Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles

Abstract
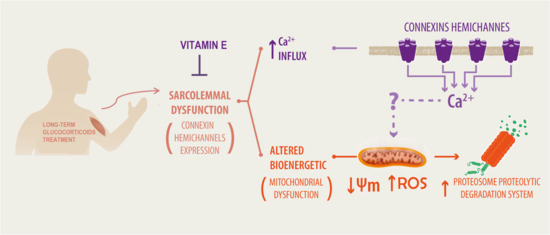
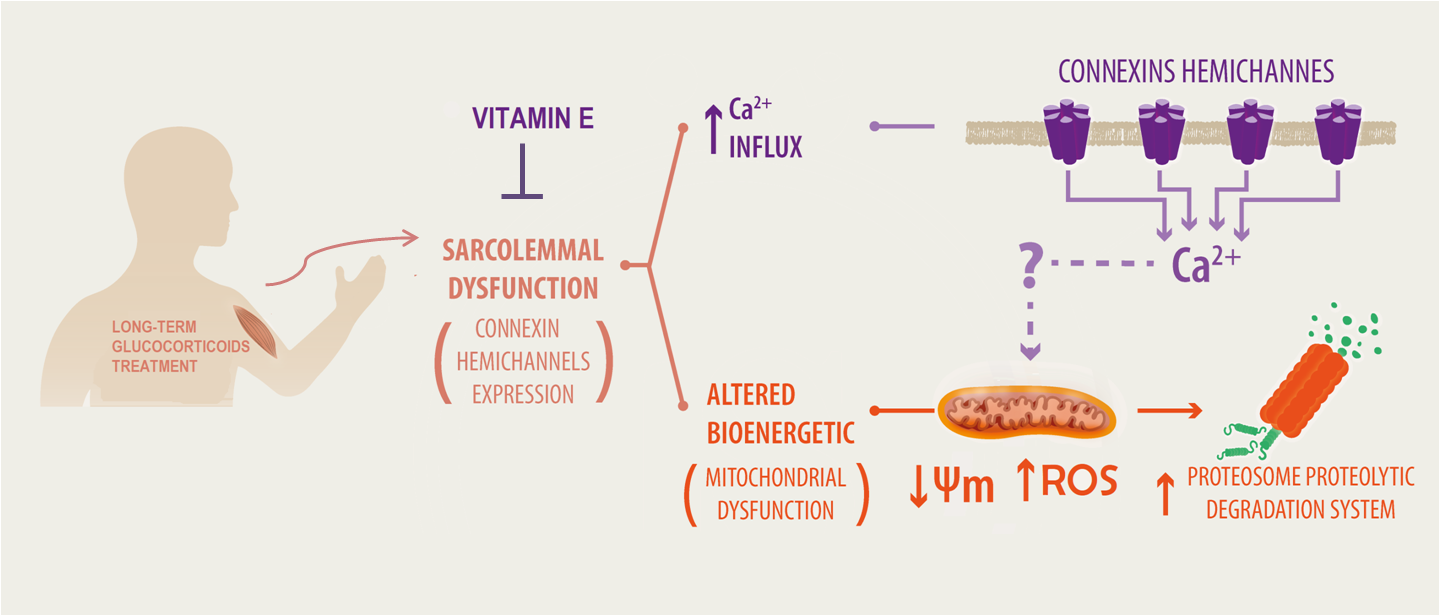{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
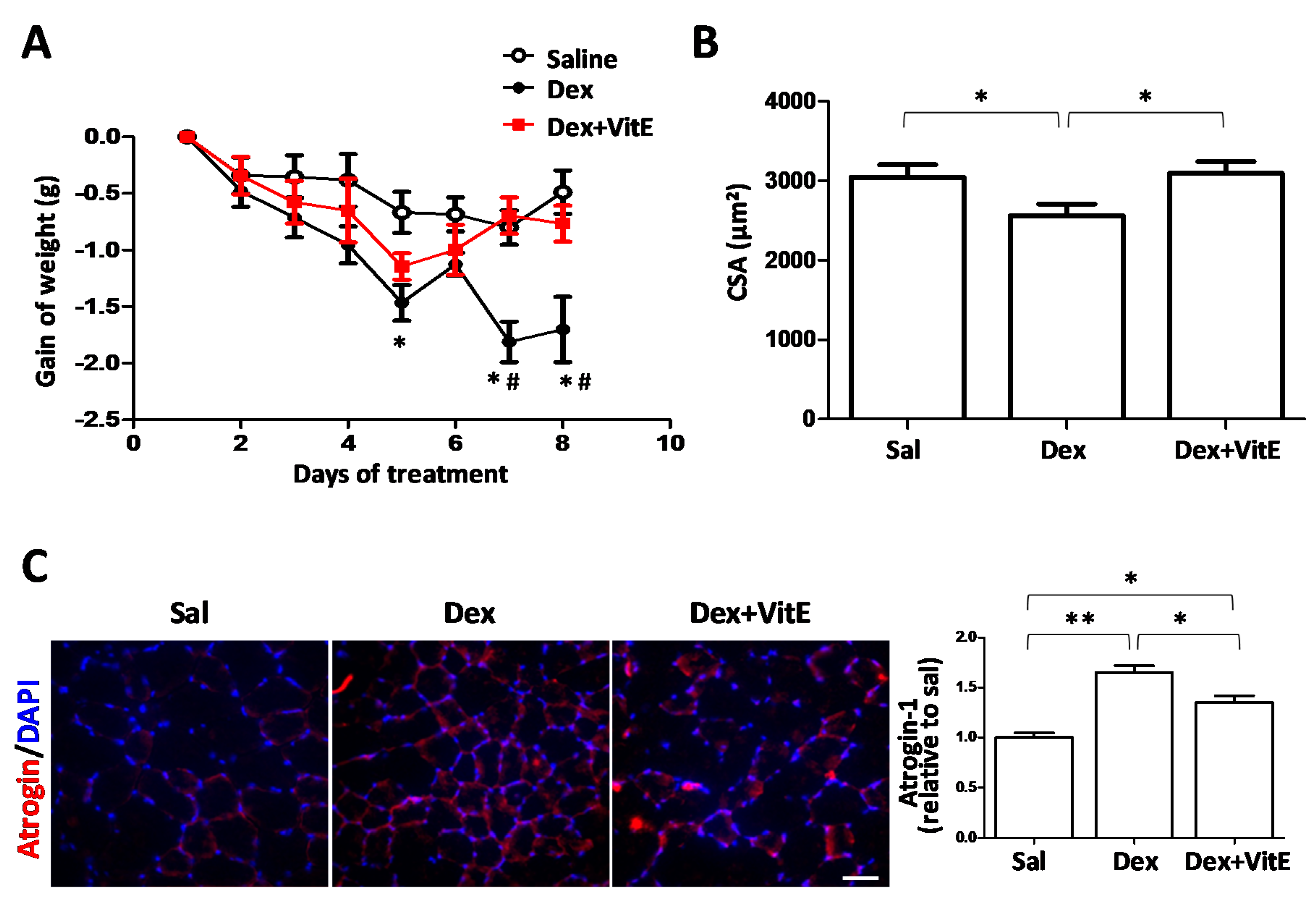
2.1. Vitamin E Mitigates Dexamethasone-Induced Weight Loss and Muscle Atrophy in Mice
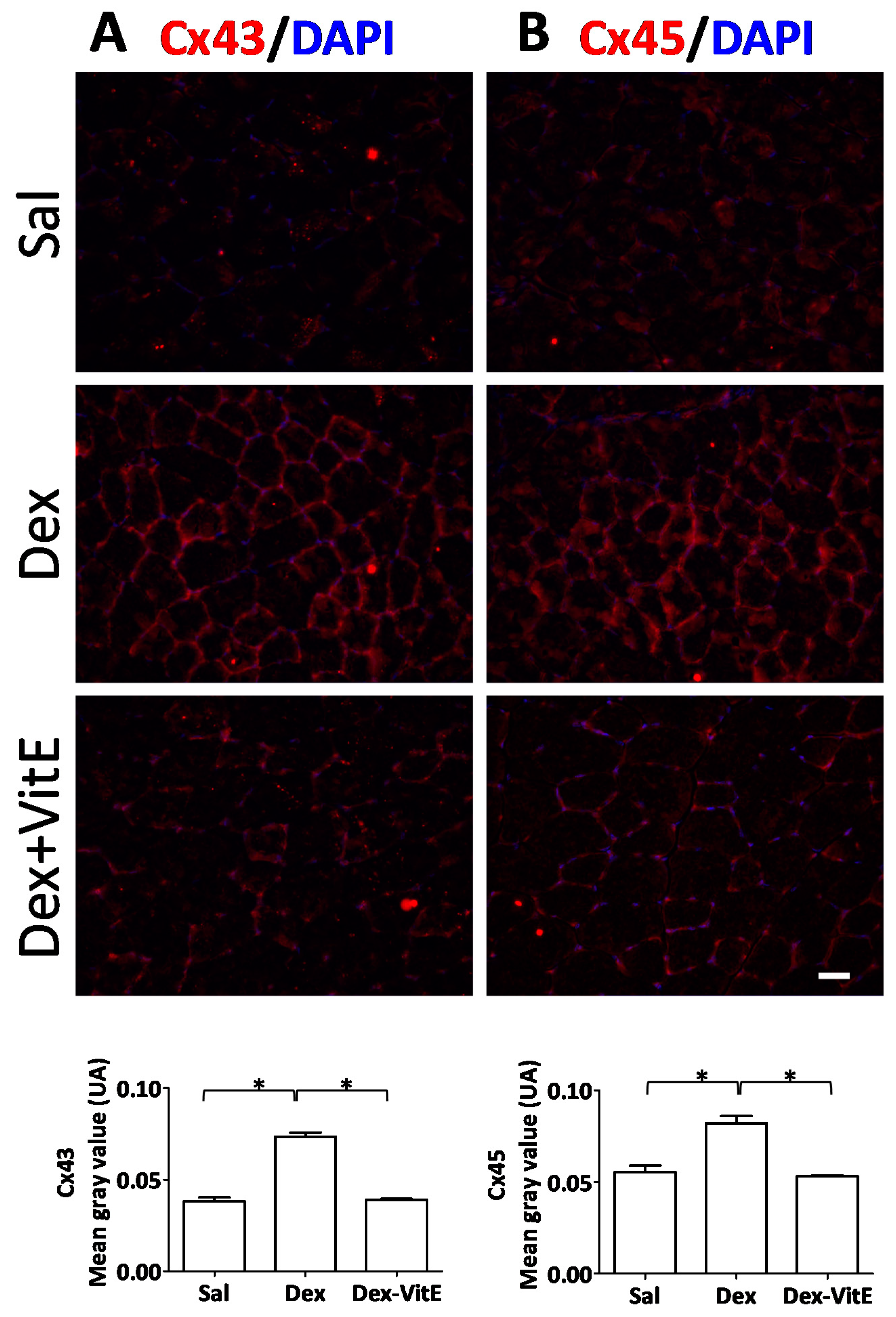
2.2. Vitamin E Prevents the Dexamethasone-Induced Increase in Connexin Immunoreactivity of Skeletal Myofibers
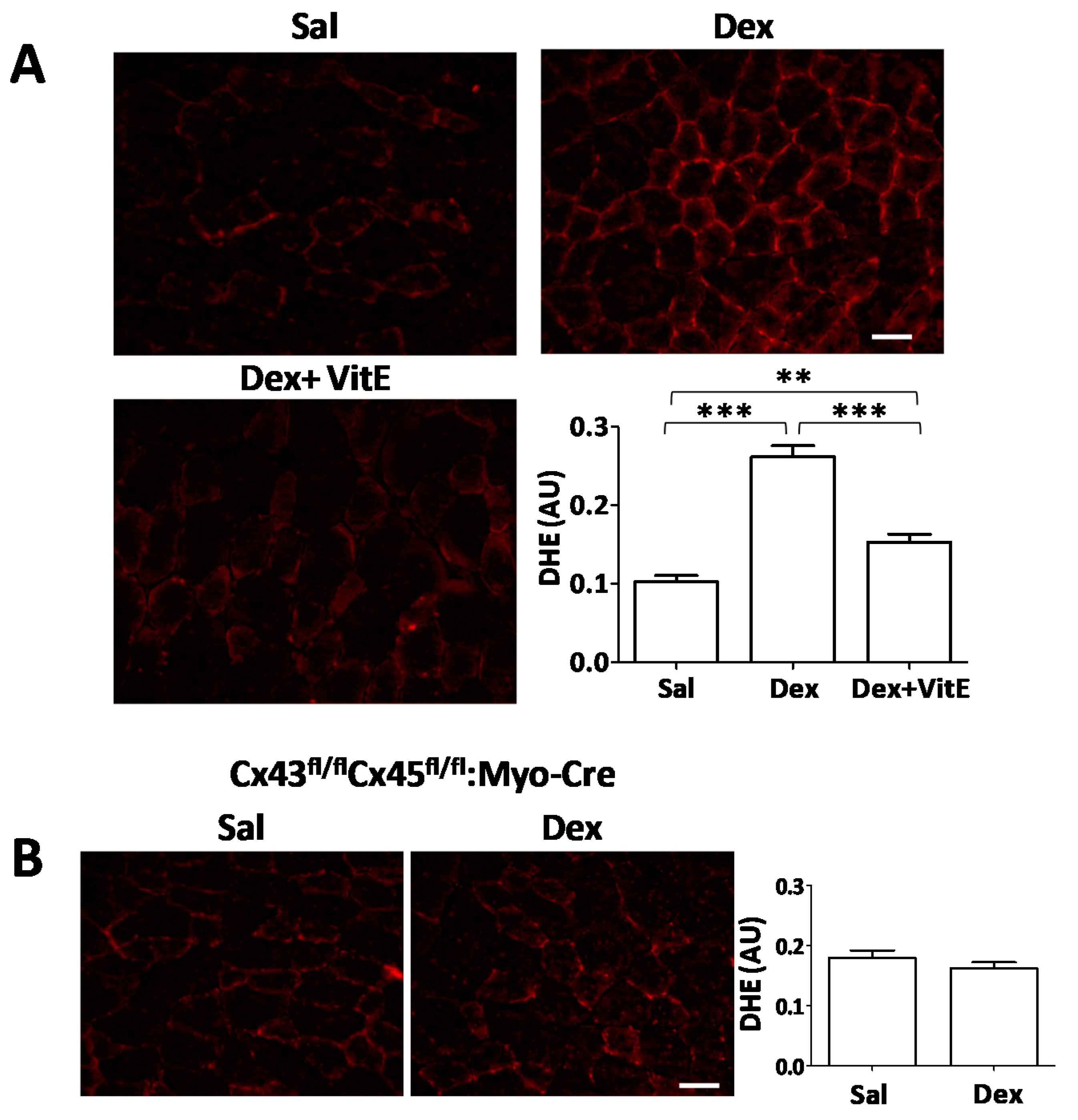
2.3. Vitamin E-Rich Diet Prevents Oxidative Stress in Myofibers of Dexamethasone-Treated Mice, and Connexin Expression is Necessary for Dexamethasone to Induce Oxidative Stress
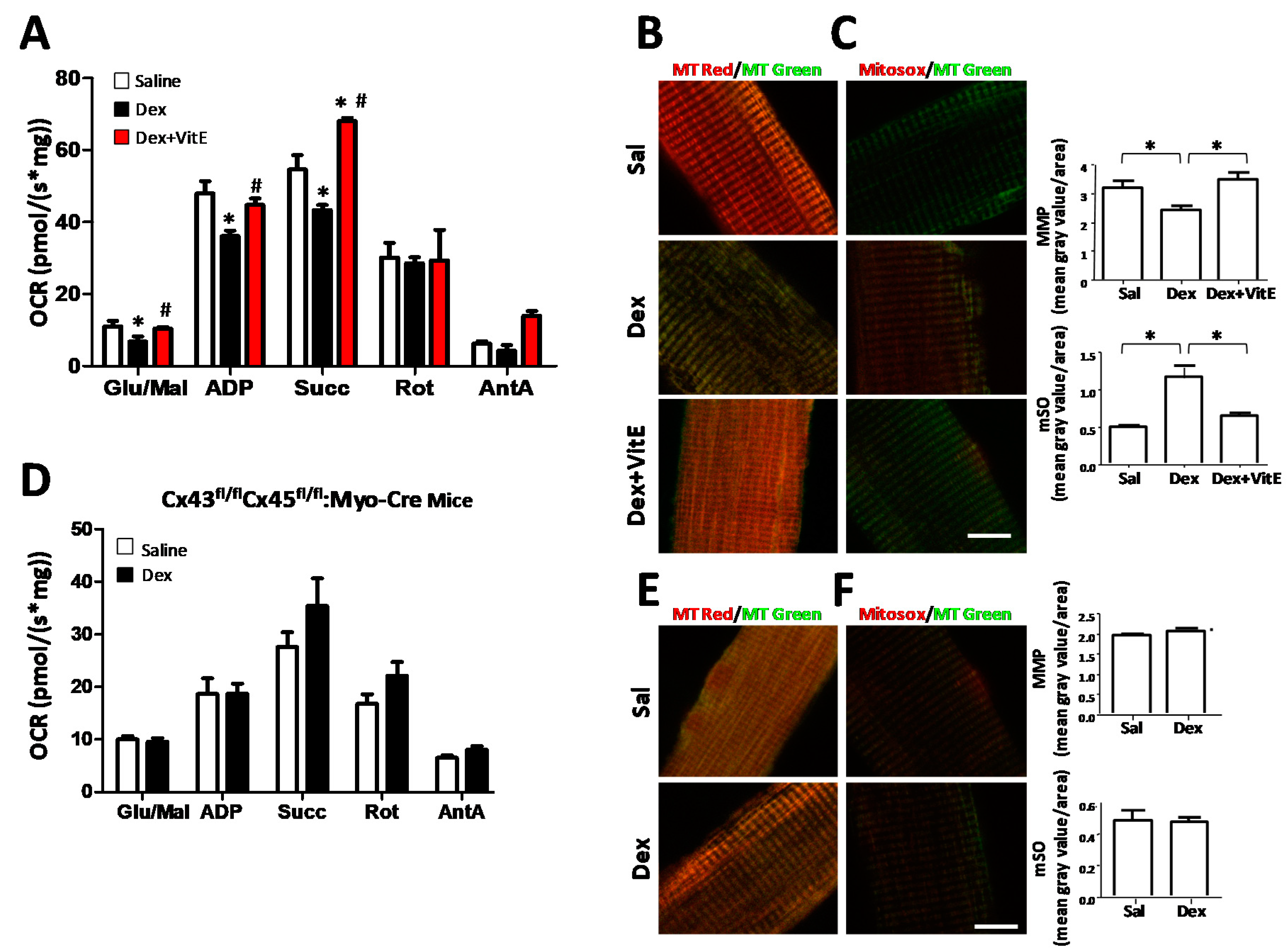
2.4. Mitochondrial Dysfunction Induced by Dexamethasone Requires the Expression of Connexins and is Prevented by Vitamin E
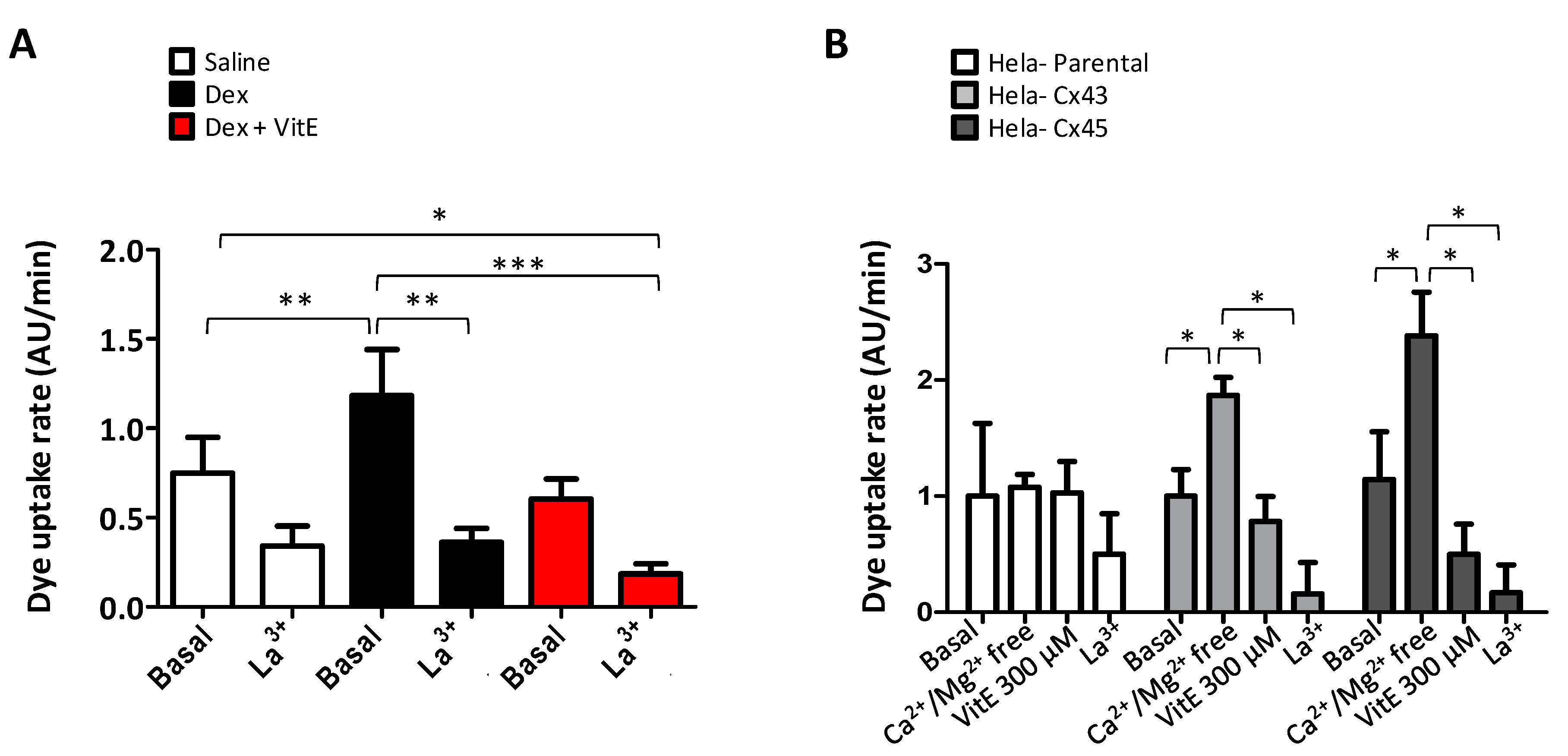
2.5. Vitamin E Blocks the connexin hemichannel activity
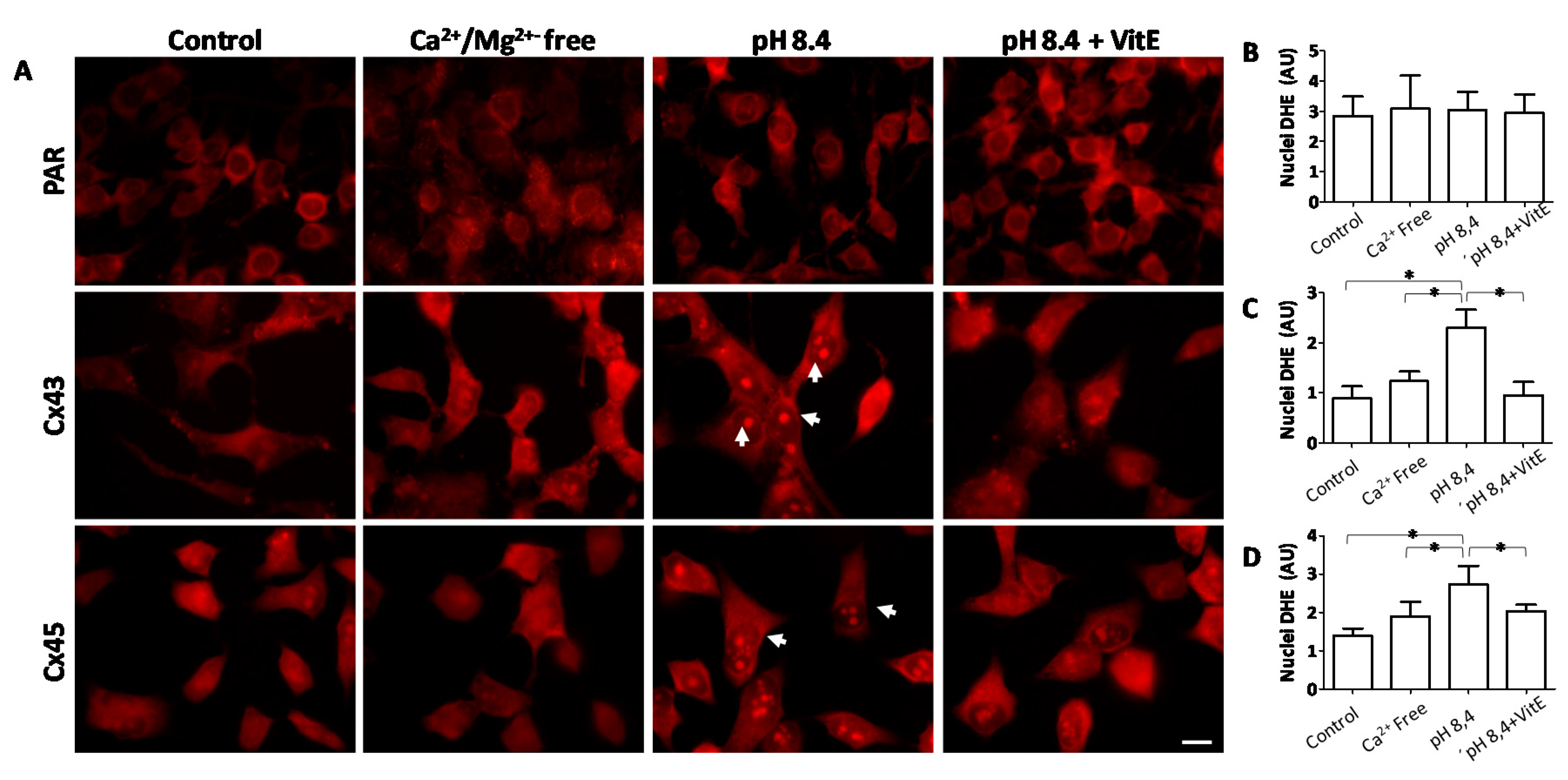
2.6. Opening of Connexin Hemichannels Promotes Generation of Reactive Oxygen Substances in HeLa Cells
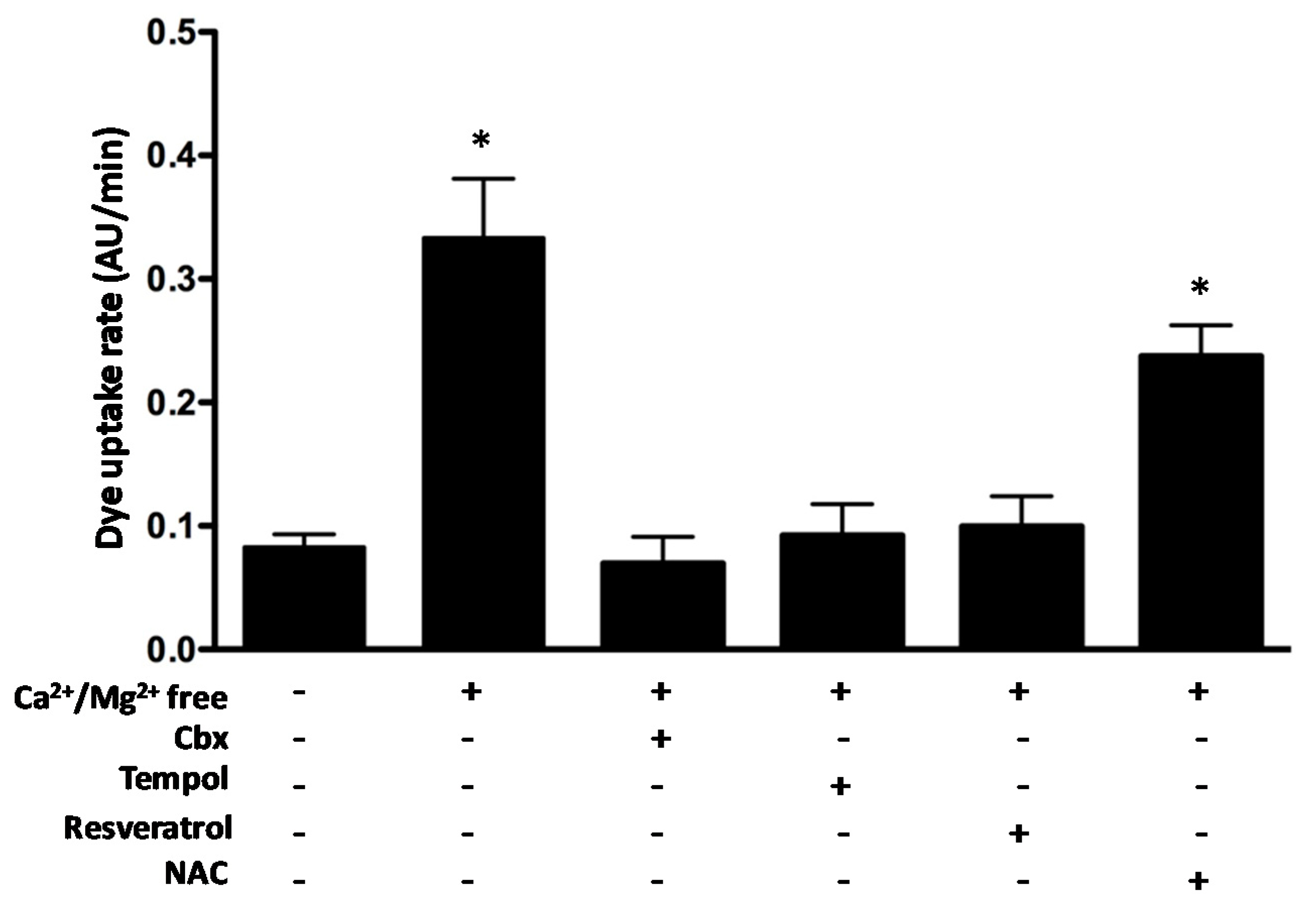
2.7. The Antioxidants Tempol and Resveratrol but Not N-acetylcysteine Block Connexin Hemichannel Activity
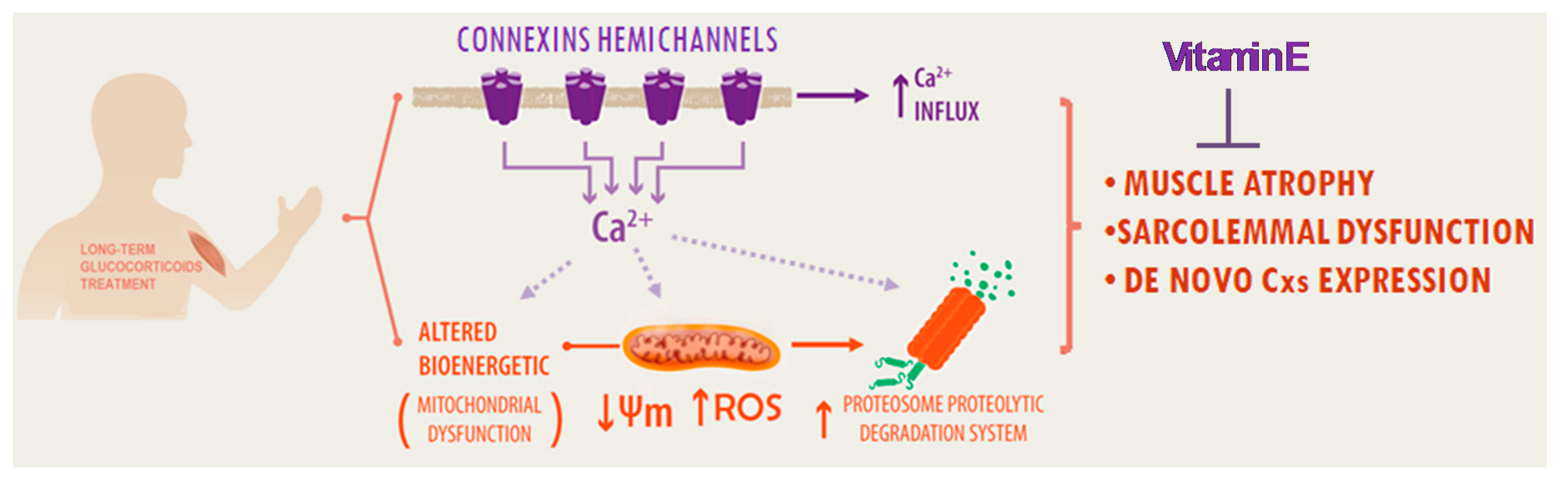
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Cell Cultures
4.4. Histological Analysis
4.5. Immunofluorescence
4.6. Skeletal Myofiber Isolation
4.7. Dye Uptake Assay
4.8. Laser Confocal Imaging
4.9. Oxygen Consumption
4.10. Detection of ROS with DHE
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PBS | Phosphate-buffered saline |
| Cx | Connexin |
| Cx HC | Connexin43 hemichannel |
| Cx45 HC | Connexin45 hemichannel |
| Etd+ | Ethidium |
| FBS | Fetal bovine serum |
| ROS | Reactive oxygen species |
| DHE | dihydroethidium |
| VitE | Vitamin E |
| Dex | Dexamethasone |
| CSA | Cross sectional area |
