Ibrutinib in Gynecological Malignancies and Breast Cancer: A Systematic Review


Abstract
1. Introduction
2. Results
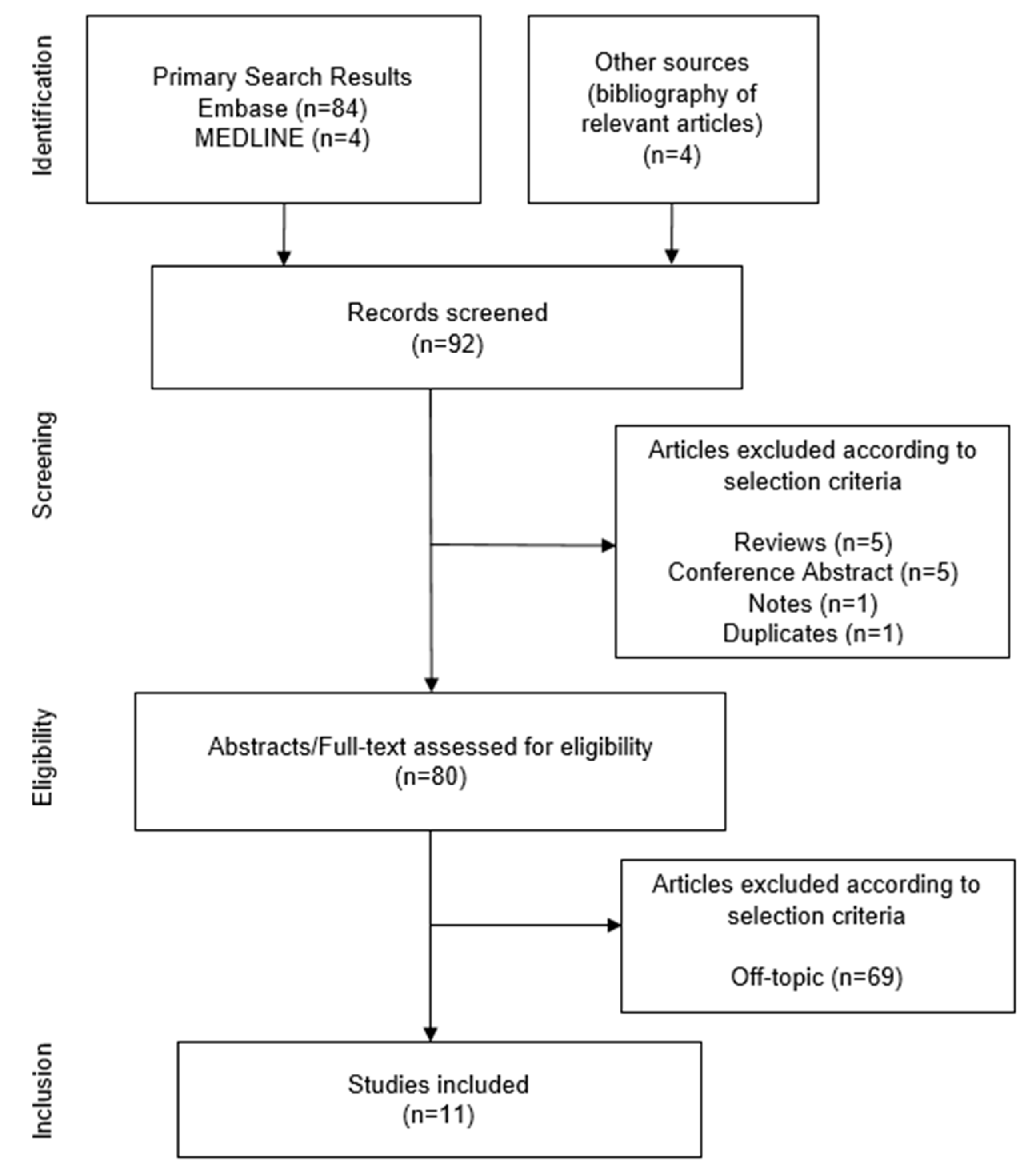
2.1. Overview
2.2. Review of Identified Studies
2.2.1. Breast Cancer
2.2.2. Gynecological Malignancies
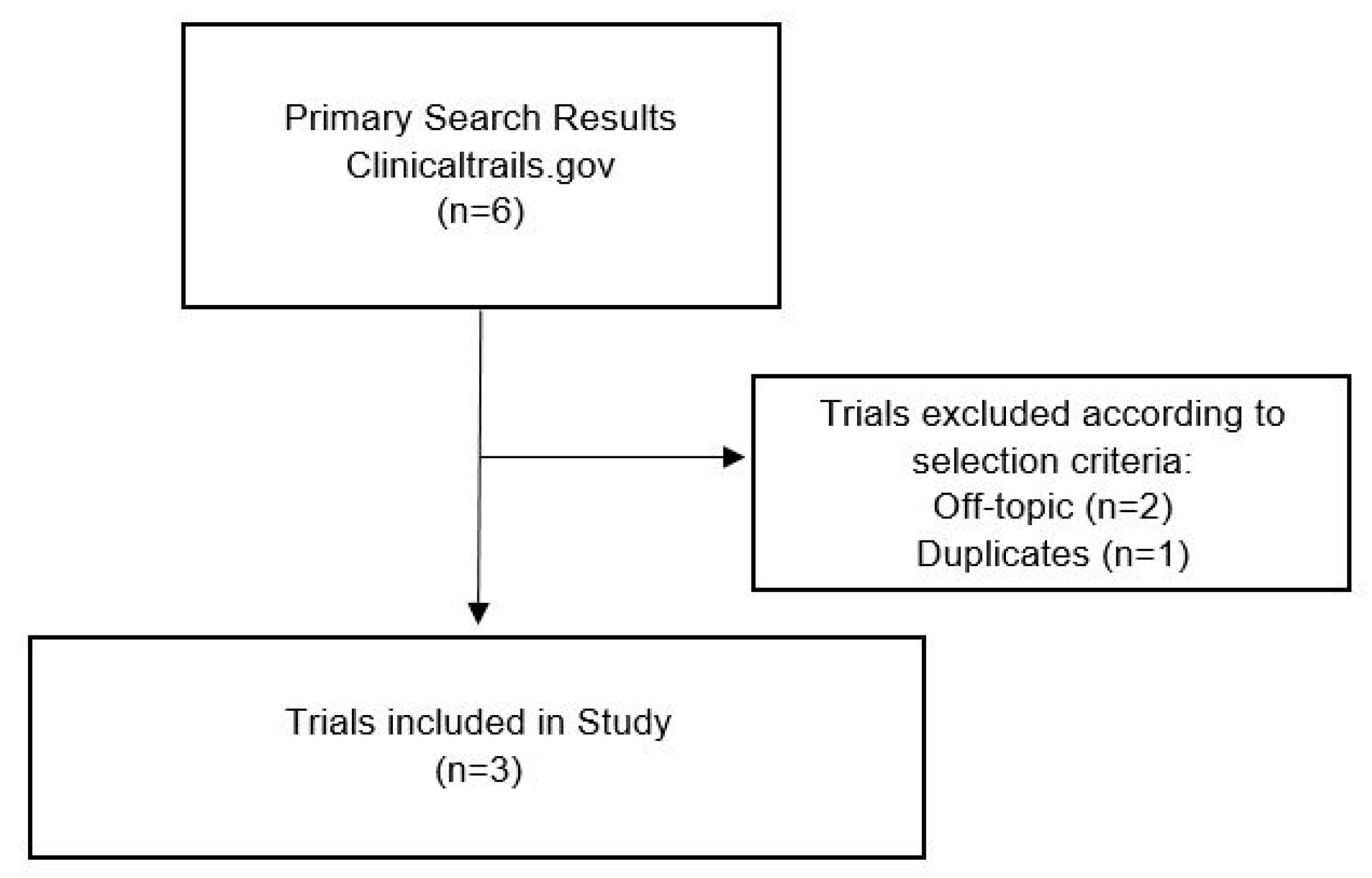
2.3. Clinical Trials
3. Discussion
4. Materials and Methods
4.1. Inclusion Criteria
4.2. Search Strategy
4.3. Data Collection Process, Data Items
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMX | Bone marrow X-linked kinase |
| BTK | Bruton’s tyrosine kinase |
| cGVHD | Chronic graft versus host disease |
| CLL | Chronic lymphocytic leukemia |
| CSC | Cancer stem cell |
| CTL | Cytotoxic T lymphocytes |
| DC | Dendritic cell |
| EGFR | Epidermal growth factor receptor |
| ER | Estrogene receptor |
| i.v. | Intravenously |
| IC50 | Half-maximal inhibitory concentration |
| IFN | Interferon |
| IL | Interleukin |
