Stable Fibroblast Growth Factor 2 Dimers with High Pro-Survival and Mitogenic Potential

 , , and
, , and
Abstract
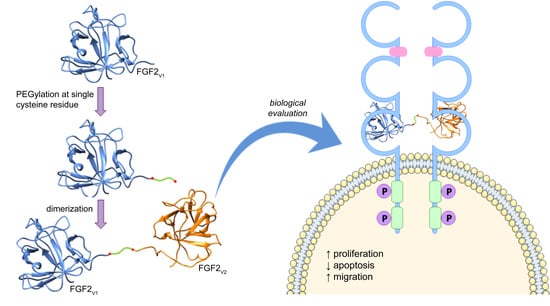
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
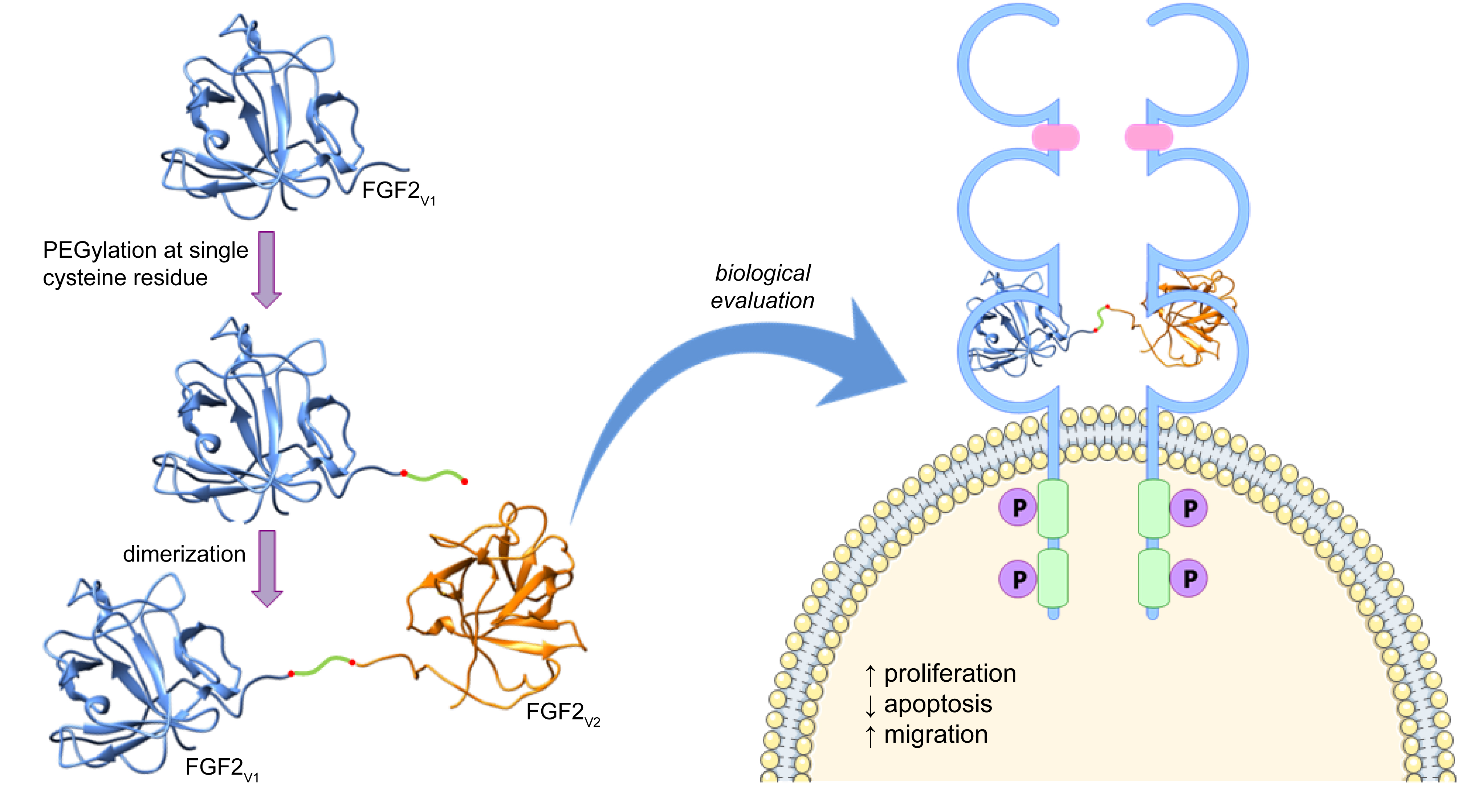
2.1. Design of FGF2 Dimers
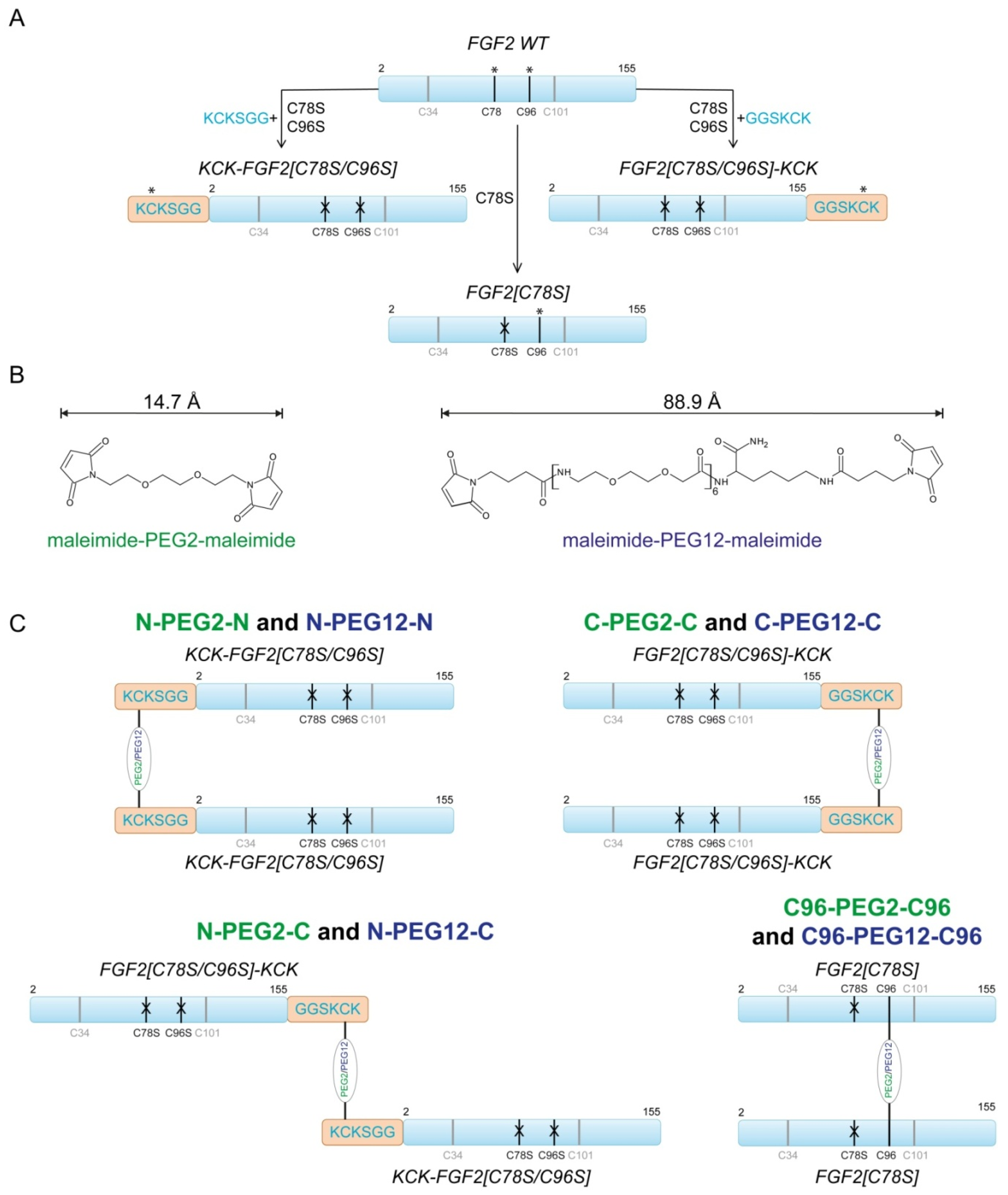
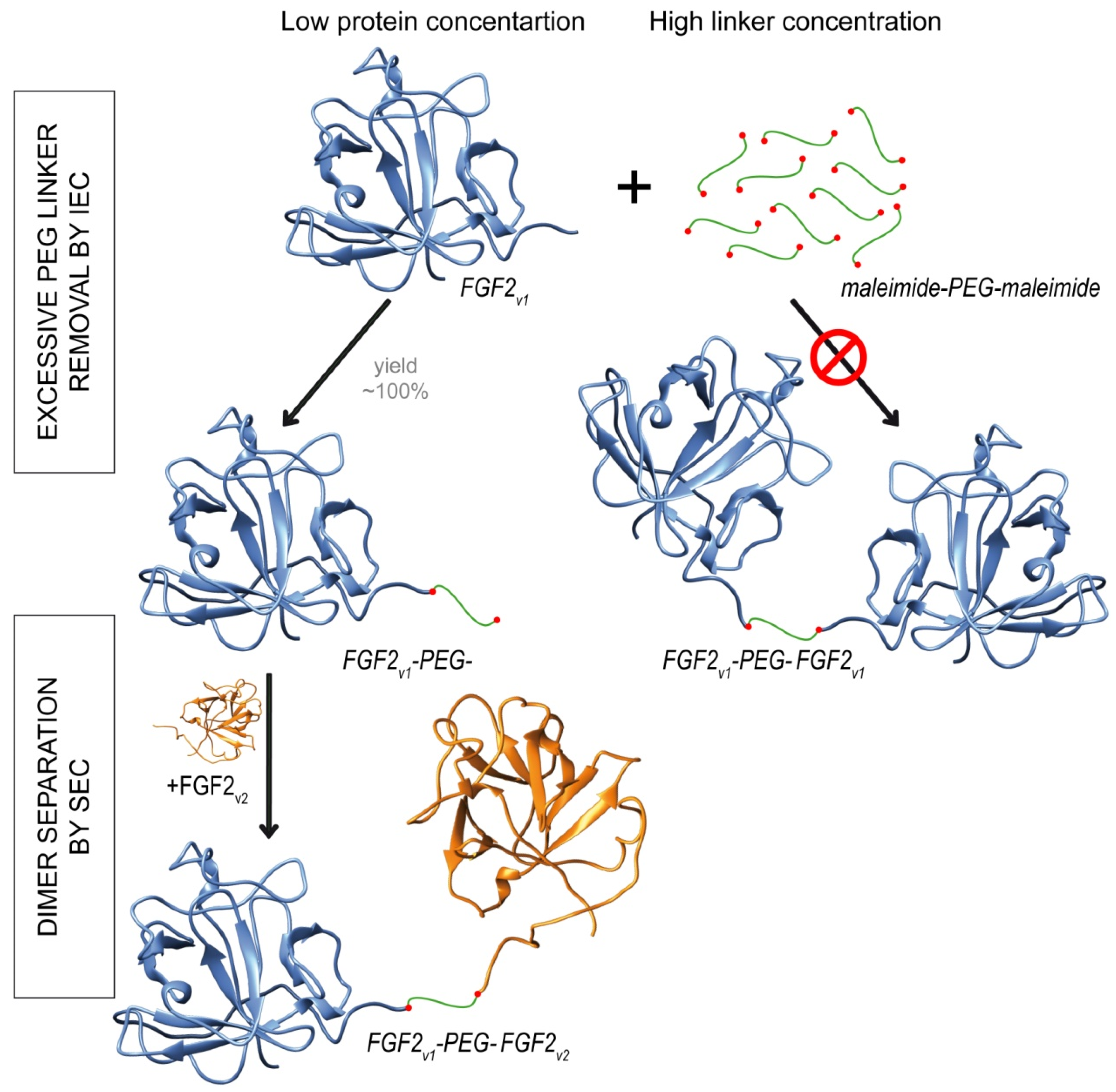
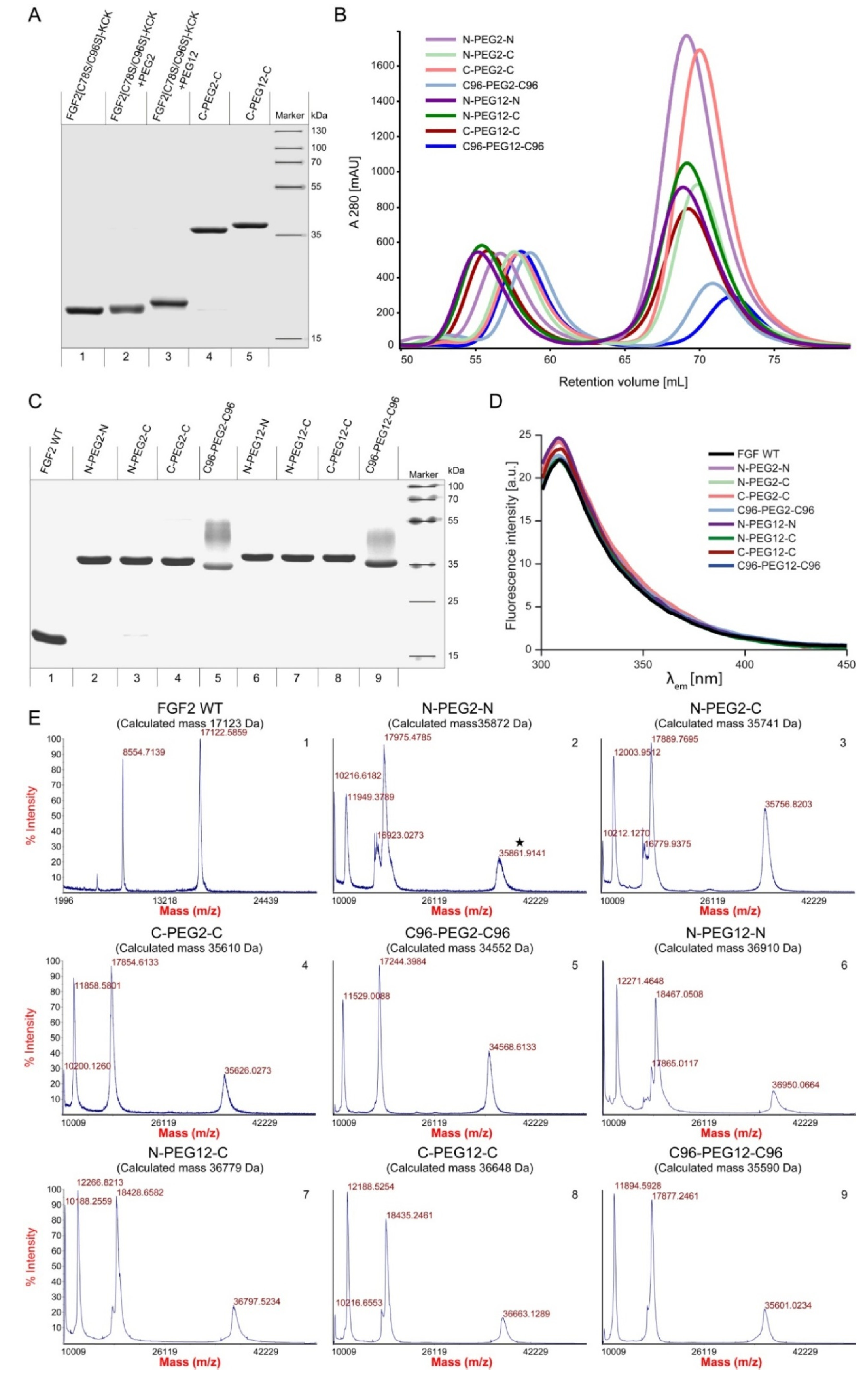
2.2. Synthesis of Dimeric FGF2 Variants
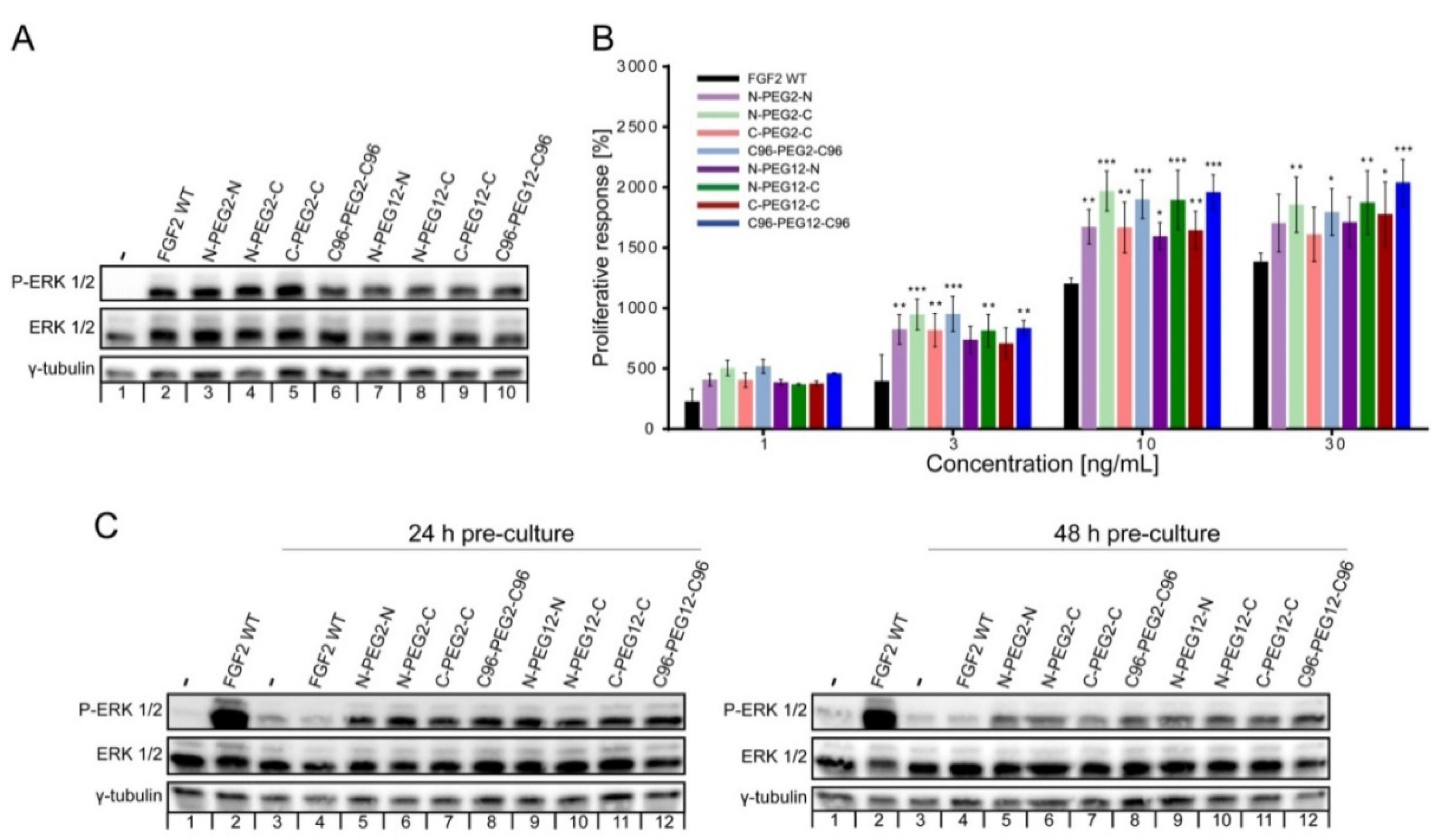
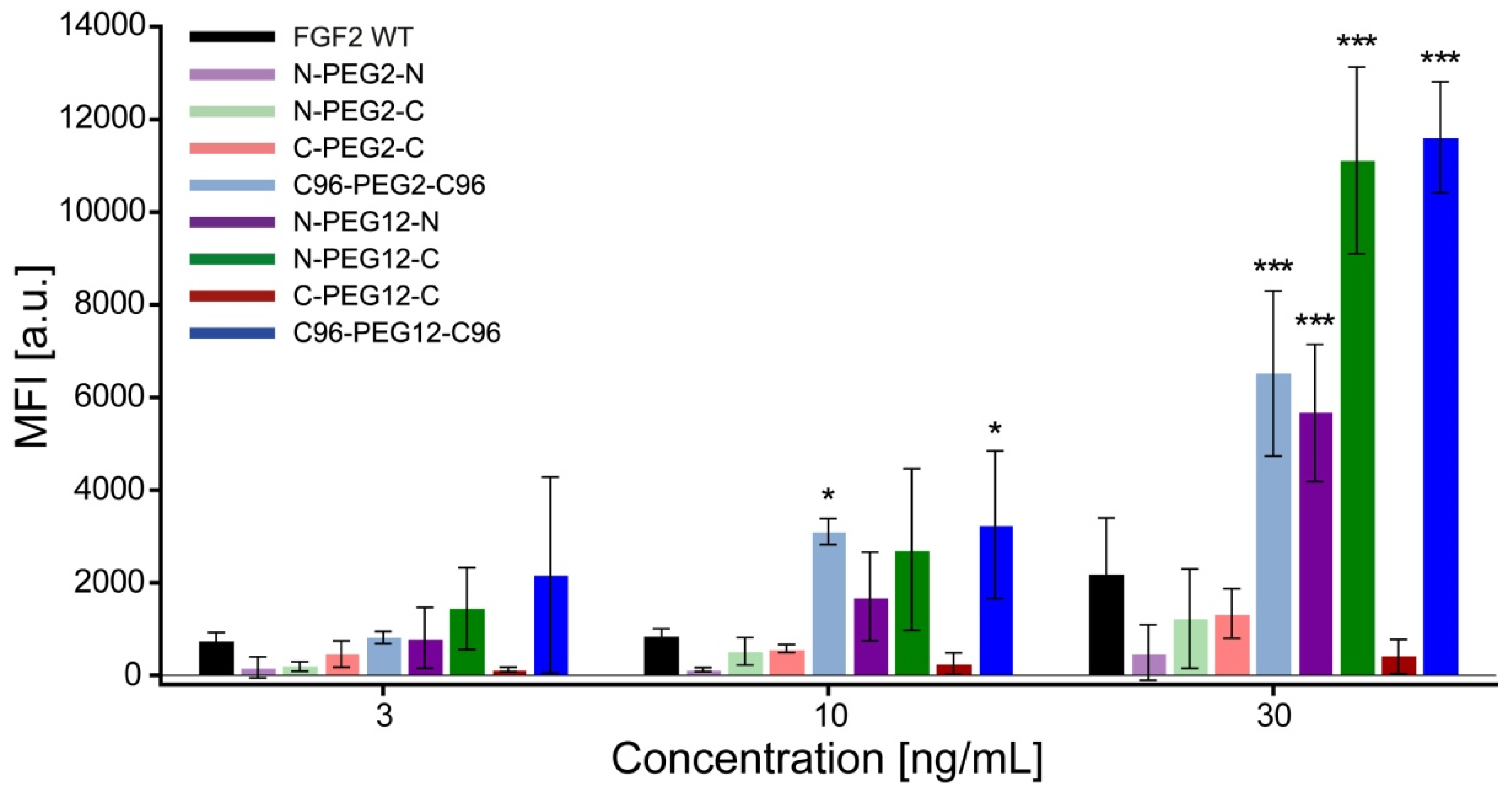
2.3. Biological Activities of FGF2 Dimer
2.4. Mitogenic Potential of FGF2 Dimers
2.5. Susceptibility of FGF2 Dimers to Degradation
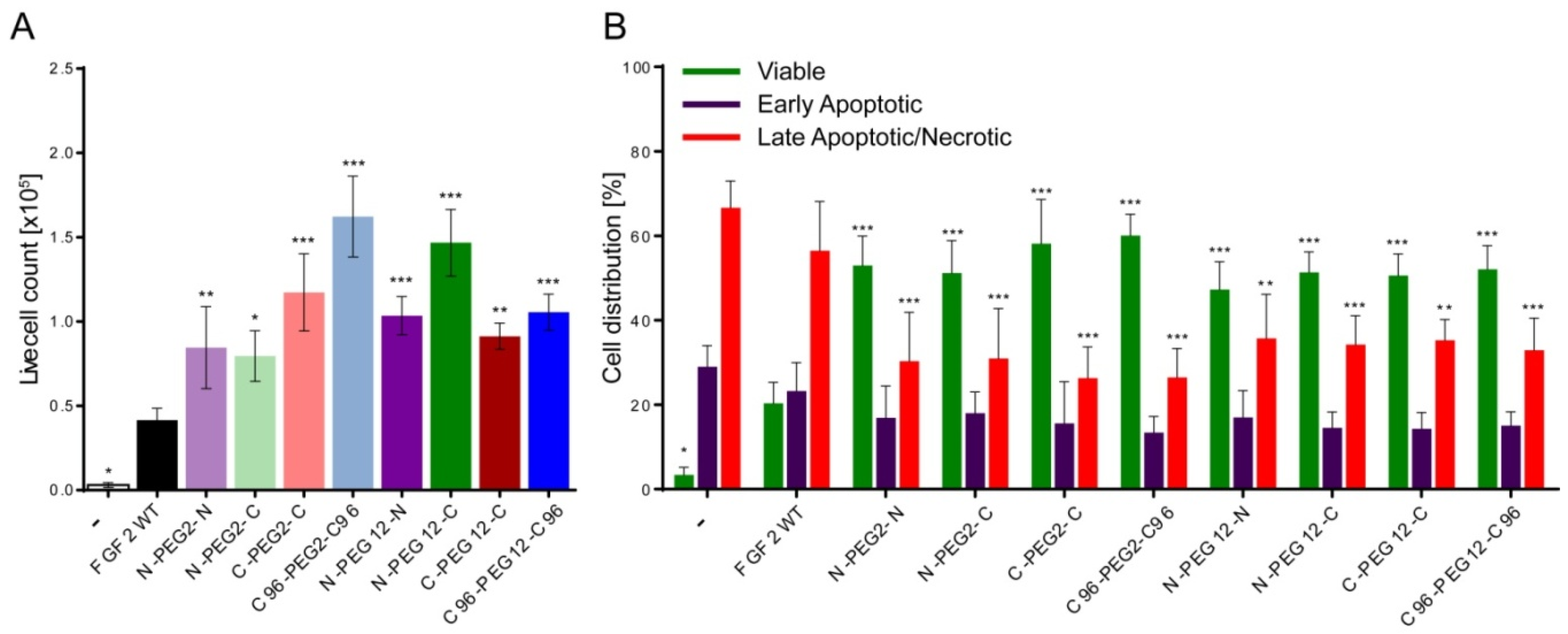
2.6. Anti-Apoptotic Activity of FGF2 Dimers
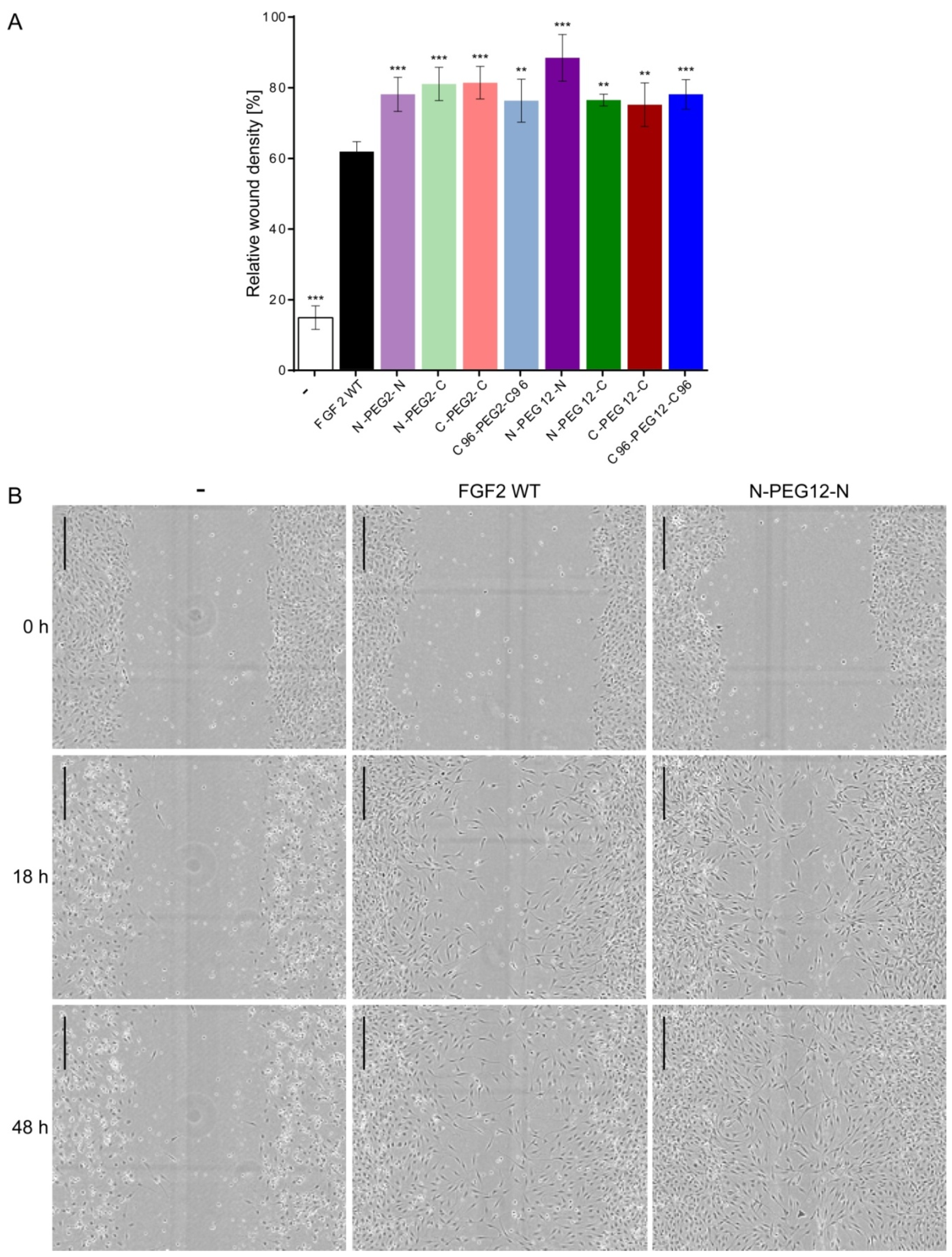
2.7. Induction of Cell Migration by FGF2 Variants
2.8. Increased Internalization of Dimeric Variants
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Lines
4.3. Protein Expression and Purification
4.4. Synthesis and Purification of maleimide-PEG12-maleimide Linker
4.5. Synthesis of FGF2 Dimers
4.6. Spectrofluorimetry
4.7. Mass Spectrometry
4.8. Activation of Signaling Pathways
4.9. Protein Stability in Cell-Conditioned Medium
4.10. Cell Proliferative Activity Assay
4.11. Cell Counting
4.12. Detection of Apoptosis
4.13. Cell Migration Assay
4.14. Protein Labeling with Fluorescent Probe
4.15. Flow Cytometric Analysis of Steady-State Internalization