| OCRs | consumption rates |
| TA | Tibialis anterior |
| SEM | standard error of the mean |
References
- Qin, J.; Du, R.; Yang, Y.Q.; Zhang, H.Q.; Li, Q.; Liu, L.; Guan, H.; Hou, J.; An, X.R. Dexamethasone-induced skeletal muscle atrophy was associated with upregulation of myostatin promoter activity. Res. Vet. Sci. 2013, 94, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Massaccesi, L.; Goi, G.; Tringali, C.; Barassi, A.; Venerando, B.; Papini, N. Dexamethasone-induced skeletal muscle atrophy increases o-glcnacylation in c2c12 cells. J. Cell. Biochem. 2016, 117, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Azzam, K.M.; Cidlowski, J.A. Glucocorticoids sensitize the innate immune system through regulation of the nlrp3 inflammasome. J. Biol. Chem. 2011, 286, 38703–38713. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Balboa, E.; Puebla, C.; Vargas, A.A.; Cisterna, B.A.; Escamilla, R.; Regueira, T.; Sáez, J.C. Dexamethasone-induced muscular atrophy is mediated by functional expression of connexin-based hemichannels. Biochim. Biophys. Acta 2016, 1862, 1891–1899. [Google Scholar] [CrossRef]
- Fischer, R.; Reinehr, R.; Lu, T.P.; Schonicke, A.; Warskulat, U.; Dienes, H.P.; Haussinger, D. Intercellular communication via gap junctions in activated rat hepatic stellate cells. Gastroenterology 2005, 128, 433–448. [Google Scholar] [CrossRef]
- Cisterna, B.A.; Cardozo, C.; Sáez, J.C. Neuronal involvement in muscular atrophy. Front. Cell. Neurosci. 2014, 8, 405. [Google Scholar] [CrossRef]
- Constantin, B.; Cronier, L. Involvement of gap junctional communication in myogenesis. Int. Rev. Cytol. 2000, 196, 1–65. [Google Scholar]
- Cea, L.A.; Cisterna, B.A.; Puebla, C.; Frank, M.; Figueroa, X.F.; Cardozo, C.; Willecke, K.; Latorre, R.; Sáez, J.C. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 16229–16234. [Google Scholar] [CrossRef]
