, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liang, S.; Yu, W.; Zhao, M.; Huang, L.; Zhao, M.; Li, X. Semaphorin 3A blocks the formation of pathologic choroidal neovascularization induced by transforming growth factor beta. Mol. Vis. 2014, 20, 1258–1270. [Google Scholar]
- Lebrun, J.J. The Dual Role of TGFbeta in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef]
- Klaassen, I.; van Geest, R.J.; Kuiper, E.J.; van Noorden, C.J.; Schlingemann, R.O. The role of CTGF in diabetic retinopathy. Exp. Eye Res. 2015, 133, 37–48. [Google Scholar] [CrossRef]
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.; Goldschmeding, R.; Schlingemann, R.O. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS ONE 2008, 3, e2675. [Google Scholar] [CrossRef]
- Cicha, I.; Goppelt-Struebe, M. Connective tissue growth factor: Context-dependent functions and mechanisms of regulation. Biofactors 2009, 35, 200–208. [Google Scholar] [CrossRef]
- Inoki, I.; Shiomi, T.; Hashimoto, G.; Enomoto, H.; Nakamura, H.; Makino, K.; Ikeda, E.; Takata, S.; Kobayashi, K.; Okada, Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002, 16, 219–221. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, Y.; Zeng, Q.; Han, Q.; Zhang, L.; Liu, M.; Li, X. Intravitreal injection of ranibizumab and CTGF shRNA improves retinal gene expression and microvessel ultrastructure in a rodent model of diabetes. Int. J. Mol. Sci. 2014, 15, 1606–1624. [Google Scholar] [CrossRef]
- Johnson, L.V.; Ozaki, S.; Staples, M.K.; Erickson, P.A.; Anderson, D.H. A potential role for immune complex pathogenesis in drusen formation. Exp. Eye Res. 2000, 70, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, H.; Liu, Z.L. Interleukin-6 receptor blockade suppresses subretinal fibrosis in a mouse model. Int. J. Ophthalmol. 2014, 7, 194–197. [Google Scholar] [CrossRef]
- Jo, Y.J.; Sonoda, K.H.; Oshima, Y.; Takeda, A.; Kohno, R.; Yamada, J.; Hamuro, J.; Yang, Y.; Notomi, S.; Hisatomi, T.; et al. Establishment of a new animal model of focal subretinal fibrosis that resembles disciform lesion in advanced age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6089–6095. [Google Scholar] [CrossRef]
- Cui, L.; Lyu, Y.; Jin, X.; Wang, Y.; Li, X.; Wang, J.; Zhang, J.; Deng, Z.; Yang, N.; Zheng, Z.; et al. miR-194 suppresses epithelial-mesenchymal transition of retinal pigment epithelial cells by directly targeting ZEB1. Ann. Transl. Med. 2019, 7, 751. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Z.; Zhang, H.; Zhang, Y.; Lin, D. The COX-2-Selective Antagonist (NS-398) Inhibits Choroidal Neovascularization and Subretinal Fibrosis. PLoS ONE 2016, 11, e0146808. [Google Scholar] [CrossRef]
- Ishikawa, K.; He, S.; Terasaki, H.; Nazari, H.; Zhang, H.; Spee, C.; Kannan, R.; Hinton, D.R. Resveratrol inhibits epithelial-mesenchymal transition of retinal pigment epithelium and development of proliferative vitreoretinopathy. Sci. Rep. 2015, 5, 16386. [Google Scholar] [CrossRef]
- Krstic, J.; Trivanovic, D.; Mojsilovic, S.; Santibanez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxidative Med. Cell Longev. 2015, 2015, 654594. [Google Scholar] [CrossRef]
- Li, G.; Li, C.X.; Xia, M.; Ritter, J.K.; Gehr, T.W.B.; Boini, K.; Li, P.L. Enhanced Epithelial-to-Mesenchymal Transition Associated with Lysosome Dysfunction in Podocytes: Role of p62/Sequestosome 1 as a Signaling Hub. Cell. Physiol. Biochem. 2015, 35, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162–2177. [Google Scholar] [PubMed]