4.16. Bio-Layer Interferometry (BLI) Analysis of Binding of Alexa Fluor 488-Labeled C-PEG12-C Dimer to FGFR1c
4.17. Evaluation of Biological Competence of AF488-Labeled C-PEG12-C Dimer
4.18. Fluorescence Microscopy: Internalization of FGF2 Dimers into Cells Overproducing FGFR1
4.19. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FGF | fibroblast growth factor |
| FGFR | fibroblast growth factor receptor |
| RTK | receptor tyrosine kinase |
| HSPG | heparan sulfate proteoglycans |
| SEC | size exclusion chromatography |
| IEC | ion exchange chromatography |
| PEG | poly(ethylene glycol) |
| Ado | 8-amino-3,6-dioxaoctanoyl |
| MMAE | monomethyl auristatin E |
References
- Yun, Y.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.-H.; Shin, U.S.; Kim, H. Fibroblast Growth Factors: Biology, Function, and Application for Tissue Regeneration. J. Tissue Eng. 2010, 1, 218142. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Schlessinger, J.; Hubbard, S.R.; Mohammadi, M. Structural Basis for FGF Receptor Dimerization and Activation. Cell 1999, 98, 641–650. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal Structure of a Ternary FGF-FGFR-Heparin Complex Reveals a Dual Role for Heparin in FGFR Binding and Dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Marianayagam, N.J.; Sunde, M.; Matthews, J.M. The power of two: Protein dimerization in biology. Trends Biochem. Sci. 2004, 29, 618–625. [Google Scholar] [CrossRef]
