Complex Regulatory Role of the TRPA1 Receptor in Acute and Chronic Airway Inflammation Mouse Models

 ,
, 
Abstract
1. Introduction
2. Results
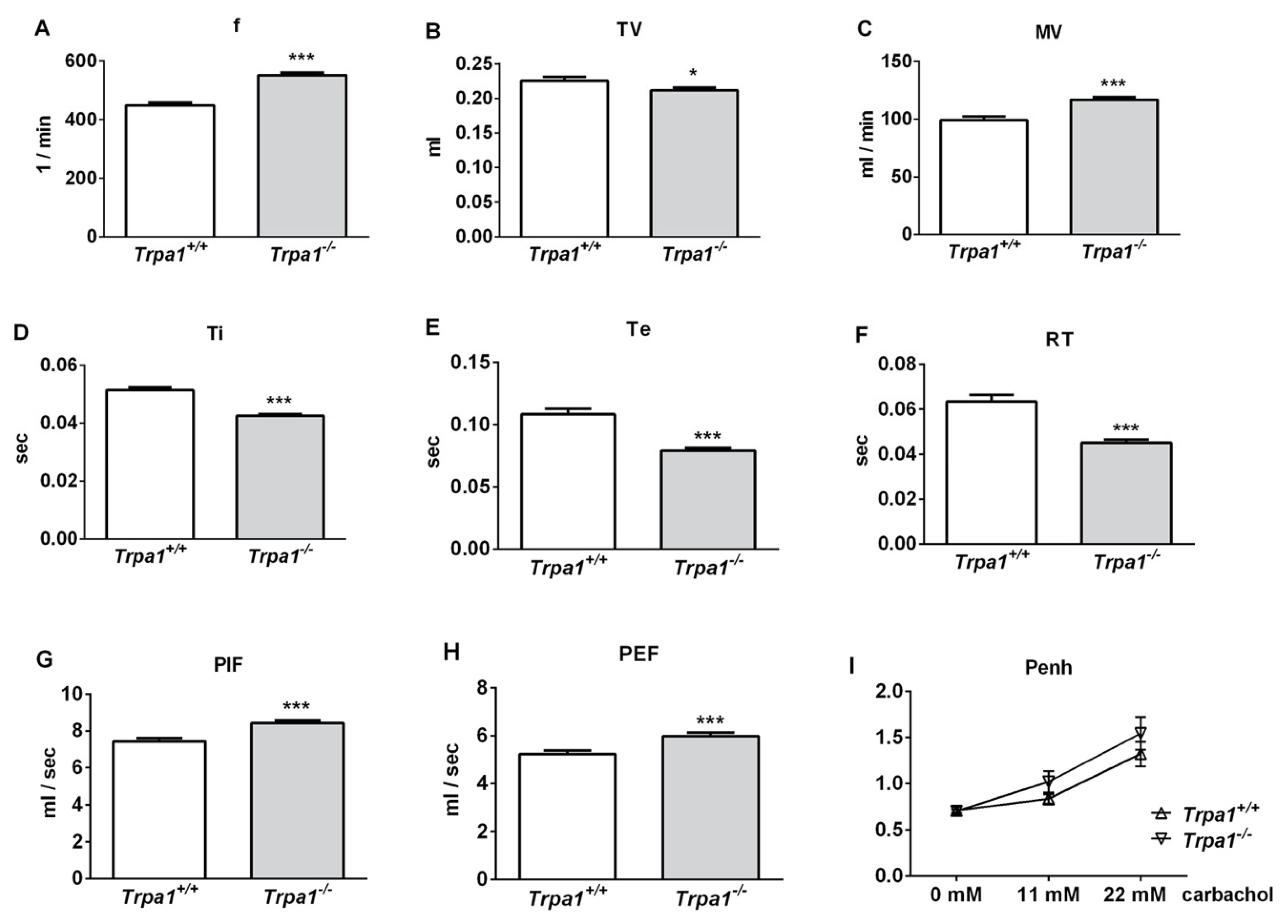
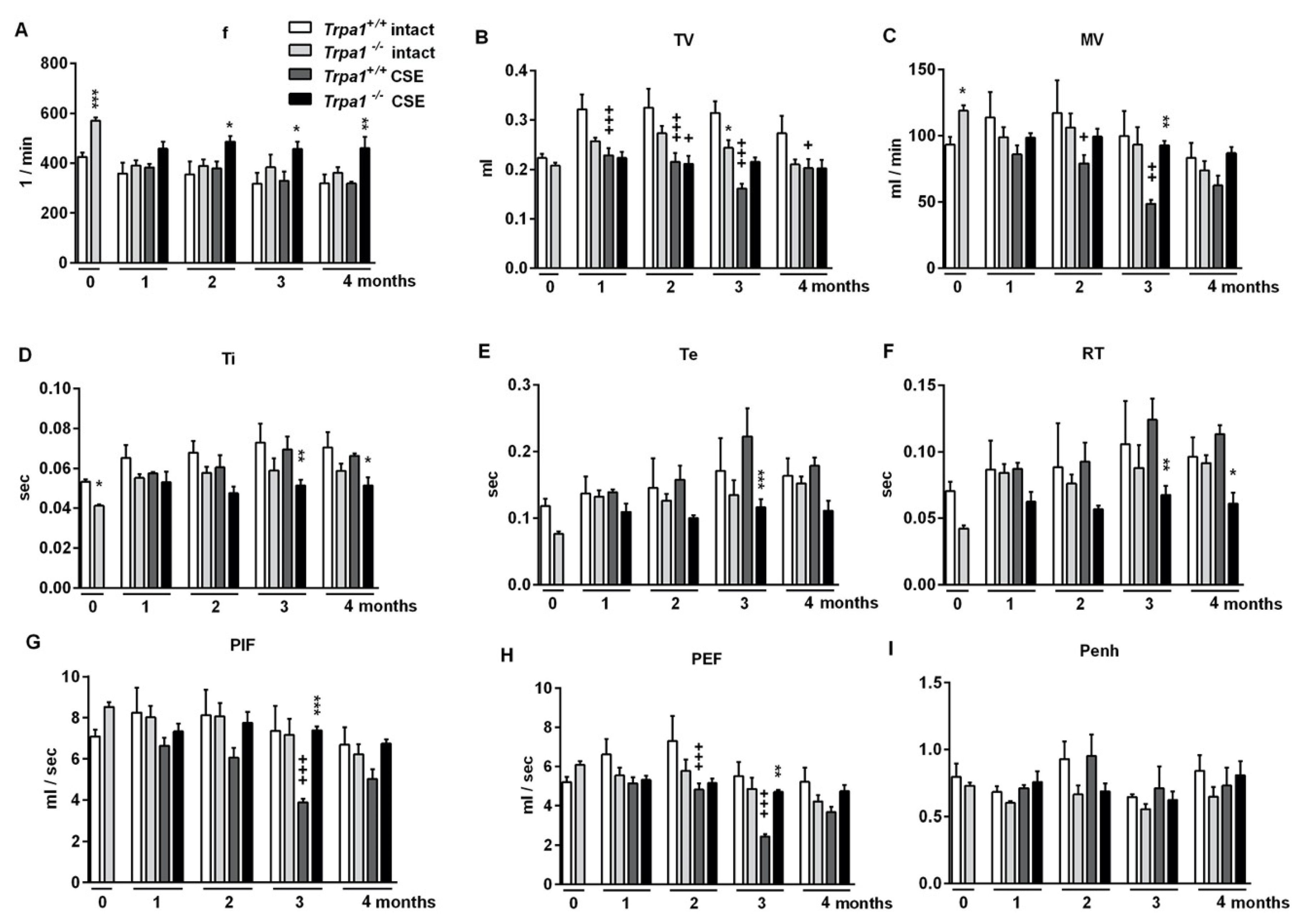
2.1. Differences in Basal Airway Function Parameters of Trpa1+/+ and Trpa1−/− Mice
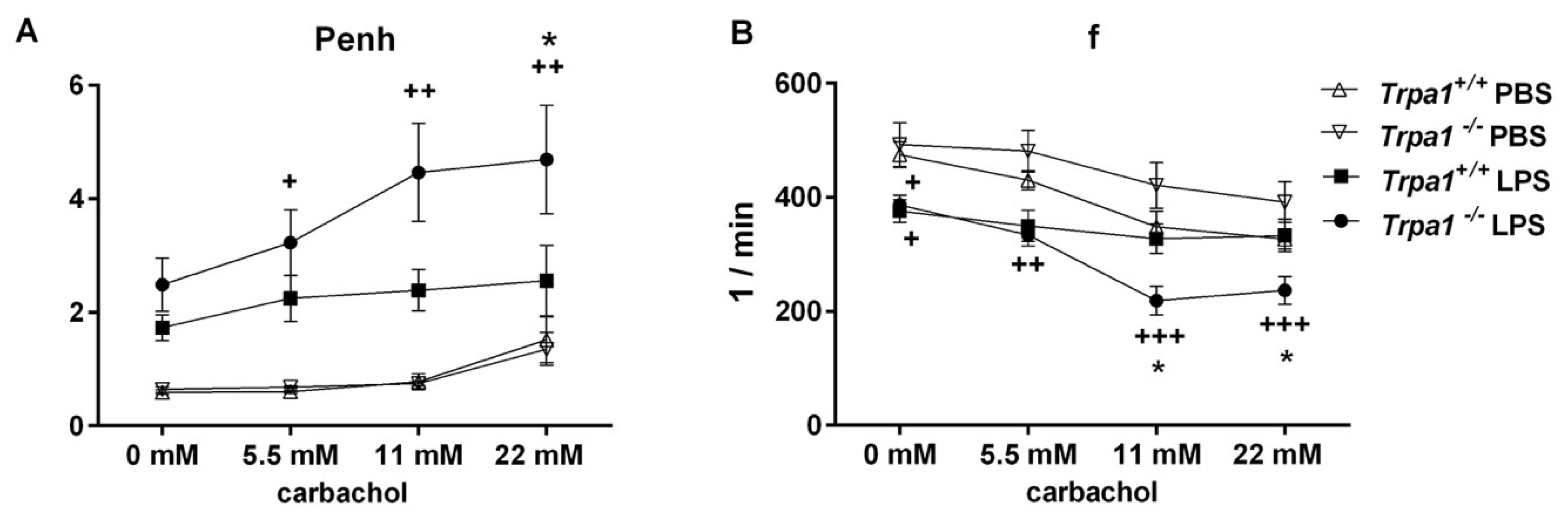
2.2. Endotoxin-Induced Bronchial Hyperreactivity is Greater in Trpa1−/− Mice
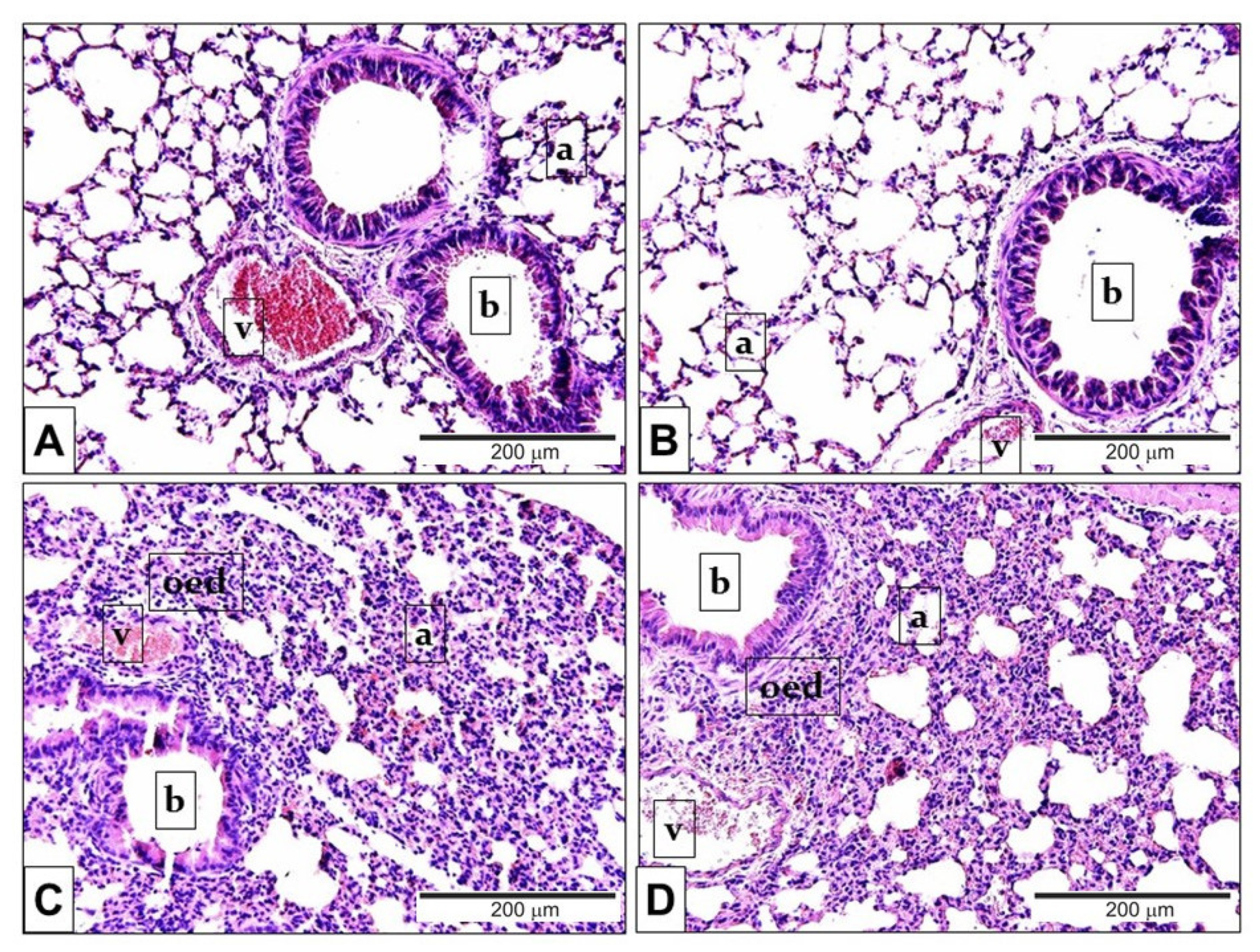
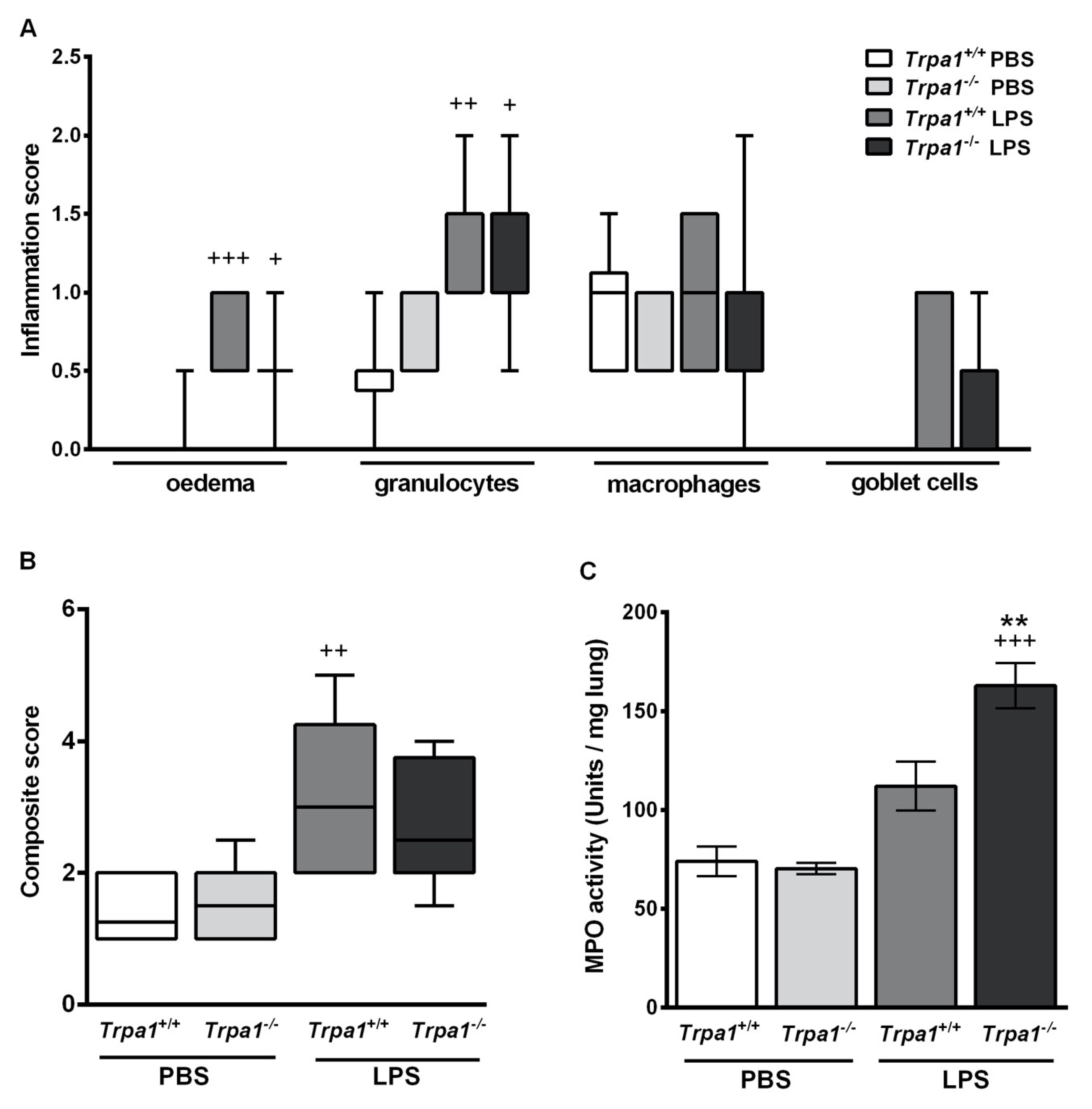
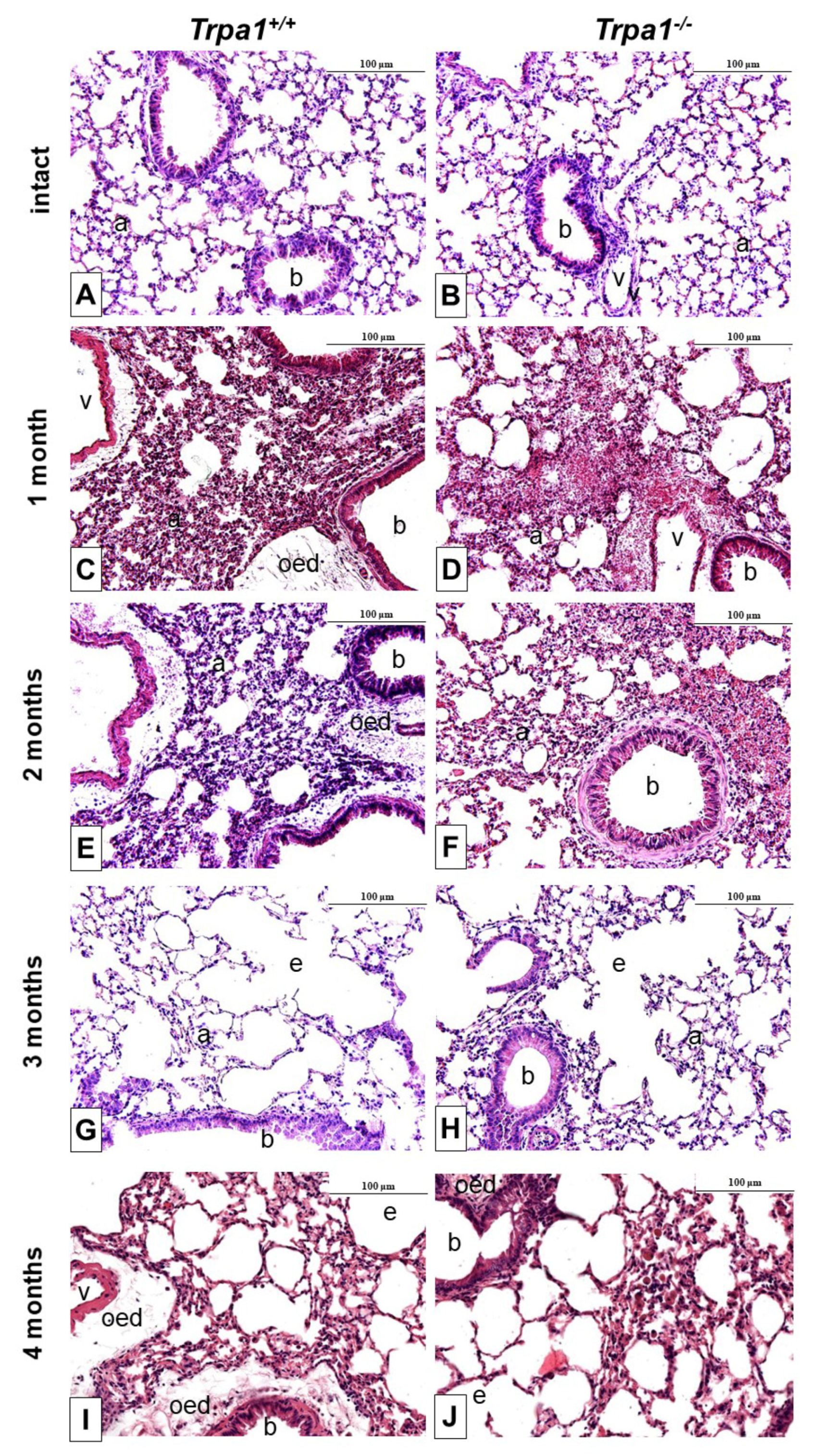
2.3. Endotoxin-Induced Histological Alterations Show No Difference in the Case of TRPA1-Deficiency
2.4. Endotoxin-Induced Myeloperoxidase Activity in the Lung is Greater in Trpa1−/− Mice
2.5. Chronic Cigarette Smoke Induces Respiratory Function Alterations in Trpa1+/+, but not in Trpa1−/− Mice
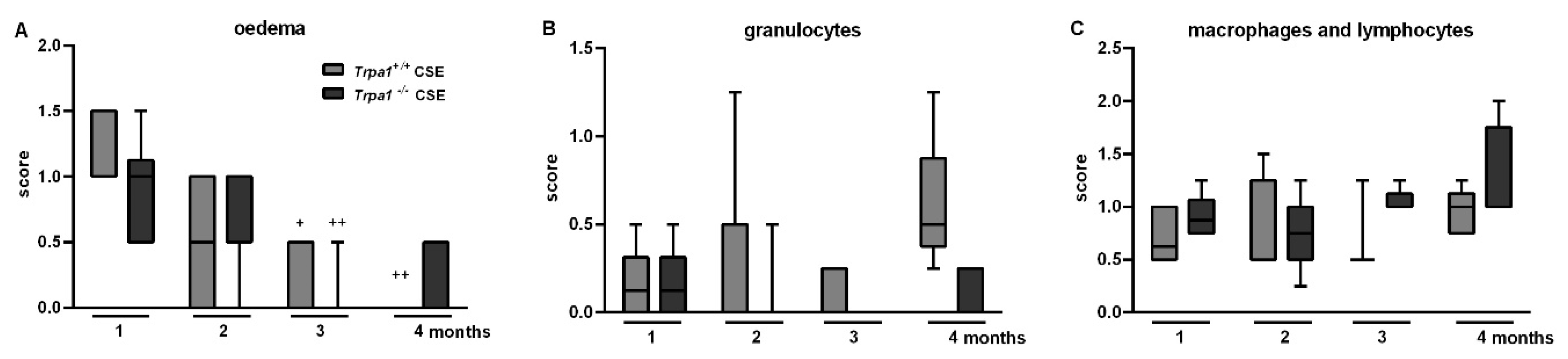
2.6. CSE-Induced Inflammatory Histopathological Alterations are Similar in Trpa1+/+ and Trpa1−/− Mice
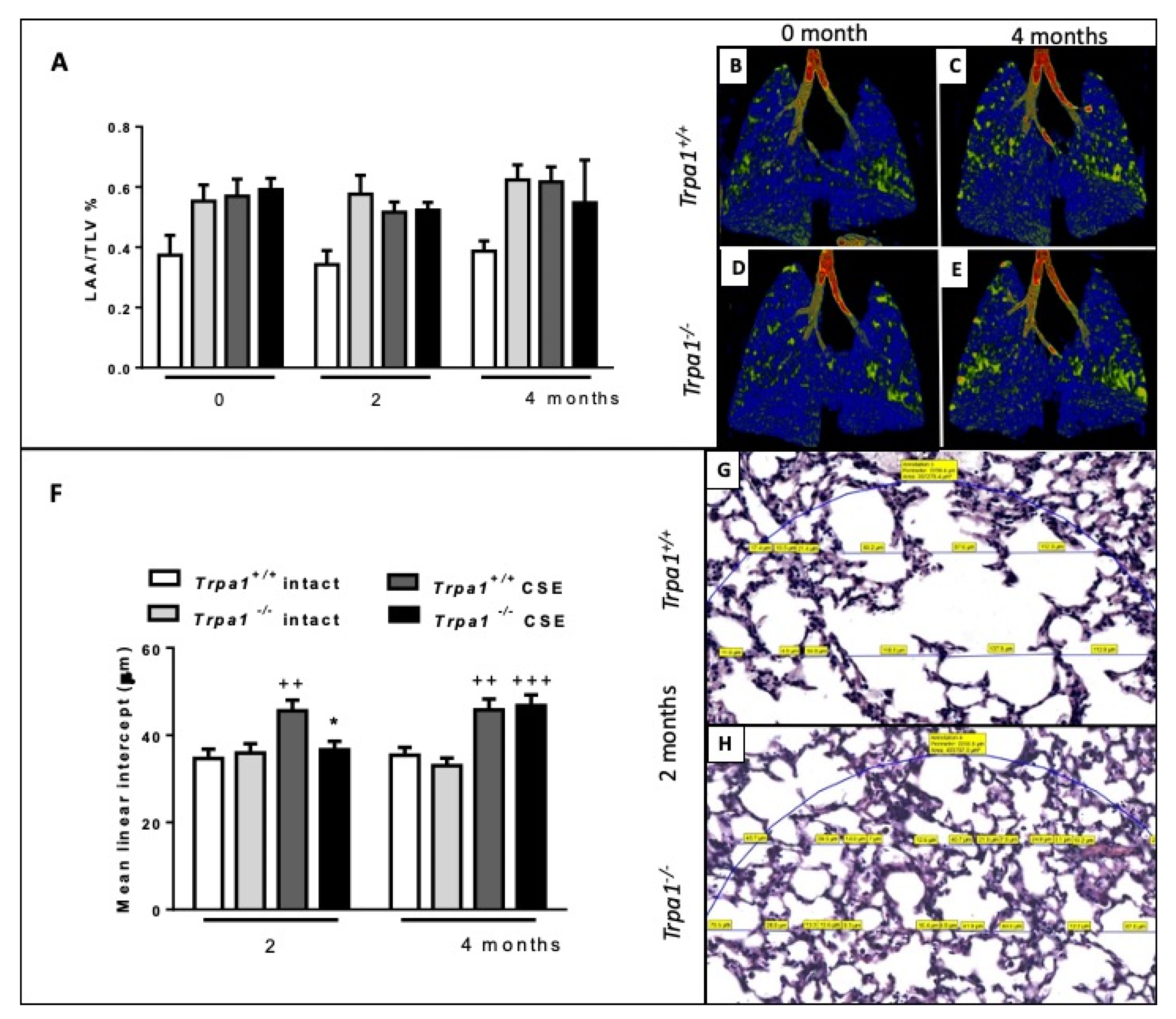
2.7. Cigarette Smoke-Induced Emphysema Develops Earlier in Trpa1+/+ Mice
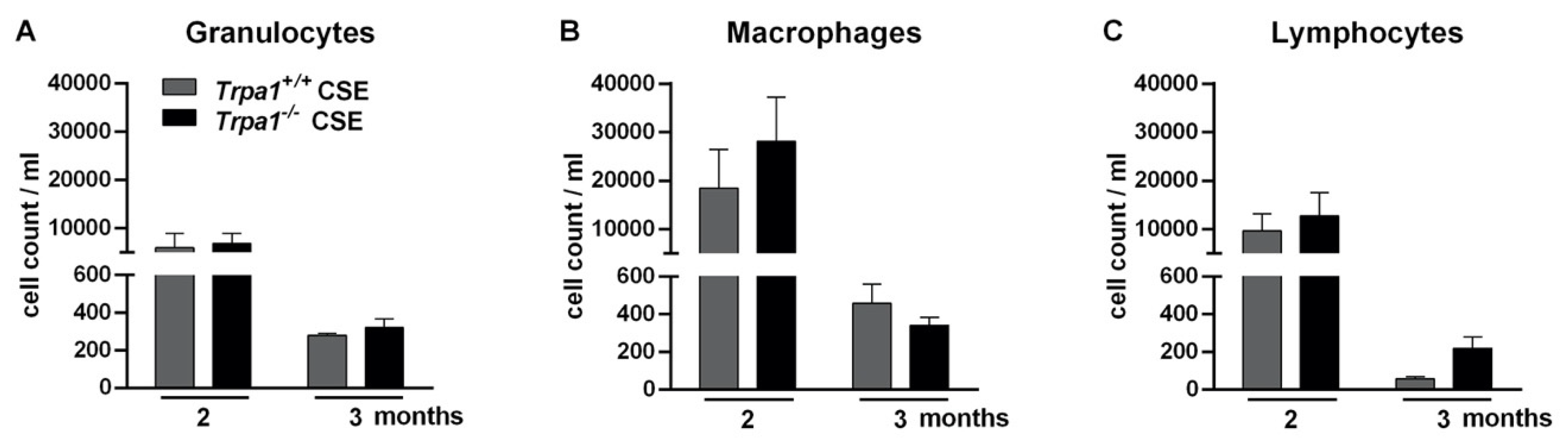
2.8. Chronic Cigarette Smoke-Induced Inflammatory Cell Accumulation in the BALF Shows No Strain Difference
3. Discussion and Conclusion
4. Methods
4.1. Animals
4.2. Ethics
4.3. Endotoxin-Induced Acute Pneumonitis
4.4. Cigarette Smoke-Induced Chronic Airway Inflammation
4.5. Investigation of Airway Function and Bronchial Responsiveness
4.6. Cell Composition Analysis of the Bronchoalveolar Lavage Fluid (BALF)
4.7. Histological Examination of the Lungs
4.8. Morphological Evaluation of the Lungs with In Vivo MicroCT Investigation
4.9. Determination of Myeloperoxidase (MPO) Activity from Lung Homogenates with Spectrophotometry
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nassenstein, C.; Kwong, K.; Taylor-Clark, T.; Kollarik, M.; MacGlashan, D.M.; Braun, A.; Undem, B.J. Expression and function of the ion channel TRPA1 in vagal afferent nerves innervating mouse lungs. J. Physiol. 2008, 586, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Högestätt, E.D. TRPA1. In Mammalian Transient Receptor Potential (TRP) Cation Channels; Nilius, B., Flockerzi, V., Eds.; Mammalian; Springer: Berlin/Heidelberg, Germany, 2014; ISBN 9783642542152. [Google Scholar]
- Gaudet, R. A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 2008, 4, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Morales, J.F.; Gracheva, E.O.; Julius, D. Cytoplasmic ankyrin repeats of transient receptor potential A1 (TRPA1) dictate sensitivity to thermal and chemical stimuli. Proc. Natl. Acad. Sci. USA 2011, 108, E1184–E1191. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, V.; Alpizar, Y.A.; Luis, E.; Tajada, S.; Denlinger, B.; Fajardo, O.; Manenschijn, J.-A.; Fernández-Pena, C.; Talavera, A.; Kichko, T.; et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Andrè, E.; Campi, B.; Materazzi, S.; Trevisani, M.; Amadesi, S.; Massi, D.; Creminon, C.; Vaksman, N.; Nassini, R.; Civelli, M.; et al. Cigarette smoke—Induced neurogenic inflammation is mediated by α,β-unsaturated aldehydes and the TRPA1 receptor in rodents. J. Clin. Investig. 2008, 118, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Bessac, B.F.; Jordt, S.-E. Breathtaking TRP Channels: TRPA1 and TRPV1 in Airway Chemosensation and Reflex Control. Physiology 2008, 23, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Baumlin, N.; Dennis, J.S.; Moore, R.; Salathe, S.F.; Whitney, P.L.; Sabater, J.; Abraham, W.M.; Kim, M.D.; Salathe, M. Electronic cigarette vapor with nicotine causes airway mucociliary dysfunction preferentially via TRPA1 receptors. Am. J. Respir. Crit. Care Med. 2019, 200, 1134–1145. [Google Scholar] [CrossRef]