- Stefansson, E.; Geirsdottir, A.; Sigurdsson, H. Metabolic physiology in age related macular degeneration. Prog. Retin. Eye Res. 2011, 30, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 100858. [Google Scholar] [CrossRef] [PubMed]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef]
- Udar, N.; Atilano, S.R.; Memarzadeh, M.; Boyer, D.S.; Chwa, M.; Lu, S.; Maguen, B.; Langberg, J.; Coskun, P.; Wallace, D.C.; et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2966–2974. [Google Scholar] [CrossRef]
- Kenney, M.C.; Atilano, S.R.; Boyer, D.; Chwa, M.; Chak, G.; Chinichian, S.; Coskun, P.; Wallace, D.C.; Nesburn, A.B.; Udar, N.S. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4289–4297. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2848–2855. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Berg, K.M.; Kapphahn, R.J.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Matoba, R.; Morizane, Y.; Shiode, Y.; Hirano, M.; Doi, S.; Toshima, S.; Araki, R.; Hosogi, M.; Yonezawa, T.; Shiraga, F. Suppressive effect of AMP-activated protein kinase on the epithelial-mesenchymal transition in retinal pigment epithelial cells. PLoS ONE 2017, 12, e0181481. [Google Scholar] [CrossRef] [PubMed]
- Shukal, D.; Bhadresha, K.; Shastri, B.; Mehta, D.; Vasavada, A.; Johar, K., Sr. Dichloroacetate prevents TGFβ-induced epithelial-mesenchymal transition of retinal pigment epithelial cells. Exp. Eye Res. 2020, 197, 108072. [Google Scholar] [CrossRef] [PubMed]
- Che, D.; Zhou, T.; Lan, Y.; Xie, J.; Gong, H.; Li, C.; Feng, J.; Hong, H.; Qi, W.; Ma, C.; et al. High glucose-induced epithelial-mesenchymal transition contributes to the upregulation of fibrogenic factors in retinal pigment epithelial cells. Int. J. Mol. Med. 2016, 38, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Feng, B.; Chen, S.; Chu, Y.; Chakrabarti, S. Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7321–7331. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. Pgc-1α repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Models Mech. 2018, 11, dmm032698. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1α Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1alpha in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Satish, S.; Philipose, H.; Rosales, M.A.B.; Saint-Geniez, M. Pharmaceutical Induction of PGC-1alpha Promotes Retinal Pigment Epithelial Cell Metabolism and Protects against Oxidative Damage. Oxidative Med. Cell Longev. 2018, 2018, 9248640. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Valapala, M.; Shang, P.; Hose, S.; Grebe, R.; Lutty, G.A.; Zigler, J.S., Jr.; Kaarniranta, K.; Handa, J.T. Lysosomes: Regulators of autophagy in the retinal pigmented epithelium. Exp. Eye Res. 2016, 144, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Kim, H.N.; Hwang, J.J.; Kim, Y.H.; Park, S.E. Lysosomal dysfunction in proteinopathic neurodegenerative disorders: Possible therapeutic roles of cAMP and zinc. Mol. Brain 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Błasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef]
- Guerra, F.; Guaragnella, N.; Arbini, A.A.; Bucci, C.; Giannattasio, S.; Moro, L. Mitochondrial Dysfunction: A Novel Potential Driver of Epithelial-to-Mesenchymal Transition in Cancer. Front. Oncol. 2017, 7, 295. [Google Scholar] [CrossRef]