- Kramer, R.H.; Karpen, J.W. Spanning binding sites on allosteric proteins with polymer-linked ligand dimers. Nature 1998, 395, 710–713. [Google Scholar] [CrossRef]
- Safran, M.; Eisenstein, M.; Aviezer, D.; Yayon, A. Oligomerization reduces heparin affinity but enhances receptor binding of fibroblast growth factor 2. Biochem. J. 2000, 345 Pt 1, 107–113. [Google Scholar] [CrossRef]
- Kwan, C.; Venkataraman, G.; Shriver, Z.; Raman, R.; Liu, D.; Qi, Y.; Varticovski, L.; Sasisekharan, R. Probing Fibroblast Growth Factor Dimerization and Role of Heparin-like Glycosaminoglycans in Modulating Dimerization and Signaling. J. Biol. Chem. 2001, 276, 23421–23429. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.G.; Wang, Y.; Paluck, S.J.; Shen, L.; Loo, J.A.; Levine, A.J.; Miller, L.S.; Maynard, H.D. Fibroblast growth factor 2 dimer with superagonist in vitro activity improves granulation tissue formation during wound healing. Biomaterials 2016, 81, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Stauber, D.J.; DiGabriele, A.D.; Hendrickson, W.A. Structural interactions of fibroblast growth factor receptor with its ligands. Proc. Natl. Acad. Sci. USA 2000, 97, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Herr, A.B.; Ornitz, D.M.; Sasisekharan, R.; Venkataraman, G.; Waksman, G. Heparin-induced self-association of fibroblast growth factor-2. Evidence for two oligomerization processes. J. Biol. Chem. 1997, 272, 16382–16389. [Google Scholar] [CrossRef]