| ITK | interleukin-2-inducible T-cell kinase |
| MDSC | Myeloid-derived suppressor cell |
| MeSH | Medical Subject Headings |
| MZL | Marginal zone lymphoma |
| NCI | National cancer institute |
| NSCLC | Non-small-cell lung cancer |
| ORR | Overall response rate |
| PARP | poly-ADP-ribose-polymerase |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PR | Progesterone receptor |
| q2w | 2-weekly, every 2 weeks |
| RLK | Redundant-resting lymphocyte kinase |
| SLL | Small lymphocytic lymphoma |
| TEC | Tec protein tyrosine kinase |
| TFK | TEC family kinase |
| TNBC | Triple-negative breast cancer |
| WM | Waldenstrom’s macroglobulinemia |
Appendix A

{kind=link}
{kind=link}
| Number | Search Terms | Search Items Medline |
|---|---|---|
| 1 | ibrutinib (Mesh) | 2026 results |
| 2 | (‘genital neoplasms, female’ [Mesh]) OR ‘breast neoplasms’ [Mesh] | 498.349 results |
| 3 | 1 AND 2 | 4 results |
| Number | Search Terms | Search Items Embase |
|---|---|---|
| 1 | ibrutinib (Emtree) | 6186 results |
| 2 | ‘female genital tract tumor’/exp OR ‘breast tumor/exp’ | 866.090 results |
| 3 | 1 AND 2 | 84 results |

References
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.-M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kinoshita, T.; Sukbuntherng, J.; Chang, B.Y.; Elias, L. Ibrutinib Inhibits ERBB Receptor Tyrosine Kinases and HER2-Amplified Breast Cancer Cell Growth. Mol. Cancer Ther. 2016, 15, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.; Chong, G.; Hawkes, E. Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, beyond Hematological Malignancies. J. Clin. Med. 2018, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Pharmacyclics Inc. Imbruvica® (ibrutinib) [package insert]. U.S. Food and Drug Administration Website. Available online: www.accessdata.fda.gov/drugsatfda_docs/label/2019/205552s029,210563s004lbl.pdf (accessed on 1 January 2020).