- Kichko, T.I.; Kobal, G.; Reeh, P.W. Cigarette smoke has sensory effects through nicotinic and TRPA1 but not TRPV1 receptors on the isolated mouse trachea and larynx. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L812–L820. [Google Scholar] [CrossRef]
- Talavera, K.; Gees, M.; Karashima, Y.; Meseguer, V.M.; Vanoirbeek, J.A.J.; Damann, N.; Everaerts, W.; Benoit, M.; Janssens, A.; Vennekens, R.; et al. Nicotine activates the chemosensory cation channel TRPA1. Nat. Neurosci. 2009, 12, 1293–1299. [Google Scholar] [CrossRef]
- Shapiro, D.; Deering-Rice, C.E.; Romero, E.G.; Hughen, R.W.; Light, A.R.; Veranth, J.M.; Reilly, C.A. Activation of Transient Receptor Potential Ankyrin-1 (TRPA1) in Lung Cells by Wood Smoke Particulate Material. Chem. Res. Toxicol. 2013, 26, 750–758. [Google Scholar] [CrossRef]
- Deering-Rice, C.E.; Memon, T.; Lu, Z.; Romero, E.G.; Cox, J.; Taylor-Clark, T.; Veranth, J.M.; Reilly, C.A. Differential Activation of TRPA1 by Diesel Exhaust Particles: Relationships between Chemical Composition, Potency, and Lung Toxicity. Chem. Res. Toxicol. 2019, 32, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Memon, T.A.; Nguyen, N.D.; Burrell, K.L.; Scott, A.F.; Almestica-Roberts, M.; Rapp, E.; Deering-Rice, C.E.; Reilly, C.A. Wood Smoke Particles Stimulate MUC5AC Overproduction by Human Bronchial Epithelial Cells through TRPA1 and EGFR Signaling. Toxicol. Sci. 2020, 174, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Trevisani, M.; Siemens, J.; Materazzi, S.; Bautista, D.M.; Nassini, R.; Campi, B.; Imamachi, N.; André, E.; Patacchini, R.; Cottrell, G.S.; et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc. Natl. Acad. Sci. USA 2007, 104, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Clark, T.E.; McAlexander, M.A.; Nassenstein, C.; Sheardown, S.A.; Wilson, S.; Thornton, J.; Carr, M.J.; Undem, B.J. Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J. Physiol. 2008, 14, 3447–3459. [Google Scholar] [CrossRef]
- Graepel, R.; Fernandes, E.S.; Aubdool, A.A.; Andersson, D.A.; Bevan, S.; Brain, S.D. 4-Oxo-2-nonenal (4-ONE): Evidence of Transient Receptor Potential Ankyrin 1-Dependent and -Independent Nociceptive and Vasoactive Responses In Vivo. J. Pharmacol. Exp. Ther. 2011, 337, 117–124. [Google Scholar] [CrossRef]
- Lin, A.-H.; Liu, M.-H.; Ko, H.-K.; Perng, D.-W.; Lee, T.-S.; Kou, Y.R. Lung Epithelial TRPA1 Transduces the Extracellular ROS into Transcriptional Regulation of Lung Inflammation Induced by Cigarette Smoke: The Role of Influxed Ca2+. Mediat. Inlfammation 2015, 2015. [Google Scholar] [CrossRef]
- Taylor-Clark, T.E.; Undem, B.J.; MacGlashan, D.W.; Ghatta, S.; Carr, M.J.; McAlexander, M.A. Prostaglandin-Induced Activation of Nociceptive Neurons via Direct Interaction with Transient Receptor Potential A1 (TRPA1). Mol. Pharmacol. 2008, 73, 274–281. [Google Scholar] [CrossRef]
- Al-Shamlan, F.; El-Hashim, A.Z. Bradykinin sensitizes the cough reflex via a B2 receptor dependent activation of TRPV1 and TRPA1 channels through metabolites of cyclooxygenase and 12-lipoxygenase. Respir. Res. 2019, 20. [Google Scholar] [CrossRef]