- Venkataraman, G.; Sasisekharan, V.; Herr, A.B.; Ornitz, D.M.; Waksman, G.; Cooney, C.L.; Langer, R.; Sasisekharan, R. Preferential self-association of basic fibroblast growth factor is stabilized by heparin during receptor dimerization and activation. Proc. Natl. Acad. Sci. USA 1996, 93, 845–850. [Google Scholar] [CrossRef]
- Davis, J.C.; Venkataraman, G.; Shriver, Z.; Raj, P.A.; Sasisekharan, R. Oligomeric self-association of basic fibroblast growth factor in the absence of heparin-like glycosaminoglycans. Biochem. J. 1999, 341 Pt 3, 613–620. [Google Scholar] [CrossRef]
- Krzyscik, M.A.; Zakrzewska, M.; Sørensen, V.; Sokolowska-Wedzina, A.; Lobocki, M.; Swiderska, K.W.; Krowarsch, D.; Wiedlocha, A.; Otlewski, J. Cytotoxic Conjugates of Fibroblast Growth Factor 2 (FGF2) with Monomethyl Auristatin E for Effective Killing of Cells Expressing FGF Receptors. ACS Omega 2017, 2, 3792–3805. [Google Scholar] [CrossRef]
- Lobocki, M.; Zakrzewska, M.; Szlachcic, A.; Krzyscik, M.A.; Sokolowska-Wedzina, A.; Otlewski, J. High-Yield Site-Specific Conjugation of Fibroblast Growth Factor 1 with Monomethylauristatin E via Cysteine Flanked by Basic Residues. Bioconjug. Chem. 2017, 28, 1850–1858. [Google Scholar] [CrossRef]