- Wei, L.; Su, Y.-K.; Lin, C.-M.; Chao, T.-Y.; Huang, S.-P.; Huynh, T.-T.; Jan, H.-J.; Whang-Peng, J.; Chiou, J.-F.; Wu, A.T.; et al. Preclinical investigation of ibrutinib, a Bruton’s kinase tyrosine (Btk) inhibitor, in suppressing glioma tumorigenesis and stem cell phenotypes. Oncotarget 2016, 7, 69961–69975. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, M.; Wang, L.; Lu, H.; Wu, S.; Dai, B.; Ou, Z.; Zhang, L.; Heymach, J.V.; Gold, K.A.; et al. Selective Antitumor Activity of Ibrutinib in EGFR-Mutant Non–Small Cell Lung Cancer Cells. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Massó-Vallés, D.; Jauset, T.; Soucek, L. Ibrutinib repurposing: From B-cell malignancies to solid tumors. Oncoscience 2016, 3, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malign. Rep. 2019, 14, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, B.T.; Ellmeier, W.; Watson, S.P. Tec regulates platelet activation by GPVI in the absence of Btk. Blood 2003, 102, 3592–3599. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.; Bolen, J.; Watson, S.P. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr. Biol. 1998, 8, 1137–1140. [Google Scholar] [CrossRef]
- Younes, A.; Thieblemont, C.; Morschhauser, F.; Flinn, I.W.; Friedberg, J.W.; Amorim, S.; Hivert, B.; Westin, J.; Vermeulen, J.; Bandyopadhyay, N.; et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: A non-randomised, phase 1b study. Lancet Oncol. 2014, 15, 1019–1026. [Google Scholar] [CrossRef]
- Jaglowski, S.M.; Jones, J.A.; Nagar, V.; Flynn, J.M.; Andritsos, L.A.; Maddocks, K.J.; Woyach, J.A.; Blum, K.A.; Grever, M.R.; Smucker, K.; et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: A phase 1b/2 study. Blood 2015, 126, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.; Cramer, P.; Demirkan, F.; Fraser, G.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): A randomised, double-blind, phase 3 study. Lancet Oncol. 2016, 17, 200–211. [Google Scholar] [CrossRef]
- A Multi-Center Study of Ibrutinib in Combination with MEDI4736 in Subjects With Relapsed or Refractory Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT02403271 (accessed on 30 April 2020).
- Trial of Ibrutinib Plus Trastuzumab in HER2-amplified Metastatic Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT03379428 (accessed on 30 April 2020).
- Ibrutinib and Nivolumab in Treating Participants with Metastatic Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT03525925 (accessed on 30 April 2020).
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-generation inhibitors of Bruton tyrosine kinase. J. Hematol. Oncol. 2016, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca. Calquence® (acalabrutinib) [package insert]. U.S. Food and Drug Administration website. Available online: www.accessdata.fda.gov/drugsatfda_docs/label/2019/210259s006s007lbl.pdf (accessed on 27 May 2020).
- Wu, J.; Zhang, M.; Liu, D. Acalabrutinib (ACP-196): A selective second-generation BTK inhibitor. J. Hematol. Oncol. 2016, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Covey, T.; Izumi, R.; Van De Kar, B.; Gulrajani, M.; Van Lith, B.; Van Hoek, M.; De Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. [Google Scholar] [CrossRef] [PubMed]
- BeiGene. Brukinsa® (zanubrutinib) [package insert]. U.S. Food and Drug Administration website. Available online: www.accessdata.fda.gov/drugsatfda_docs/label/2019/213217s000lbl.pdf (accessed on 27 May 2020).
- Liclican, A.; Serafini, L.; Xing, W.; Czerwieniec, G.; Steiner, B.; Wang, T.; Brendza, K.M.; Lutz, J.D.; Keegan, K.S.; Ray, A.S.; et al. Biochemical characterization of tirabrutinib and other irreversible inhibitors of Bruton’s tyrosine kinase reveals differences in on—and off—arget inhibition. Biochim. Biophys. Acta BBA Gen. Subj. 2020, 1864, 129531. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef] [PubMed]
- Study of Evobrutinib in Participants with Relapsing Multiple Sclerosis (RMS). Available online: https://ClinicalTrials.gov/show/NCT04032158 (accessed on 27 May 2020).