- Zou, J.; Liu, Y.; Li, B.; Zheng, Z.; Ke, X.; Hao, Y.; Li, X.; Li, X.; Liu, F.; Zhang, Z. Autophagy attenuates endothelial-to-mesenchymal transition by promoting Snail degradation in human cardiac microvascular endothelial cells. Biosci. Rep. 2017, 37, BSR20171049. [Google Scholar] [CrossRef]
- Ishikawa, M.; Sawada, Y.; Yoshitomi, T. Structure and function of the interphotoreceptor matrix surrounding retinal photoreceptor cells. Exp. Eye Res. 2015, 133, 3–18. [Google Scholar] [CrossRef]
- Pauleikhoff, D.; Harper, C.A.; Marshall, J.; Bird, A.C. Aging changes in Bruch’s membrane. A histochemical and morphologic study. Ophthalmology 1990, 97, 171–178. [Google Scholar] [CrossRef]
- Marshall, G.E.; Konstas, A.G.; Reid, G.G.; Edwards, J.G.; Lee, W.R. Type IV collagen and laminin in Bruch’s membrane and basal linear deposit in the human macula. Br. J. Ophthalmol. 1992, 76, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Johnson, M. Structure, Function, and Pathology of Bruch’s Membrane. Retina 2012, 1, 465–481. [Google Scholar]
- Sun, K.; Cai, H.; Tezel, T.H.; Paik, D.; Gaillard, E.R.; Del Priore, L.V. Bruch’s membrane aging decreases phagocytosis of outer segments by retinal pigment epithelium. Mol. Vis. 2007, 13, 2310–2319. [Google Scholar] [PubMed]
- Ramrattan, R.S.; van der Schaft, T.L.; Mooy, C.M.; de Bruijn, W.C.; Mulder, P.G.; de Jong, P.T. Morphometric analysis of Bruch’s membrane, the choriocapillaris, and the choroid in aging. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2857–2864. [Google Scholar]
- Moore, D.J.; Hussain, A.A.; Marshall, J. Age-related variation in the hydraulic conductivity of Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1290–1297. [Google Scholar]
- Del Priore, L.V.; Tezel, T.H. Reattachment rate of human retinal pigment epithelium to layers of human Bruch’s membrane. Arch. Ophthalmol. 1998, 116, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Del Priore, L.V. Bruch membrane aging alters the gene expression profile of human retinal pigment epithelium. Curr. Eye Res. 2006, 31, 181–189. [Google Scholar] [CrossRef]
- Gullapalli, V.K.; Sugino, I.K.; Van Patten, Y.; Shah, S.; Zarbin, M.A. Impaired RPE survival on aged submacular human Bruch’s membrane. Exp. Eye Res. 2005, 80, 235–248. [Google Scholar] [CrossRef]
- Wang, H.; Ninomiya, Y.; Sugino, I.K.; Zarbin, M.A. Retinal pigment epithelium wound healing in human Bruch’s membrane explants. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2199–2210. [Google Scholar] [CrossRef]
- Tezel, T.H.; Kaplan, H.J.; Del Priore, L.V. Fate of human retinal pigment epithelial cells seeded onto layers of human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1999, 40, 467–476. [Google Scholar]
- Thomas, M.A.; Dickinson, J.D.; Melberg, N.S.; Ibanez, H.E.; Dhaliwal, R.S. Visual results after surgical removal of subfoveal choroidal neovascular membranes. Ophthalmology 1994, 101, 1384–1396. [Google Scholar] [CrossRef]
- Heller, J.P.; Martin, K.R. Enhancing RPE Cell-Based Therapy Outcomes for AMD: The Role of Bruch’s Membrane. Transl. Vis. Sci. Technol. 2014, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Strzalka-Mrozik, B.; Grzybowski, A.; Mazurek, U.; Romaniuk, W. Age-related macular degeneration and changes in the extracellular matrix. Med. Sci. Monit. 2014, 20, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Puklin, J.E.; Frank, R.N.; Zhang, N.L. Ultrastructural immunocytochemistry of subretinal neovascular membranes in age-related macular degeneration. Ophthalmology 1992, 99, 1368–1376. [Google Scholar] [CrossRef]