- Pozsgai, G.; Hajna, Z.; Bagoly, T.; Boros, M.; Kemény, Á.; Materazzi, S.; Nassini, R.; Helyes, Z.; Szolcsányi, J.; Pintér, E. The role of transient receptor potential ankyrin 1 (TRPA1) receptor activation in hydrogen-sulphide-induced CGRP-release and vasodilation. Eur. J. Pharmacol. 2012, 689, 56–64. [Google Scholar] [CrossRef]
- Hajna, Z.; Sághy, É.; Payrits, M.; Aubdool, A.A.; Szőke, É.; Pozsgai, G.; Bátai, I.Z.; Nagy, L.; Filotás, D.; Helyes, Z.; et al. Capsaicin-Sensitive Sensory Nerves Mediate the Cellular and Microvascular Effects of H2S via TRPA1 Receptor Activation and Neuropeptide Release. J. Mol. Med. 2016, 60, 157–170. [Google Scholar] [CrossRef]
- Bandell, M.; Story, G.M.; Hwang, S.W.; Viswanath, V.; Eid, S.R.; Petrus, M.J.; Earley, T.J.; Patapoutian, A. Noxious Cold Ion Channel TRPA1 Is Activated by Pungent Compounds and Bradykinin. Neuron 2004, 41, 849–857. [Google Scholar] [CrossRef]
- Hinman, A.; Chuang, H.; Bautista, D.M.; Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 2006, 103, 19564–19568. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. [Google Scholar] [CrossRef]
- Nilius, B.; Prenen, J.; Owsianik, G. Irritating channels: The case of TRPA1. J. Physiol. 2011, 589, 1543–1549. [Google Scholar] [CrossRef]
- Chen, J.; Hackos, D.H. TRPA1 as a drug target—Promise and challenges. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 451–463. [Google Scholar] [CrossRef]
- Grace, M.; Birrell, M.A.; Dubuis, E.; Maher, S.A.; Belvisi, M.G. Transient receptor potential channels mediate the tussive response to prostaglandin E2 and bradykinin. Thorax 2012, 67, 891–900. [Google Scholar] [CrossRef]
- Wang, S.; Dai, Y.; Fukuoka, T.; Yamanaka, H.; Kobayashi, K.; Obata, K.; Cui, X.; Tominaga, M.; Noguchi, K. Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: A molecular mechanism of inflammatory pain. Brain 2008, 131, 1241–1251. [Google Scholar] [CrossRef]
- Meents, J.E.; Fischer, M.J.M.; McNaughton, P.A. Sensitization of TRPA1 by Protein Kinase A. PLoS ONE 2017, 12, e0170097. [Google Scholar] [CrossRef]
- Sun, H.; Meeker, S.; Undem, B.J. Role of TRP channels in Gq-coupled Protease-activated Receptor 1-mediated activation of Mouse Nodose Pulmonary C-fibers. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L192–L199. [Google Scholar] [CrossRef]
- Kádková, A.; Synytsya, V.; Krusek, J.; Zímová, L.; Vlachová, V. Molecular Basis of TRPA1 Regulation in Nociceptive Neurons. A Review. Physiol. Res. 2017, 66, 425–439. [Google Scholar] [CrossRef]
- Geppetti, P.; Nassini, R.; Materazzi, S.; Benemei, S. The concept of neurogenic inflammation. BJU Int. 2008, 101, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Szolcsányi, J. Capsaicin-sensitive sensory nerve terminals with local and systemic efferent functions: Facts and scopes of an unorthodox neuroregulatory mechanism. Prog. Brain Res. 1996, 113, 343–359. [Google Scholar] [PubMed]
- Helyes, Z.; Pintér, E.; Sándor, K.; Elekes, K.; Bánvölgyi, Á.; Keszthelyi, D.; Szőke, É.; Tóth, D.M.; Sándor, Z.; Kereskai, L.; et al. Impaired defense mechanism against inflammation, hyperalgesia, and airway hyperreactivity in somatostatin 4 receptor gene-deleted mice. Proc. Natl. Acad. Sci. USA 2009, 106, 13088–13093. [Google Scholar] [CrossRef]
- Fernandes, E.S.; Fernandes, M.A.; Keeble, J.E. The functions of TRPA1 and TRPV1: Moving away from sensory nerves. Br. J. Pharmacol. 2012, 166, 510–521. [Google Scholar] [CrossRef]
- Kun, J.; Perkecz, A.; Knie, L.; Sétáló, G., Jr.; Tornóczki, T.; Pintér, E.; Bán, Á. TRPA1 receptor is upregulated in human oral lichen planus. Oral. Dis. 2017, 23, 189–198. [Google Scholar] [CrossRef]