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Berek, J.S.; Chen, L.-M.; Cristea, M.; DeRosa, M.; et al. NCCN Guidelines Insights: Ovarian Cancer, Version 1.2019. J. Natl. Compr. Cancer Netw. 2019, 17, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, P.; Scatena, R. The Role of CA 125 as Tumor Marker: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919849753. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Sessa, C.; Du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO consensus conference recommendations on ovarian cancer: Pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Ovarian Cancer (Version 1.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf (accessed on 29 March 2020).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Ewald, F. Ibrutinib (ImbruvicaTM) potently inhibits ErbB receptor phosphorylation and cell viability of ErbB2-positive breast cancer cells. Investig. New Drugs 2014, 32, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Sagiv-Barfi, I.; Kohrt, H.E.K.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. [Google Scholar] [CrossRef] [PubMed]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.L.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Eifert, C.; Wang, X.; Kokabee, L.; Kourtidis, A.; Jain, R.; Gerdes, M.J.; Conklin, D.S. A novel isoform of the B cell tyrosine kinase BTK protects breast cancer cells from apoptosis. Genes Chromosom. Cancer 2013, 52, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wong, J.; Sevinsky, C.J.; Kokabee, L.; Khan, F.; Sun, Y.; Conklin, D.S. Bruton’s Tyrosine Kinase Inhibitors Prevent Therapeutic Escape in Breast Cancer Cells. Mol. Cancer Ther. 2016, 15, 2198–2208. [Google Scholar] [CrossRef] [PubMed]
- Di, J.; Zheng, B.; Kong, Q.; Jiang, Y.; Liu, S.; Yang, Y.; Han, X.; Sheng, Y.; Zhang, Y.; Cheng, L.; et al. Prioritization of candidate cancer drugs based on a drug functional similarity network constructed by integrating pathway activities and drug activities. Mol. Oncol. 2019, 13, 2259–2277. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.J.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Varikuti, S.; Singh, B.; Volpedo, G.; Ahirwar, D.K.; Jha, B.K.; Saljoughian, N.; Viana, A.G.; Verma, C.; Hamza, O.; Halsey, G.; et al. Ibrutinib treatment inhibits breast cancer progression and metastasis by inducing conversion of myeloid-derived suppressor cells to dendritic cells. Br. J. Cancer 2020, 122, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, A.; Koffi, Y.; Diab, M.; Le Guennec, D.; Rougé, S.; Aldekwer, S.; Goncalves-Mendes, N.; Talvas, J.; Farges, M.; Caldefie-Chezet, F.; et al. The EO771 mammary cancer cell line displays a luminal B phenotype and it is sensitive to anti-estrogen treatments. Biorxiv 2019. [Google Scholar] [CrossRef]
- Zucha, M.A.; Wu, A.T.; Lee, W.H.; Wang, L.S.; Lin, W.W.; Yuan, C.C.; Yeh, C.T. Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib suppresses stem-like traits in ovarian cancer. Oncotarget 2015, 6, 13255–13268. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Higa, A.; Hoshi, H.; Hiyama, G.; Takahashi, N.; Ryufuku, M.; Morisawa, G.; Yanagisawa, Y.; Ito, E.; Imai, J.I.; et al. Evaluation of anticancer agents using patient-derived tumor organoids characteristically similar to source tissues. Oncol. Rep. 2018, 40, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Azzam, D.J.; Al-Ali, H.; Volmar, C.H.; Brothers, S.P.; Ince, T.A.; Wahlestedt, C. Ovarian Cancer Treatment Stratification Using Ex Vivo Drug Sensitivity Testing. Anticancer Res. 2019, 39, 4023–4030. [Google Scholar] [CrossRef] [PubMed]