- Vargas, A.A.; Cisterna, B.A.; Saavedra-Leiva, F.; Urrutia, C.; Cea, L.A.; Vielma, A.H.; Gutierrez-Maldonado, S.E.; Martin, A.J.; Pareja-Barrueto, C.; Escalona, Y.; et al. On biophysical properties and sensitivity to gap junction blockers of connexin 39 hemichannels expressed in hela cells. Front. Physiol. 2017, 8, 38. [Google Scholar] [CrossRef]
- Ramachandran, S.; Xie, L.H.; John, S.A.; Subramaniam, S.; Lal, R. A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS ONE 2007, 2, e712. [Google Scholar] [CrossRef]
- Chi, Y.; Zhang, X.; Zhang, Z.; Mitsui, T.; Kamiyama, M.; Takeda, M.; Yao, J. Connexin43 hemichannels contributes to the disassembly of cell junctions through modulation of intracellular oxidative status. Redox Biol. 2016, 9, 198–209. [Google Scholar] [CrossRef]
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Bennett, M.V.; Sáez, J.C. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc. Natl. Acad. Sci. USA 2007, 104, 8322–8327. [Google Scholar] [CrossRef]
- Fang, X.; Huang, T.; Zhu, Y.; Yan, Q.; Chi, Y.; Jiang, J.X.; Wang, P.; Matsue, H.; Kitamura, M.; Yao, J. Connexin43 hemichannels contribute to cadmium-induced oxidative stress and cell injury. Antioxid. Redox Signal. 2011, 14, 2427–2439. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid. Med. Cell. Longev. 2018, 2018, 2063179. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Kavazis, A.N.; DeRuisseau, K.C. Mechanisms of disuse muscle atrophy: Role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R337–R344. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Smuder, A.J.; Judge, A.R. Oxidative stress and disuse muscle atrophy: Cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 240–245. [Google Scholar] [CrossRef]
- Oshima, Y.; Kuroda, Y.; Kunishige, M.; Matsumoto, T.; Mitsui, T. Oxidative stress-associated mitochondrial dysfunction in corticosteroid-treated muscle cells. Muscle Nerve 2004, 30, 49–54. [Google Scholar] [CrossRef]