- Bertin, S.; Aoki-Nonaka, Y.; Lee, J.; de Jong, P.R.; Kim, P.; Han, T.; Yu, T.; To, K.; Takahashi, N.; Boland, B.S.; et al. The TRPA1 ion channel is expressed in CD4+ T cells and restrains T cell-mediated colitis through inhibition of TRPV1. Gut 2017, 66, 1584–1596. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Majhi, R.K.; Tiwari, A.; Acharya, T.; Kumar, P.S.; Saha, S.; Kumar, A.; Goswami, C.; Chattopadhyay, S. Transient receptor potential ankyrin1 channel is endogenously expressed in T cells and is involved in immune functions. Biosci. Rep. 2019, 39, 1–16. [Google Scholar] [CrossRef]
- Atoyan, R.; Shnader, D.; Botchkareva, N. Non-Neuronal Expression of Transient Receptor Potential Type A1 (TRPA1) in Human Skin. J. Invest. Dermatol. 2009, 129, 2312–2315. [Google Scholar] [CrossRef]
- Kemény, Á.; Kodji, X.; Horváth, S.; Komlódi, R.; Szőke, É.; Sándor, Z.; Perkecz, A.; Gyömörei, C.; Sétáló, G.; Kelemen, B.; et al. TRPA1 acts in a protective manner in imiquimod-induced psoriasiform dermatitis in mice. J. Investig. Dermatol. 2018, 138, 1774–1784. [Google Scholar] [CrossRef]
- Streng, T.; Axelsson, H.E.; Hedlund, P.; Andersson, D.A.; Jordt, S.-E.; Bevan, S.; Andersson, K.-E.; Högestätt, E.D.; Zygmunt, P.M. Distribution and Function of the Hydrogen Sulfide—Sensitive TRPA1 Ion Channel in Rat Urinary Bladder. Eur. Urol. 2008, 53, 391–400. [Google Scholar] [CrossRef]
- Earley, S.; Gonzales, A.L.; Crnich, R. Endithelium-Dependent Cerebral Artery Dilation Mediated by TRPA1 and Ca2+-Activated K+ Channels. Circ. Res. 2009, 104, 987–994. [Google Scholar] [CrossRef]
- Nummenmaa, E.; Hämäläinen, M.; Moilanen, L.J.; Paukkeri, E.-L.; Nieminen, R.M.; Moilanen, T.; Vuolteenaho, K.; Moilanen, E. Transient receptor potential ankyrin 1 (TRPA1) is functionally expressed in primary human osteoarthritic chondrocytes. Arthritis Res. Ther. 2016, 18, 185. [Google Scholar] [CrossRef]
- Jaquemar, D.; Schenker, T.; Trueb, B. An Ankyrin-like Protein with Transmembrane Domains Is Specifically Lost after Oncogenic Transformation of Human Fibroblasts. J. Biol. Chem. 1999, 274, 7325–7333. [Google Scholar] [CrossRef]
- Stokes, A.; Wakano, C.; Koblan-Huberson, M.; Adra, C.N.; Fleig, A.; Turner, H. TRPA1 is a substrate for de-ubiquitination by the tumor suppressor CYLD. Cell. Signal. 2006, 18, 1584–1594. [Google Scholar] [CrossRef]
- Mukhopadhyay, I.; Kulkarni, A.; Khairatkar-Joshi, N. Blocking TRPA1 in Respiratory Disorders: Does It Hold a Promise? Pharmaceuticals 2016, 9, 70. [Google Scholar] [CrossRef]
- Nassini, R.; Pedretti, P.; Moretto, N.; Fusi, C.; Carnini, C.; Facchinetti, F.; Viscomi, A.R.; Pisano, A.R.; Stokesberry, S.; Brunmark, C.; et al. Transient Receptor Potential Ankyrin 1 Channel Localized to Non-Neuronal Airway Cells Promotes Non-Neurogenic Inflammation. PLoS ONE 2012, 7, e42454. [Google Scholar] [CrossRef]
- Kannler, M.; Lüling, R.; Yildirim, A.Ö.; Gudermann, T.; Steinritz, D.; Dietrich, A. TRPA1 channels: Expression in non-neuronal murine lung tissues and dispensability for hyperoxia-induced alveolar epithelial hyperplasia. Pflügers Arch. Eur. J. Physiol. 2018, 470, 1231–1241. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Y.; Xu, M.; Zhang, H.; Chen, Y.; Fan, K.; Adcock, I.M.; Li, F. Free Radical Biology and Medicine Roles of TRPA1 and TRPV1 in cigarette smoke -induced airway epithelial cell injury model. Free Radic. Biol. Med. 2019, 134, 229–238. [Google Scholar] [CrossRef]