- Boilly, B.; Vercoutter-Edouart, A.S.; Hondermarck, H.; Nurcombe, V.; Le Bourhis, X. FGF signals for cell proliferation and migration through different pathways. Cytokine Growth Factor Rev. 2000, 11, 295–302. [Google Scholar] [CrossRef]
- LaVallee, T.M.; Prudovsky, I.A.; McMahon, G.A.; Hu, X.; Maciag, T. Activation of the MAP Kinase Pathway by FGF-1 Correlates with Cell Proliferation Induction While Activation of the Src Pathway Correlates with Migration. J. Cell Biol. 1998, 141, 1647–1658. [Google Scholar] [CrossRef]
- Szlachcic, A.; Sochacka, M.; Czyrek, A.; Opalinski, L.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. Low Stability of Integrin-Binding Deficient Mutant of FGF1 Restricts Its Biological Activity. Cells 2019, 8, 899. [Google Scholar] [CrossRef]
- Bruno, M.; Rizzo, I.M.; Romero-Guevara, R.; Bernacchioni, C.; Cencetti, F.; Donati, C.; Bruni, P. Sphingosine 1-phosphate signaling axis mediates fibroblast growth factor 2-induced proliferation and survival of murine auditory neuroblasts. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 814–824. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, F.; Yu, X.; Chen, C.; Meng, J.; Zhong, R.; Zhang, Y.; Zhu, D. Linc-RAM is required for FGF2 function in regulating myogenic cell differentiation. RNA Biol. 2018, 15, 404–412. [Google Scholar] [CrossRef]