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998, 275, C1–C24. [Google Scholar] [CrossRef]
- Kim, H.; Jang, M.; Park, R.; Jo, D.; Choi, I.; Choe, J.; Oh, W.K.; Park, J. Conessine treatment reduces dexamethasone-induced muscle atrophy by regulating murf1 and atrogin-1 expression. J. Microbiol. Biotechnol. 2018, 28, 520–526. [Google Scholar] [CrossRef]
- Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. Traf6 inhibition rescues dexamethasone-induced muscle atrophy. Int. J. Mol. Sci. 2014, 15, 11126–11141. [Google Scholar] [CrossRef]
- Orzechowski, A.; Ostaszewski, P.; Wilczak, J.; Jank, M.; Balasinska, B.; Wareski, P.; Fuller, J., Jr. Rats with a glucocorticoid-induced catabolic state show symptoms of oxidative stress and spleen atrophy: The effects of age and recovery. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2002, 49, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Konno, S. Hydroxyl radical formation in skeletal muscle of rats with glucocorticoid-induced myopathy. Neurochem. Res. 2005, 30, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Griendling, K.K.; Harrison, D.G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 2007, 49, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Chen, Y.; Li, A.S.; Reid, M.B. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific e2 and e3 proteins in skeletal muscle myotubes. Am. J. Physiol. Cell Physiol. 2003, 285, C806–C812. [Google Scholar] [CrossRef]
- Powers, S.K.; Smuder, A.J.; Criswell, D.S. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid. Redox Signal. 2011, 15, 2519–2528. [Google Scholar] [CrossRef]
- Smuder, A.J.; Kavazis, A.N.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic. Biol. Med. 2010, 49, 1152–1160. [Google Scholar] [CrossRef]
- Nakazawa, K.; Liu, M.; Inoue, K.; Ohno, Y. Potent inhibition by trivalent cations of atp-gated channels. Eur. J. Pharm. 1997, 325, 237–243. [Google Scholar] [CrossRef]