- Yap, M.J.G.; Ueda, T.; Takeda, N.; Fukumitsu, K.; Fukuda, S.; Uemura, T.; Tajiri, T.; Ohkubo, H.; Maeno, K.; Ito, Y.; et al. Cytokine An inflammatory stimulus sensitizes TRPA1 channel to increase cytokine release in human lung fibroblasts. Cytokine 2020, 129, 155027. [Google Scholar] [CrossRef]
- Nilius, B.; Appendino, G.; Owsianik, G. The transient receptor potential channel TRPA1: From gene to pathophysiology. Pflügers Arch. Eur. J. Physiol. 2012, 464, 425–458. [Google Scholar] [CrossRef]
- López-Requena, A.; Boonen, B.; van Greven, L.; Hellings, P.W.; Alpizar, Y.A.; Talavera, K. Roles of Neuronal TRP Channels in Neuroimmune Interactions. In Neurobiology of TRP Channels; Emir, T.L.R., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2017. [Google Scholar]
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian Transient Receptor Potential TRPA1 Channels: From Structure to Disease. Physiol. Rev. 2020, 100, 725–803. [Google Scholar] [CrossRef]
- Chen, J.; McGaraughty, S.; Kym, P. TRPA1 in drug discovery. In TRP Channels in Drug Discovery; Szallasi, A., Bíró, T., Eds.; Humana Press, Springer: Totowa, NJ, USA, 2012; pp. 43–59. [Google Scholar]
- Viana, F. TRPA1 channels: Molecular sentinels of cellular stress and tissue damage. J. Physiol. 2016, 594, 4151–4169. [Google Scholar] [CrossRef]
- Grace, M.S.; Baxter, M.; Dubuis, E.; Birrell, M.A.; Belvisi, M.G. Transient receptor potential (TRP) channels in the airway: Role in airway disease. Br. J. Pharmacol. 2014, 171, 2593–2607. [Google Scholar] [CrossRef]
- Banner, H.K.; Igney, F.; Poll, C. TRP channels: Emerging targets for respiratory disease. Pharmacol. Ther. 2011, 130, 371–384. [Google Scholar] [CrossRef]
- Yang, H.; Li, S. Transient Receptor Potential Ankyrin 1 (TRPA1) Channel and Neurogenic Inflammation in Pathogenesis of Asthma. Med. Sci. Monit. 2016, 22, 2917–2923. [Google Scholar] [CrossRef]
- Bonvini, S.J.; Belvisi, M.G. Cough and airway disease: The role of ion channels. Pulm. Pharmacol. Ther. 2017, 47, 21–28. [Google Scholar] [CrossRef]
- Wallace, H. Airway Pathogenesis Is Linked to TRP Channels. In Neurobiology of TRP Channels; Emir, T.L.R., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2017. [Google Scholar]
- Prandini, P.; De Logu, F.; Fusi, C.; Provezza, L.; Nassini, R.; Montagner, G.; Materazzi, S.; Munari, S.; Gilioli, E.; Bezzerri, V.; et al. Transient Receptor Potential Ankyrin 1 Channels Modulate Inflammatory Response in Respiratory Cells from Patients with Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 645–656. [Google Scholar] [CrossRef]
- Dietrich, A. Modulators of Transient Receptor Potential (TRP) Channels as Therapeutic Options in Lung Disease. Pharmaceuticals 2019, 12, 23. [Google Scholar] [CrossRef]
- Kemény, Á.; Csekő, K.; Szitter, I.; Varga, Z.V.; Bencsik, P.; Kiss, K.; Halmosi, R.; Deres, L.; Erős, K.; Perkecz, A.; et al. Integrative characterization of chronic cigarette smoke-induced cardiopulmonary comorbidities in a mouse model. Environ. Pollut. 2017, 229, 746–759. [Google Scholar] [CrossRef]
- Caceres, A.I.; Brackmann, M.; Elia, M.D.; Bessac, B.F.; del Camino, D.; D’Amours, M.; Witek, J.S.; Fanger, C.M.; Chong, J.A.; Hayward, N.J.; et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc. Natl. Acad. Sci. USA 2009, 106, 9099–9104. [Google Scholar] [CrossRef]