- Berglof, A.; Hamasy, A.; Meinke, S.; Palma, M.; Krstić, A.; Månsson, R.; Kimby, E.; Österborg, A.; Smith, C.I.E. Targets for Ibrutinib Beyond B Cell Malignancies. Scand. J. Immunol. 2015, 82, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.M.; Kooijman, J.J.; De Roos, J.A.M.; Prinsen, M.B.W.; Dylus, J.; Willemsen-Seegers, N.; Kawase, Y.; Sawa, M.; De Man, J.; Van Gerwen, S.J.C.; et al. Combined Cellular and Biochemical Profiling to Identify Predictive Drug Response Biomarkers for Kinase Inhibitors Approved for Clinical Use between 2013 and 2017. Mol. Cancer Ther. 2019, 18, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Pan, W.-T.; Ma, Y.-B.; Xu, X.-L.; Gao, Y.; He, Y.-Q.; Wei, L.; Zhang, J.-W. BMX activates Wnt/β-catenin signaling pathway to promote cell proliferation and migration in breast cancer. Breast Cancer 2019, 27, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, N.; Zheng, P.-S.; Yang, W.-T. BMX/Etk promotes cell proliferation and tumorigenicity of cervical cancer cells through PI3K/AKT/mTOR and STAT3 pathways. Oncotarget 2017, 8, 49238–49252. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.L.; Storey, A. BMX Negatively Regulates BAK Function, Thereby Increasing Apoptotic Resistance to Chemotherapeutic Drugs. Cancer Res. 2015, 75, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Horna, P.; Sotomayor, E.M. Cellular and molecular mechanisms of tumor-induced T-cell tolerance. Curr. Cancer Drug Targets 2007, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Loose, D.; Van De Wiele, C. The Immune System and Cancer. Cancer Biother. Radiopharm. 2009, 24, 369–376. [Google Scholar] [CrossRef] [PubMed]

| First Author [Ref.] | Year | Study Type | Model | Histology | Outcome Measures | Main Outcomes |
|---|---|---|---|---|---|---|
| Grabinski | 2014 | Preclinical: in vitro | Cell culture | Breast cancer (HER2+) | Cell viability, phosphorylation of receptor tyrosine kinases | Significant reduction in the viability of HER2+ cell lines due to ibrutinib. IC50 values at nanomolar concentrations. Synergistic cell viability reduction of ibrutinib and PI3K/mTOR inhibitor dactolisib. Reduced MAPK and AKT phosphorylation. |
| Sagiv-Barfi | 2015 | Preclinical: in vitro + in vivo (animal) | Cell culture, xenograft mouse model | Breast cancer (TNBC) | Cell viability, tumor size, no. of metastases; Animal survival | Combination of ibrutinib and anti–PD-L1 inhibits growth of TNBC. The combination therapy generates specific antitumor T cells. |
| Chen | 2016 | Preclinical: in vitro + in vivo (animal) | Cell culture, xenograft mouse model | Breast cancer (Her2+) | Cell viability, tumor size, drug exposure | Inhibition of growth and suppression of key signaling pathways in HER2+ breast cancer cell lines. Clinically achievable drug levels suppress HER2+ human breast cancer growth in xenograft mouse models. |
| Stiff | 2016 | Preclinical: in vitro + in vivo (animal) | Cell culture, xenograft mouse model | Breast cancer (TNBC) | BTK expression, MDSCs frequency in vivo (by IHC), tumor size | Human and murine MDSCs express BTK. Ibrutinib modulates MDSCs’ cell function and generation and diminishes MDSCs in tumor-bearing mice. Ibrutinib potentially enhances immune-based therapies in solid malignancies. |
| Wang X | 2016 | Preclinical: in vitro + in vivo (mice) | Cell culture, xenograft mouse model | Breast cancer (Her2+) | Cell viability, tumor size | Ibrutinib is more potent in inhibiting HER2+ cell than lapatinib. Ibrutinib blocks EGFR, HER2, ErbB3, ErbB4 at its downstream effectors. |
| Di | 2019 | Preclinical: bioinformatics | Drug functional similarity network | Breast cancer, ovarian cancer | Prior score, false discovery rate | Ibrutinib ranks 3rd in the prioritized list of candidate breast cancer drugs and is considered to have great potential effects. |
| Hong | 2019 | Phase Ib/II | Human | Breast cancer (TNBC, HER2+), pancreatic cancer, NSCLC | Overall response rate, progression-free survival, overall survival | Recommended phase 2 dose: 560 mg ibrutinib daily, durvalumab 10 mg/kg i.v. q2w. ORR: 3% for breast cancer; 2% for pancreatic cancer; 0% for NSCLC. Limited antitumor activity; acceptable safety profile. |
| Varikuti | 2020 | Preclinical: in vivo (animal) | Cell culture, xenograft mouse model | Breast cancer (Luminal B) | Cell viability, tumor size, metastasis count, cell maturation analysis, T-cell proliferation and effector function | Ibrutinib inhibits tumor growth in vitro. Treated mice suffer lower tumor burden and less metastases. MDSCs switch phenotype to mature dendritic cells in vitro and less MDSCs and more DCs in vivo. Ibrutinib induces antitumor Th1 and CTL response. |