- Riquelme, M.A.; Jiang, J.X. Elevated intracellular ca(2+) signals by oxidative stress activate connexin 43 hemichannels in osteocytes. Bone Res. 2013, 1, 355–361. [Google Scholar] [CrossRef]
- Orellana, J.A.; Díaz, E.; Schalper, K.A.; Vargas, A.A.; Bennett, M.V.; Sáez, J.C. Cation permeation through connexin 43 hemichannels is cooperative, competitive and saturable with parameters depending on the permeant species. Biochem. Biophys. Res. Commun. 2011, 409, 603–609. [Google Scholar] [CrossRef]
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 hemichannels mediate the ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef] [PubMed]
- Sáez, J.C.; Schalper, K.A.; Retamal, M.A.; Orellana, J.A.; Shoji, K.F.; Bennett, M.V. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 2010, 316, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Fang, Q.; Xu, T.; Wu, C.; Xu, L.; Wang, L.; Yang, X.; Yu, S.; Zhang, Q.; Ding, F.; et al. Mechanistic role of reactive oxygen species and therapeutic potential of antioxidants in denervation- or fasting-induced skeletal muscle atrophy. Front. Physiol. 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Rizvi, S.; Raza, S.T.; Ahmed, F.; Ahmad, A.; Abbas, S.; Mahdi, F. The role of vitamin e in human health and some diseases. Sultan Qaboos Univ. Med. J. 2014, 14, e157–e165. [Google Scholar]
- Cea, L.A.; Balboa, E.; Vargas, A.A.; Puebla, C.; Branes, M.C.; Escamilla, R.; Regueira, T.; Saez, J.C. De novo expression of functional connexins 43 and 45 hemichannels increases sarcolemmal permeability of skeletal myofibers during endotoxemia. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 2765–2773. [Google Scholar] [CrossRef]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Fernandez, P.; Escamilla, R.; Lagos, C.F.; Matus, M.F.; Vilos, C.; Cea, L.A.; Barnafi, E.; et al. Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat. Commun. 2020, 11, 1073. [Google Scholar] [CrossRef]