- Chau, K.Y.; Sivaprasad, S.; Patel, N.; Donaldson, T.A.; Luthert, P.J.; Chong, N.V. Plasma levels of matrix metalloproteinase-2 and -9 (MMP-2 and MMP-9) in age-related macular degeneration. Eye (Lond.) 2007, 21, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Marin-Castano, M.E.; Csaky, K.G.; Cousins, S.W. Nonlethal oxidant injury to human retinal pigment epithelium cells causes cell membrane blebbing but decreased MMP-2 activity. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3331–3340. [Google Scholar] [CrossRef]
- Marin-Castano, M.E.; Striker, G.E.; Alcazar, O.; Catanuto, P.; Espinosa-Heidmann, D.G.; Cousins, S.W. Repetitive nonlethal oxidant injury to retinal pigment epithelium decreased extracellular matrix turnover in vitro and induced sub-RPE deposits in vivo. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4098–4112. [Google Scholar] [CrossRef]
- Steen, B.; Sejersen, S.; Berglin, L.; Seregard, S.; Kvanta, A. Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2194–2200. [Google Scholar]
- Plantner, J.J.; Jiang, C.; Smine, A. Increase in interphotoreceptor matrix gelatinase A (MMP-2) associated with age-related macular degeneration. Exp. Eye Res. 1998, 67, 637–645. [Google Scholar] [CrossRef]
- Behzadian, M.A.; Wang, X.L.; Windsor, L.J.; Ghaly, N.; Caldwell, R.B. TGF-beta increases retinal endothelial cell permeability by increasing MMP-9: Possible role of glial cells in endothelial barrier function. Investig. Ophthalmol. Vis. Sci. 2001, 42, 853–859. [Google Scholar]
- Elner, S.G.; Elner, V.M.; Kindzelskii, A.L.; Horino, K.; Davis, H.R.; Todd, R.F., III; Glagov, S.; Petty, H.R. Human RPE cell lysis of extracellular matrix: Functional urokinase plasminogen activator receptor (uPAR), collagenase and elastase. Exp. Eye Res. 2003, 76, 585–595. [Google Scholar] [CrossRef]
- Siren, V.; Myohanen, H.; Vaheri, A.; Immonen, I. Transforming growth factor beta induces urokinase receptor expression in cultured retinal pigment epithelial cells. Ophthalmic Res. 1999, 31, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, K.; Kodama, A.; Okada, K.; Iwata, M.; Yoshida, K.; Kusaka, S.; Matsumoto, C.; Kaji, H.; Shimomura, Y. TGF-β2 promotes RPE cell invasion into a collagen gel by mediating urokinase-type plasminogen activator (uPA) expression. Exp. Eye Res. 2013, 115, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ozkaya, A.; Erdogan, G.; Tarakcioglu, H.N. Submacular hemorrhage secondary to age-related macular degeneration managed with vitrectomy, subretinal injection of tissue plasminogen activator, hemorrhage displacement with liquid perfluorocarbon, gas tamponade, and face-down positioning. Saudi J. Ophthalmol. 2018, 32, 269–274. [Google Scholar] [CrossRef]
- Lambert, V.; Munaut, C.; Carmeliet, P.; Gerard, R.D.; Declerck, P.J.; Gils, A.; Claes, C.; Foidart, J.M.; Noel, A.; Rakic, J.M. Dose-dependent modulation of choroidal neovascularization by plasminogen activator inhibitor type I: Implications for clinical trials. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2791–2797. [Google Scholar] [CrossRef]