- Raemdonck, K.; de Alba, J.; Birrell, M.A.; Grace, M.; Maher, S.A.; Irvin, C.G.; Fozard, J.R.; O’Byrne, P.M.; Belvisi, M.G. A role for sensory nerves in the late asthmatic response. Thorax 2012, 67, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Geppetti, P.; Patacchini, R.; Nassini, R.; Materazzi, S. Cough: The Emerging Role of the TRPA1 Channel. Lung 2010, 188, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Nassini, R.; Materazzi, S.; De Siena, G.; De Cesaris, F.; Geppetti, P. Transient receptor potential channels as novel drug targets in respiratory diseases. Curr. Opin. Investig. Drugs 2010, 11, 535–542. [Google Scholar]
- Mazzone, S.B.; Undem, B.J. Vagal afferent innervation of the airways in health and disease. Physiol. Rev. 2016, 96, 975–1024. [Google Scholar] [CrossRef]
- Undem, B.J.; Kollarik, M. The role of vagal afferent nerves in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 355–360. [Google Scholar] [CrossRef]
- Canning, B.J. Afferent nerves regulating the cough reflex: Mechanisms and Mediators of Cough in Disease. Otolaryngol. Clin. N. Am. 2010, 43, 15–25. [Google Scholar] [CrossRef]
- Boonen, B.; Alpizar, Y.A.; Meseguer, V.M.; Talavera, K. TRP Channels as Sensors of Bacterial Endotoxins. Toxins (Basel) 2018, 10, 326. [Google Scholar] [CrossRef]
- Mazgaeen, L.; Gurung, P. Recent Advances in Lipopolysaccharide Recognition Systems. Int. J. Mol. Sci. 2020, 21, 379. [Google Scholar]
- Mendes, S.J.F.; Sousa, F.I.A.B.; Pereira, D.M.S.; Ferro, T.A.F.; Pereira, I.C.P.; Silva, B.L.R.; Pinheiro, A.J.M.C.R.; Mouchrek, A.Q.S.; Monteiro-Neto, V.; Costa, S.K.P.; et al. Cinnamaldehyde modulates LPS-induced systemic inflammatory response syndrome through TRPA1-dependent and independent mechanisms. Int. Immunopharmacol. 2016, 34, 60–70. [Google Scholar] [CrossRef]
- Yin, S.; Wang, P.; Xing, R.; Zhao, L.; Li, X.; Zhang, L.; Xiao, Y. Transient Receptor Potential Ankyrin 1 (TRPA1) Mediates Lipopolysaccharide (LPS)-Induced Inflammatory Responses in Primary Human Osteoarthritic Fibroblast-Like Synoviocytes. Inflammation 2018, 41, 700–709. [Google Scholar] [CrossRef]
- Soldano, A.; Alpizar, Y.A.; Boonen, B.; Franco, L.; López-Requena, A.; Liu, G.; Mora, N.; Yaksi, E.; Voets, T.; Vennekens, R.; et al. Gustatory-mediated avoidance of bacterial lipopolysaccharides via TRPA1 activation in Drosophila. Elife 2016, 5, e13133. [Google Scholar] [CrossRef]
- Startek, J.B.; Talavera, K.; Voets, T.; Alpizar, Y.A. Differential interactions of bacterial lipopolysaccharides with lipid membranes: Implications for TRPA1-mediated chemosensation. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Elekes, K.; Helyes, Z.; Németh, J.; Sándor, K.; Pozsgai, G.; Kereskai, L.; Börzsei, R.; Pintér, E.; Szabó, Á.; Szolcsányi, J. Role of capsaicin-sensitive afferents and sensory neuropeptides in endotoxin-induced airway inflammation and consequent bronchial hyperreactivity in the mouse. Regul. Pept. 2007, 141, 44–54. [Google Scholar] [CrossRef]
- Helyes, Z.; Elekes, K.; Németh, J.; Pozsgai, G.; Sándor, K.; Kereskai, L.; Börzsei, R.; Pintér, E.; Szabó, Á.; Szolcsányi, J. Role of transient receptor potential vanilloid 1 receptors in endotoxin-induced airway inflammation in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, 1173–1181. [Google Scholar] [CrossRef]
- Lee, L.-Y.; Hsu, C.-C.; Lin, Y.-J.; Lin, R.-L.; Khosravi, M. Interaction between TRPA1 and TRPV1: Synergy on pulmonary sensory nerves. Pulm. Pharmacol. Ther. 2015, 35, 87–93. [Google Scholar] [CrossRef]
- Marsh, B.J.; Fryer, A.D.; Jacoby, D.B.; Drake, M.G. TRPA1 causes rapid bronchodilation via non-epithelial PGE2. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L943–L952. [Google Scholar] [CrossRef]