- Li, K.; Chi, Y.; Gao, K.; Yan, Q.; Matsue, H.; Takeda, M.; Kitamura, M.; Yao, J. Connexin43 hemichannel-mediated regulation of connexin43. PLoS ONE 2013, 8, e58057. [Google Scholar] [CrossRef]
- Figueroa, V.A.; Retamal, M.A.; Cea, L.A.; Salas, J.D.; Vargas, A.A.; Verdugo, C.A.; Jara, O.; Martinez, A.D.; Sáez, J.C. Extracellular gentamicin reduces the activity of connexin hemichannels and interferes with purinergic ca(2+) signaling in hela cells. Front. Cell Neurosci. 2014, 8, 265. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Riquelme, M.A.; Werner, S.; Jiang, J.X. Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J. Bone Min. Res. 2013, 28, 1611–1621. [Google Scholar] [CrossRef]
- Lewandowski, M.; Gwozdzinski, K. Nitroxides as antioxidants and anticancer drugs. Int. J. Mol. Sci. 2017, 18, 2490. [Google Scholar] [CrossRef]
- Oliveira, A.L.B.; Monteiro, V.V.S.; Navegantes-Lima, K.C.; Reis, J.F.; Gomes, R.S.; Rodrigues, D.V.S.; Gaspar, S.L.F.; Monteiro, M.C. Resveratrol role in autoimmune disease-a mini-review. Nutrients 2017, 9, 1306. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ros: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Pozzan, T.; Rizzuto, R.; Volpe, P.; Meldolesi, J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994, 74, 595–636. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Sáez, J.C. Connexin hemichannels explain the ionic imbalance and lead to atrophy in denervated skeletal muscles. Biochim. Biophys. Acta 2016, 1862, 2168–2176. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Palacios-Prado, N.; Retamal, M.A.; Shoji, K.F.; Martinez, A.D.; Sáez, J.C. Connexin hemichannel composition determines the fgf-1-induced membrane permeability and free [ca2+]i responses. Mol. Biol. Cell 2008, 19, 3501–3513. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, atp, and ros: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Hasselgren, P.O. Glucocorticoids and muscle catabolism. Curr. Opin. Clin. Nutr. Metab. Care 1999, 2, 201–205. [Google Scholar] [CrossRef]