- Pepper, M.S.; Sappino, A.P.; Montesano, R.; Orci, L.; Vassalli, J.D. Plasminogen activator inhibitor-1 is induced in migrating endothelial cells. J. Cell Physiol. 1992, 153, 129–139. [Google Scholar] [CrossRef]
- Basu, A.; Menicucci, G.; Maestas, J.; Das, A.; McGuire, P. Plasminogen activator inhibitor-1 (PAI-1) facilitates retinal angiogenesis in a model of oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4974–4981. [Google Scholar] [CrossRef]
- Swaney, J.S.; Moreno, K.M.; Gentile, A.M.; Sabbadini, R.A.; Stoller, G.L. Sphingosine-1-phosphate (S1P) is a novel fibrotic mediator in the eye. Exp. Eye Res. 2008, 87, 367–375. [Google Scholar] [CrossRef]
- Penn, J.S.; Rajaratnam, V.S. Inhibition of retinal neovascularization by intravitreal injection of human rPAI-1 in a rat model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5423–5429. [Google Scholar] [CrossRef][Green Version]
- Lambert, V.; Munaut, C.; Noel, A.; Frankenne, F.; Bajou, K.; Gerard, R.; Carmeliet, P.; Defresne, M.P.; Foidart, J.M.; Rakic, J.M. Influence of plasminogen activator inhibitor type 1 on choroidal neovascularization. FASEB J. 2001, 15, 1021–1027. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Lagares, D. Mechano-therapeutics: Targeting Mechanical Signaling in Fibrosis and Tumor Stroma. Pharmacol. Ther. 2020, 107575. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Maberley, D.; Samad, A.; Ma, P.; Ning, A.; Matsubara, J.A.; Baciu, P. Expression of integrins on human choroidal neovascular membranes. J. Ocul. Biol. Dis. Inform. 2009, 2, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Han, Q.H.; Hu, D.; Tian, L.; Guo, C.M.; Du, H.J.; Zhang, P.; Wang, Y.S.; Hui, Y.N. Mechanical force enhances MMP-2 activation via p38 signaling pathway in human retinal pigment epithelial cells. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Goswami, R.; Zhang, D.X.; Rahaman, S.O. TRPV4 regulates matrix stiffness and TGFbeta1-induced epithelial-mesenchymal transition. J. Cell Mol. Med. 2019, 23, 761–774. [Google Scholar] [CrossRef]
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Krishna, V.S.; Singh, H.; Tan, T.Z.; et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci. Rep. 2016, 6, 27903. [Google Scholar] [CrossRef]
- Okada, Y.; Shirai, K.; Miyajima, M.; Reinach, P.S.; Yamanaka, O.; Sumioka, T.; Kokado, M.; Tomoyose, K.; Saika, S. Loss of TRPV4 Function Suppresses Inflammatory Fibrosis Induced by Alkali-Burning Mouse Corneas. PLoS ONE 2016, 11, e0167200. [Google Scholar] [CrossRef]
- Arredondo Zamarripa, D.; Noguez Imm, R.; Bautista Cortes, A.M.; Vazquez Ruiz, O.; Bernardini, M.; Fiorio Pla, A.; Gkika, D.; Prevarskaya, N.; Lopez-Casillas, F.; Liedtke, W.; et al. Dual contribution of TRPV4 antagonism in the regulatory effect of vasoinhibins on blood-retinal barrier permeability: Diabetic milieu makes a difference. Sci. Rep. 2017, 7, 13094. [Google Scholar] [CrossRef]
- Phuong, T.T.T.; Redmon, S.N.; Yarishkin, O.; Winter, J.M.; Li, D.Y.; Križaj, D. Calcium influx through TRPV4 channels modulates the adherens contacts between retinal microvascular endothelial cells. J. Physiol. 2017, 595, 6869–6885. [Google Scholar] [CrossRef]
- Bu, S.C.; Kuijer, R.; Li, X.R.; Hooymans, J.M.; Los, L.I. Idiopathic epiretinal membrane. Retina 2014, 34, 2317–2335. [Google Scholar] [CrossRef] [PubMed]