- Alpizar, Y.A.; Boonen, B.; Sanchez, A.; Jung, C.; López-Requena, A.; Naert, R.; Steelant, B.; Luyts, K.; Plata, C.; De Vooght, V.; et al. TRPV4 activation triggers protective responses to bacterial lipopolysaccharides in airway epithelial cells. Nat. Commun. 2017, 8, 1059. [Google Scholar] [CrossRef]
- Chao, L.K.; Hua, K.-F.; Hsu, H.-Y.; Cheng, S.-S.; Lin, I.-F.; Chen, C.-J.; Chen, S.-T.; Chang, S.-T. Cinnamaldehyde inhibits pro-inflammatory cytokines secretion from monocytes/macrophages through suppression of intracellular signaling. Food Chem. Toxicol. 2008, 46, 220–231. [Google Scholar] [CrossRef]
- Pulli, B.; Ali, M.; Forghani, R.; Schob, S.; Hsieh, K.L.C.; Wojtkiewicz, G.; Linnoila, J.J.; Chen, J.W. Measuring Myeloperoxidase Activity in Biological Samples. PLoS ONE 2013, 8, e67976. [Google Scholar] [CrossRef]
- Dupont, L.L.; Alpizar, Y.A.; Brusselle, G.G.; Bracke, K.R.; K, T.; Joos, G.F.; Maes, T. Expression of transient receptor potential (TRP) channels in a murine model of cigarette smoke exposure. In Proceedings of the C34 Insights into COPD Pathogenesis from Pre-Clinical Studies, San Diego, CA, USA, 16–21 May 2014. [Google Scholar]
- Nie, Y.; Huang, C.; Zhong, S.; Wortley, M.A.; Luo, Y.; Luo, W.; Xie, Y.; Lai, K.; Zhong, N. Cigarette smoke extract (CSE) induces transient receptor potential ankyrin 1 (TRPA1) expression via activation of HIF1αin A549 cells. Free Radic. Biol. Med. 2016, 99, 498–507. [Google Scholar] [CrossRef]
- Ács, K.; Bencsik, T.; Böszörményi, A.; Kocsis, B.; Horváth, G. Essential Oils and Their Vapors as Potential Antibacterial Agents Against Respiratory Tract Pathogens. Nat. Pord. Commun. 2016, 11, 1709–1712. [Google Scholar] [CrossRef]
- Bautista, D.M.; Jordt, S.-E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 Mediates the Inflammatory Actions of Environmental Irritants and Proalgesic Agents. Cells 2006, 124, 1269–1282. [Google Scholar] [CrossRef]
- Westphal, O.; Jahn, K. Bacterial lipopolysaccharides: Extraction with phenolwater and further applications of the procedure. In Methods in Carbohydrate Chemistry; Whistler, R.L., Ed.; Academic Press: New York, NY, USA, 1965; pp. 83–91. [Google Scholar]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamai, U.; Günter, S.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; Di Padova, F.; et al. Bacterial endotoxin: Molecular rekationships of sturcutre to activity and function. FASEB J. 1994, 8, 217–225. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.-Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Haddad, E.-B.; Birrell, M.; McCluskie, K.; Ling, A.; Webber, S.E.; Foster, M.L.; Belvisi, M.G. Role of p38 MAP kinase in LPS-induced airway inflammation in the rat. Br. J. Pharmacol. 2001, 132, 1715–1724. [Google Scholar] [CrossRef]
- Togbe, D.; Schnyder-Candrian, S.; Schnyder, B.; Doz, E.; Noulin, N.; Janot, L.; Secher, T.; Gasse, P.; Lima, C.; Coelho, F.R.; et al. Toll-like receptor and tumour necrosis factor dependent endotoxin-induced acute lung injury. Int. J. Exp. Path. 2007, 88, 387–391. [Google Scholar] [CrossRef]
- Helyes, Z.; Hajna, Z. Endotoxin-induced airway inflammation and asthma models. In TRP Channels in Drug Discovery; Szallasi, A., Bíró, T., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 301–342. ISBN 978-1-62703-076-2. [Google Scholar]
- Lefort, J.; Motreff, L.; Vargaftig, B.B. Airway Administration of Escherichia coli Endotoxin to Mice Induces Glucocorticosteroid-Resistant Bronchoconstriction and Vasopermeation. Am. J. Respir. Cell Mol. Biol. 2001, 24, 345–351. [Google Scholar] [CrossRef]