- Hyatt, H.; Deminice, R.; Yoshihara, T.; Powers, S.K. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Arch. Biochem. Biophys. 2019, 662, 49–60. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Mitochondrial quality control and muscle mass maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Smuder, A.J.; Min, K.; Kwon, O.S.; Szeto, H.H.; Powers, S.K. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J. Appl. Physiol. 2013, 115, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Hudson, M.B.; Nelson, W.B.; Talbert, E.E.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Smuder, A.J. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 2011, 39, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, Y.; Wang, X.; Fan, Y.; Qin, C.; Shi, L.; Tang, Y.; Cao, K.; Li, H.; Long, J. Mitochondrial dysfunction launches dexamethasone-induced skeletal muscle atrophy via ampk/foxo3 signaling. Mol. Pharm. 2016, 13, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Hwang, J.W.; Yun, C.K.; Lee, Y.; Choi, Y.S. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci. Rep. 2018, 8, 3330. [Google Scholar] [CrossRef]
- Riquelme, M.A.; Cea, L.A.; Vega, J.L.; Boric, M.P.; Monyer, H.; Bennett, M.V.; Frank, M.; Willecke, K.; Saez, J.C. The atp required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 2013, 75, 594–603. [Google Scholar] [CrossRef]
- Elfgang, C.; Eckert, R.; Lichtenberg-Frate, H.; Butterweck, A.; Traub, O.; Klein, R.A.; Hulser, D.F.; Willecke, K. Specific permeability and selective formation of gap junction channels in connexin-transfected hela cells. J. Cell Biol. 1995, 129, 805–817. [Google Scholar] [CrossRef]