- Pierro, L.; Zampedri, E.; Milani, P.; Gagliardi, M.; Isola, V.; Pece, A. Spectral domain OCT versus time domain OCT in the evaluation of macular features related to wet age-related macular degeneration. Clin. Ophthalmol. 2012, 6, 219–223. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takahashi, E.; Fukushima, A.; Haga, A.; Inomata, Y.; Ito, Y.; Fukushima, M.; Tanihara, H. Effects of mechanical stress and vitreous samples in retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 2016, 470, 569–574. [Google Scholar] [CrossRef]
- Sarks, S.H. Ageing and degeneration in the macular region: A clinico-pathological study. Br. J. Ophthalmol. 1976, 60, 324–341. [Google Scholar] [CrossRef] [PubMed]
- Lutty, G.; Grunwald, J.; Majji, A.B.; Uyama, M.; Yoneya, S. Changes in choriocapillaris and retinal pigment epithelium in age-related macular degeneration. Mol. Vis. 1999, 5, 35. [Google Scholar] [PubMed]
- McLeod, D.S.; Grebe, R.; Bhutto, I.; Merges, C.; Baba, T.; Lutty, G.A. Relationship between RPE and choriocapillaris in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4982–4991. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Kurihara, T.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc. Natl. Acad. Sci. USA 2009, 106, 18751–18756. [Google Scholar] [CrossRef]
- Marneros, A.G.; Fan, J.; Yokoyama, Y.; Gerber, H.P.; Ferrara, N.; Crouch, R.K.; Olsen, B.R. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459. [Google Scholar] [CrossRef]
- Gaudric, A.; Sterkers, M.; Coscas, G. Retinal detachment after choroidal ischemia. Am. J. Ophthalmol. 1987, 104, 364–372. [Google Scholar] [CrossRef]
- Saito, Y.; Tano, Y. Retinal pigment epithelial lesions associated with choroidal ischemia in preeclampsia. Retina 1998, 18, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.; Abend, S.; McHugh, K.J.; Saint-Geniez, M. Identification of a synergistic interaction between endothelial cells and retinal pigment epithelium. J. Cell Mol. Med. 2017, 21, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Lehmann, G.L.; Ginsberg, M.; Nolan, D.J.; Bareja, R.; Elemento, O.; Salfati, Z.; Alam, N.M.; Prusky, G.T.; Llanos, P.; et al. Concerted regulation of retinal pigment epithelium basement membrane and barrier function by angiocrine factors. Nat. Commun. 2017, 8, 15374. [Google Scholar] [CrossRef] [PubMed]
- Ohlmann, A.; Scholz, M.; Koch, M.; Tamm, E.R. Epithelial-mesenchymal transition of the retinal pigment epithelium causes choriocapillaris atrophy. Histochem. Cell Biol. 2016, 146, 769–780. [Google Scholar] [CrossRef]
- Suzuki, M.; Kamei, M.; Itabe, H.; Yoneda, K.; Bando, H.; Kume, N.; Tano, Y. Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Mol. Vis. 2007, 13, 772–778. [Google Scholar] [PubMed]
- Yamada, Y.; Tian, J.; Yang, Y.; Cutler, R.G.; Wu, T.; Telljohann, R.S.; Mattson, M.P.; Handa, J.T. Oxidized low density lipoproteins induce a pathologic response by retinal pigmented epithelial cells. J. Neurochem. 2008, 105, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Gnanaguru, G.; Choi, A.R.; Amarnani, D.; D’Amore, P.A. Oxidized Lipoprotein Uptake Through the CD36 Receptor Activates the NLRP3 Inflammasome in Human Retinal Pigment Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4704–4712. [Google Scholar] [CrossRef]
- Kozlowski, M.R. RPE cell senescence: A key contributor to age-related macular degeneration. Med. Hypotheses 2012, 78, 505–510. [Google Scholar] [CrossRef]