- Hajna, Z.; Borbély, É.; Kemény, Á.; Botz, B.; Kereskai, L.; Szolcsányi, J.; Pintér, E.; Paige, C.J.; Berger, A.; Helyes, Z. Hemokinin-1 is an important mediator of endotoxin-induced acute airway inflammation in the mouse. Peptides 2015, 64, 1–7. [Google Scholar] [CrossRef]
- Ma, W.; Cui, W.; Lin, Q. Improved immnunophenotyping of lymphocytes in bronchoalveolar lavage fluid (BALF) by flow cytometry. Clin. Chim. Acta 2001, 313, 133–138. [Google Scholar] [CrossRef]
- Zeldin, D.C.; Wohlford-Lenane, C.; Chulada, P.; Bradbury, J.A.; Scarborough, P.E.; Roggli, V.; Langenbach, R.; Schwartz, D.A. Airway Inflammation and Responsiveness in Prostaglandin H Synthase – Deficient Mice Exposed to Bacterial Lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 2001, 25, 457–465. [Google Scholar] [CrossRef]
- Helyes, Z.; Kemény, Á.; Csekő, K.; Szőke, É.; Elekes, K.; Mester, M.; Sándor, K.; Perkecz, A.; Kereskai, L.; Márk, L.; et al. Marijuana smoke induces severe pulmonary hyperresponsiveness, inflammation, and emphysema in a predictive mouse model not via CB1 receptor activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L267–L277. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.R.; Rodriguez, D.; Russo, M.; Campa, A. Macrophage Activation Includes High Intracellular Myeloperoxidase Activity. Biochem. Biophys. Res. Commun. 2002, 292, 869–873. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endotoxin-Induced Acute Inflammation | Trpa1+/+ | Trpa1−/− | Trpa1+/+ vs. Trpa1−/− LPS-Treated Inflamed |
| Compared to PBS-Treated Control | |||
| Bronchial hyperreactivity | ns. | ↑(++) | * |
| Myeloperoxidase activity | ns. | ↑(+++) | ** |
| Inflammatory histopathological changes | ↑(++) | ns | ns |
| CSE-induced Chronic Inflammation | Trpa1+/+ | Trpa1−/− | Trpa1+/+ vs. Trpa1−/− CSE-Treated Inflamed |
| Compared to Intact Control | |||
| TV, MV, PIF, PEF | ↓(++/+++) | ns | * |
| Oedema | ↑ | ↑ | ns. |
| Inflammatory cell infiltration | ↑ | ↑ | ns. |
| Emphysema | earlier | later | * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajna, Z.; Csekő, K.; Kemény, Á.; Kereskai, L.; Kiss, T.; Perkecz, A.; Szitter, I.; Kocsis, B.; Pintér, E.; Helyes, Z. Complex Regulatory Role of the TRPA1 Receptor in Acute and Chronic Airway Inflammation Mouse Models. Int. J. Mol. Sci. 2020, 21, 4109. https://doi.org/10.3390/ijms21114109
Hajna Z, Csekő K, Kemény Á, Kereskai L, Kiss T, Perkecz A, Szitter I, Kocsis B, Pintér E, Helyes Z. Complex Regulatory Role of the TRPA1 Receptor in Acute and Chronic Airway Inflammation Mouse Models. International Journal of Molecular Sciences. 2020; 21(11):4109. https://doi.org/10.3390/ijms21114109
Chicago/Turabian StyleHajna, Zsófia, Kata Csekő, Ágnes Kemény, László Kereskai, Tamás Kiss, Anikó Perkecz, István Szitter, Béla Kocsis, Erika Pintér, and Zsuzsanna Helyes. 2020. "Complex Regulatory Role of the TRPA1 Receptor in Acute and Chronic Airway Inflammation Mouse Models" International Journal of Molecular Sciences 21, no. 11: 4109. https://doi.org/10.3390/ijms21114109
APA StyleHajna, Z., Csekő, K., Kemény, Á., Kereskai, L., Kiss, T., Perkecz, A., Szitter, I., Kocsis, B., Pintér, E., & Helyes, Z. (2020). Complex Regulatory Role of the TRPA1 Receptor in Acute and Chronic Airway Inflammation Mouse Models. International Journal of Molecular Sciences, 21(11), 4109. https://doi.org/10.3390/ijms21114109