- Awan, B.; Turkov, D.; Schumacher, C.; Jacobo, A.; McEnerney, A.; Ramsey, A.; Xu, G.; Park, D.; Kalomoiris, S.; Yao, W.; et al. FGF2 Induces Migration of Human Bone Marrow Stromal Cells by Increasing Core Fucosylations on N-Glycans of Integrins. Stem Cell Rep. 2018, 11, 325–333. [Google Scholar] [CrossRef]
- Nunes, Q.M.; Li, Y.; Sun, C.; Kinnunen, T.K.; Fernig, D.G. Fibroblast growth factors as tissue repair and regeneration therapeutics. PeerJ 2016, 4, e1535. [Google Scholar] [CrossRef]
- Akita, S.; Akino, K.; Imaizumi, T.; Hirano, A. Basic fibroblast growth factor accelerates and improves second-degree burn wound healing. Wound Repair Regen. 2008, 16, 635–641. [Google Scholar] [CrossRef]
- Matsumine, H. Treatment of Skin Avulsion Injuries with Basic Fibroblast Growth Factor. Plast. Reconstr. Surg. Glob. Open 2015, 3, e371. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Oka, H.; Jingushi, S.; Izumi, T.; Fukunaga, M.; Sato, K.; Matsushita, T.; Nakamura, K. A local application of recombinant human fibroblast growth factor 2 for tibial shaft fractures: A randomized, placebo-controlled trial. J. Bone Miner. Res. 2010, 25, 2735–2743. [Google Scholar] [CrossRef]
- Kitamura, M.; Akamatsu, M.; Machigashira, M.; Hara, Y.; Sakagami, R.; Hirofuji, T.; Hamachi, T.; Maeda, K.; Yokota, M.; Kido, J.; et al. FGF-2 Stimulates Periodontal Regeneration. J. Dent. Res. 2011, 90, 35–40. [Google Scholar] [CrossRef]