- Wu, T.; Xu, W.; Wang, Y.; Tao, M.; Hu, Z.; Lv, B.; Hui, Y.; Du, H. OxLDL enhances choroidal neovascularization lesion through inducing vascular endothelium to mesenchymal transition process and angiogenic factor expression. Cell Signal. 2020, 70, 109571. [Google Scholar] [CrossRef]
- Fukushima, A.; Takahashi, E.; Saruwatari, J.; Tanihara, H.; Inoue, T. The angiogenic effects of exosomes secreted from retinal pigment epithelial cells on endothelial cells. Biochem. Biophys. Rep. 2020, 22, 100760. [Google Scholar] [CrossRef]
- Bastiaans, J.; van Meurs, J.C.; van Holten-Neelen, C.; Nagtzaam, N.M.; van Hagen, P.M.; Chambers, R.C.; Hooijkaas, H.; Dik, W.A. Thrombin induces epithelial-mesenchymal transition and collagen production by retinal pigment epithelial cells via autocrine PDGF-receptor signaling. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8306–8314. [Google Scholar] [CrossRef] [PubMed]
- McLenachan, S.; Hao, E.; Zhang, D.; Zhang, L.; Edel, M.; Chen, F. Bioengineered Bruch’s-like extracellular matrix promotes retinal pigment epithelial differentiation. Biochem. Biophys. Rep. 2017, 10, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E. The role of the atherosclerotic process in the pathogenesis of age-related macular degeneration. Am. J. Ophthalmol. 2000, 130, 658–663. [Google Scholar] [CrossRef]
- Nackman, G.B.; Karkowski, F.J.; Halpern, V.J.; Gaetz, H.P.; Tilson, M.D. Elastin degradation products induce adventitial angiogenesis in the Anidjar/Dobrin rat aneurysm model. Surgery 1997, 122, 39–44. [Google Scholar] [CrossRef]
- Maminishkis, A.; Chen, S.; Jalickee, S.; Banzon, T.; Shi, G.; Wang, F.E.; Ehalt, T.; Hammer, J.A.; Miller, S.S. Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3612–3624. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, S.; Spee, C.; Barron, E.; Ryan, S.J.; Kannan, R.; Hinton, D.R. A protocol for the culture and differentiation of highly polarized human retinal pigment epithelial cells. Nat. Protoc. 2009, 4, 662–673. [Google Scholar] [CrossRef]
- Toops, K.A.; Tan, L.X.; Lakkaraju, A. A detailed three-step protocol for live imaging of intracellular traffic in polarized primary porcine RPE monolayers. Exp. Eye Res. 2014, 124, 74–85. [Google Scholar] [CrossRef]
- Das, S.; Becker, B.N.; Hoffmann, F.M.; Mertz, J.E. Complete reversal of epithelial to mesenchymal transition requires inhibition of both ZEB expression and the Rho pathway. BMC Cell Biol. 2009, 10, 94. [Google Scholar] [CrossRef]
- Furihata, T.; Kawamatsu, S.; Ito, R.; Saito, K.; Suzuki, S.; Kishida, S.; Saito, Y.; Kamiichi, A.; Chiba, K. Hydrocortisone enhances the barrier properties of HBMEC/ciβ, a brain microvascular endothelial cell line, through mesenchymal-to-endothelial transition-like effects. Fluids Barriers CNS 2015, 12, 7. [Google Scholar] [CrossRef]
- Tezel, T.H.; Del Priore, L.V. Reattachment to a substrate prevents apoptosis of human retinal pigment epithelium. Graefes Arch. Clin. Exp. Ophthalmol. 1997, 235, 41–47. [Google Scholar] [CrossRef]