- Balboa, E.; Saavedra-Leiva, F.; Cea, L.A.; Vargas, A.A.; Ramírez, V.; Escamilla, R.; Sáez, J.C.; Regueira, T. Sepsis-induced channelopathy in skeletal muscles is associated with expression of non-selective channels. Shock 2018, 49, 221–228. [Google Scholar] [CrossRef]
- Roelofs, B.A.; Ge, S.X.; Studlack, P.E.; Polster, B.M. Low micromolar concentrations of the superoxide probe mitosox uncouple neural mitochondria and inhibit complex iv. Free Radic. Biol. Med. 2015, 86, 250–258. [Google Scholar] [CrossRef]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. Mitosox-based flow cytometry for detecting mitochondrial ros. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef]
- Lassnig, B.; Stadlmann, S.; Rieger, G.; Haffner, B.; Lemieux, H.; Gnaiger, E. Selected media and chemicals for respirometry with mitochondria and permeabilized cells. Mito. Phys. Netw. 2008, 12, 1–9. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balboa, E.; Saavedra, F.; Cea, L.A.; Ramírez, V.; Escamilla, R.; Vargas, A.A.; Regueira, T.; Sáez, J.C. Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles. Int. J. Mol. Sci. 2020, 21, 4094. https://doi.org/10.3390/ijms21114094
Balboa E, Saavedra F, Cea LA, Ramírez V, Escamilla R, Vargas AA, Regueira T, Sáez JC. Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles. International Journal of Molecular Sciences. 2020; 21(11):4094. https://doi.org/10.3390/ijms21114094
Chicago/Turabian StyleBalboa, Elisa, Fujiko Saavedra, Luis A. Cea, Valeria Ramírez, Rosalba Escamilla, Aníbal A. Vargas, Tomás Regueira, and Juan C. Sáez. 2020. "Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles" International Journal of Molecular Sciences 21, no. 11: 4094. https://doi.org/10.3390/ijms21114094
APA StyleBalboa, E., Saavedra, F., Cea, L. A., Ramírez, V., Escamilla, R., Vargas, A. A., Regueira, T., & Sáez, J. C. (2020). Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles. International Journal of Molecular Sciences, 21(11), 4094. https://doi.org/10.3390/ijms21114094