- Uchi, H.; Igarashi, A.; Urabe, K.; Koga, T.; Nakayama, J.; Kawamori, R.; Tamaki, K.; Hirakata, H.; Ohura, T.; Furue, M. Clinical efficacy of basic fibroblast growth factor (bFGF) for diabetic ulcer. Eur. J. Dermatol. 2009, 19, 461–468. [Google Scholar] [CrossRef]
- Kumagai, M.; Marui, A.; Tabata, Y.; Takeda, T.; Yamamoto, M.; Yonezawa, A.; Tanaka, S.; Yanagi, S.; Ito-Ihara, T.; Ikeda, T.; et al. Safety and efficacy of sustained release of basic fibroblast growth factor using gelatin hydrogel in patients with critical limb ischemia. Heart Vessel. 2016, 31, 713–721. [Google Scholar] [CrossRef]
- Carter, E.P.; Fearon, A.E.; Grose, R.P. Careless talk costs lives: Fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015, 25, 221–233. [Google Scholar] [CrossRef]
- Estapé, D.; van denHeuvel, J.; Rinas, U. Susceptibility towards intramolecular disulphide-bond formation affects conformational stability and folding of human basic fibroblast growth factor. Biochem. J. 1998, 335, 343–349. [Google Scholar] [CrossRef]
- Sanz, J.M.; Gimenez-Gallego, G. A partly Folded State of Acidic Fibroblast Growth Factor at Low Ph. Eur. J. Biochem. 1997, 246, 328–335. [Google Scholar] [CrossRef]
- Galvan, L. Effects of heparin on wound healing*1. J. WOCN 1996, 23, 224–226. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef]
- Schmitz, K.; Schildhaus, H.-U. Clinical significance of FGFR1 gene amplification in lung cancer patients. Lung Cancer Manag. 2014, 3, 305–314. [Google Scholar] [CrossRef]