| First Author [Ref.] | Year | Study Type | Model | Histology | Outcome Measures | Main Outcomes |
|---|---|---|---|---|---|---|
| Zucha | 2015 | Pre-clinical: in vitro | Human tissue samples, cell culture, spheroids | Ovarian cancer | Cell viability, BTK expression | BTK is a histological biomarker and a prognostic predictor of ovarian cancer. Ovarian CSCs express BTK and contribute to cisplatin resistance. BTK inhibition targets CSCs and reduces their survival against cisplatin. Cisplatin–ibrutinib combination has synergistic effects in eliminating ovarian cancer cells. |
| Tamura | 2018 | Preclinical: in vitro | Cell culture, patient-derived tumor organoids | Endometrial cancer | Tumor size, inhibitory concentration | Ibrutinib inhibits the growth of carboplatin/paclitaxel-resistant cells at lower concentrations than carboplatin, paclitaxel, methotrexate, and vindesine. Less efficacy in clear-cell adenocarcinoma cell lines. |
| Di | 2019 | Preclinical: bioinformatics | Drug functional similarity network | Ovarian cancer, breast cancer | Prior score, false discovery rate | Low rank in ovarian cancer drug candidates. |
| Lohse | 2019 | Preclinical: in vitro | Drug sensitivity testing in patient-derived cell lines | Ovarian cancer | Modified drug sensitivity scoring | Weak effect on endometrioid and papillary-serous cell line. No effect on clear-cell cancer lines. |
| Ref. | Title | Phase/ Status | Brief Summary | Primary Outcomes |
|---|---|---|---|---|
| NCT02403271 [15] | A Multi-Center Study of Ibrutinib in Combination with MEDI4736 [Durvalumab] in Subjects With Relapsed or Refractory Solid Tumors | Phase Ib/II; completed 1 (Ref.: Results section, Hong et al., 2019 [41]) | A phase 1b/2, multi-center study to assess the safety and efficacy of ibrutinib in combination with durvalumab (MEDI4736) in participants with relapsed or refractory solid tumors. | Safety and tolerability, dosage, overall response rate per RECIST 1.1 |
| NCT03379428 [16] | Trial of Ibrutinib Plus Trastuzumab in HER2-Amplified Metastatic Breast Cancer | Phase I/II; recruiting 1 | Open-label dose-escalation study to evaluate the maximum-tolerated dose and dose-limiting side effects of daily oral ibrutinib in combination with trastuzumab i.v. q3w, in patients with HER2-amplified metastatic breast cancer with progress after prior therapy with T-DM1. | Maximum-tolerated dose, clinical benefit rate |
| NCT03525925 [17] | Ibrutinib and Nivolumab in Treating Participants with Metastatic Solid Tumors | Phase I; active, not recruiting 1 | A phase I trial investigating the effect of ibrutinib and nivolumab on circulating levels of MDSCs in patients with metastatic solid tumors and assessing the safety of the study combination. | Circulating levels of myeloid-derived suppressor cells |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metzler, J.M.; Burla, L.; Fink, D.; Imesch, P. Ibrutinib in Gynecological Malignancies and Breast Cancer: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 4154. https://doi.org/10.3390/ijms21114154
Metzler JM, Burla L, Fink D, Imesch P. Ibrutinib in Gynecological Malignancies and Breast Cancer: A Systematic Review. International Journal of Molecular Sciences. 2020; 21(11):4154. https://doi.org/10.3390/ijms21114154
Chicago/Turabian StyleMetzler, Julian Matthias, Laurin Burla, Daniel Fink, and Patrick Imesch. 2020. "Ibrutinib in Gynecological Malignancies and Breast Cancer: A Systematic Review" International Journal of Molecular Sciences 21, no. 11: 4154. https://doi.org/10.3390/ijms21114154
APA StyleMetzler, J. M., Burla, L., Fink, D., & Imesch, P. (2020). Ibrutinib in Gynecological Malignancies and Breast Cancer: A Systematic Review. International Journal of Molecular Sciences, 21(11), 4154. https://doi.org/10.3390/ijms21114154