- Tezel, T.H.; Del Priore, L.V.; Kaplan, H.J. Reengineering of aged Bruch’s membrane to enhance retinal pigment epithelium repopulation. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3337–3348. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.J.; Sadda, S.R.; Tso, M.O.; Humayan, M.S.; de Juan, E., Jr.; Binder, S. Autologous transplantation of retinal pigment epithelium after mechanical debridement of Bruch’s membrane. Curr. Eye Res. 2003, 26, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Del Priore, L.V.; Geng, L.; Tezel, T.H.; Kaplan, H.J. Extracellular matrix ligands promote RPE attachment to inner Bruch’s membrane. Curr. Eye Res. 2002, 25, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Cronstein, B.N. Methotrexate—How does it really work? Nat. Rev. Rheumatol. 2010, 6, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Gangaputra, S.; Newcomb, C.W.; Liesegang, T.L.; Kacmaz, R.O.; Jabs, D.A.; Levy-Clarke, G.A.; Nussenblatt, R.B.; Rosenbaum, J.T.; Suhler, E.B.; Thorne, J.E.; et al. Methotrexate for ocular inflammatory diseases. Ophthalmology 2009, 116, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, S.; Hendler, K.; Siegal, T.; Shalom, E.; Pe’er, J. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Br. J. Ophthalmol. 2008, 92, 383–388. [Google Scholar] [CrossRef]
- Sadaka, A.; Sisk, R.A.; Osher, J.M.; Toygar, O.; Duncan, M.K.; Riemann, C.D. Intravitreal methotrexate infusion for proliferative vitreoretinopathy. Clin. Ophthalmol. 2016, 10, 1811–1817. [Google Scholar] [CrossRef]
- ClinicalTrials.gov U.S. National Library Medicine. The GUARD Trial—Part 1: A Phase 3 Clinical Trial for Prevention of Proliferative Vitreoretinopathy. Available online: https://clinicaltrials.gov/ct2/show/NCT04136366 (accessed on 12 June 2020).
- Kurup, S.K.; Gee, C.; Greven, C.M. Intravitreal methotrexate in therapeutically resistant exudative age-related macular degeneration. Acta Ophthalmol. 2010, 88, e145–e146. [Google Scholar] [CrossRef]
- Lam, J.D.; Oh, D.J.; Wong, L.L.; Amarnani, D.; Park-Windhol, C.; Sanchez, A.V.; Cardona-Velez, J.; McGuone, D.; Stemmer-Rachamimov, A.O.; Eliott, D.; et al. Identification of RUNX1 as a Mediator of Aberrant Retinal Angiogenesis. Diabetes 2017, 66, 1950–1956. [Google Scholar] [CrossRef]
- Espinosa-Heidmann, D.G.; Reinoso, M.A.; Pina, Y.; Csaky, K.G.; Caicedo, A.; Cousins, S.W. Quantitative enumeration of vascular smooth muscle cells and endothelial cells derived from bone marrow precursors in experimental choroidal neovascularization. Exp. Eye Res. 2005, 80, 369–378. [Google Scholar] [CrossRef]
- Little, K.; Ma, J.H.; Yang, N.; Chen, M.; Xu, H. Myofibroblasts in macular fibrosis secondary to neovascular age-related macular degeneration—The potential sources and molecular cues for their recruitment and activation. EBioMedicine 2018, 38, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yang, S.; Liang, J.; Zhai, Y.; Shen, M.; Sun, J.; Feng, Y.; Lu, X.; Zhu, H.; Wang, F.; et al. Choroidal pericytes promote subretinal fibrosis after experimental photocoagulation. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. https://doi.org/10.3390/ijms21124271
Shu DY, Butcher E, Saint-Geniez M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2020; 21(12):4271. https://doi.org/10.3390/ijms21124271
Chicago/Turabian StyleShu, Daisy Y., Erik Butcher, and Magali Saint-Geniez. 2020. "EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration" International Journal of Molecular Sciences 21, no. 12: 4271. https://doi.org/10.3390/ijms21124271
APA StyleShu, D. Y., Butcher, E., & Saint-Geniez, M. (2020). EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. International Journal of Molecular Sciences, 21(12), 4271. https://doi.org/10.3390/ijms21124271