- Ducry, L.; Stump, B. Antibody–Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef]
- Li, N.; Hill, K.S.; Elferink, L.A. Analysis of Receptor Tyrosine Kinase Internalization Using Flow Cytometry. In Membrane Trafficking; Humana Press: Totowa, NJ, USA, 2008; pp. 305–317. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nawrocka, D.; Krzyscik, M.A.; Opaliński, Ł.; Zakrzewska, M.; Otlewski, J. Stable Fibroblast Growth Factor 2 Dimers with High Pro-Survival and Mitogenic Potential. Int. J. Mol. Sci. 2020, 21, 4108. https://doi.org/10.3390/ijms21114108
Nawrocka D, Krzyscik MA, Opaliński Ł, Zakrzewska M, Otlewski J. Stable Fibroblast Growth Factor 2 Dimers with High Pro-Survival and Mitogenic Potential. International Journal of Molecular Sciences. 2020; 21(11):4108. https://doi.org/10.3390/ijms21114108
Chicago/Turabian StyleNawrocka, Daria, Mateusz Adam Krzyscik, Łukasz Opaliński, Malgorzata Zakrzewska, and Jacek Otlewski. 2020. "Stable Fibroblast Growth Factor 2 Dimers with High Pro-Survival and Mitogenic Potential" International Journal of Molecular Sciences 21, no. 11: 4108. https://doi.org/10.3390/ijms21114108
APA StyleNawrocka, D., Krzyscik, M. A., Opaliński, Ł., Zakrzewska, M., & Otlewski, J. (2020). Stable Fibroblast Growth Factor 2 Dimers with High Pro-Survival and Mitogenic Potential. International Journal of Molecular Sciences, 21(11), 4108. https://doi.org/10.3390/ijms21114108