1. Introduction
The current gold standard for the evaluation of chemical toxicity is based on animal testing. Increasingly, however, high-throughput in vitro experiments and in silico approaches are becoming valuable complements to animal experiments for identifying the mechanisms underlying chemical-induced toxicity [
1,
2,
3,
4,
5,
6,
7]. Two major questions in toxicology have yet to be satisfactorily resolved. First, does the cellular response induced by a toxic chemical (i.e., toxicant) in vitro correlate with the injury response induced by that toxicant in vivo? Second, how well does the toxicant-induced response in a non-human animal translate to that in a human? Interspecies comparisons are complex due to differences in genetics and bioavailability [
8,
9,
10]. Many compounds may be toxic for one species but benign for others [
11]. For example, theobromine, the compound that gives chocolate its bitter taste, can be lethal for dogs and cats, but not for humans, who have an enzyme that easily metabolizes it [
12].
One way to try to bridge the gap between in vivo and in vitro results is to analyze the genomic response induced by a toxicant, i.e., toxicogenomics. In the field of toxicogenomics, a common assumption is that toxicity is associated with a change in the expression of either a single gene or a set of genes (i.e., a gene module) [
13,
14,
15,
16], and that chemical exposures leading to the same injury endpoint cause similar changes in gene expression.
Using gene expression data from the Open Toxicogenomics Project-Genomics Assisted Toxicity Evaluation System (TG-GATEs) database, which contains data from Sprague–Dawley rats exposed to different chemicals for 4 to 29 days [
17], we derived 11 and 8 chemical-induced injury modules (i.e., gene modules, each uniquely associated with a specific organ-injury phenotype) for the liver and kidney, respectively, based on the histopathological injury phenotypes documented in the same database [
18]. In a subsequent study, we validated these injury modules in vivo by treating Sprague–Dawley rats with thioacetamide [
19], an organosulfur compound extensively used in animal studies as a fibrosis-promoting liver toxicant [
20,
21,
22,
23]. Thioacetamide is highly toxic because it is rapidly metabolized in vivo by cytochrome P450 and flavin-containing monooxygenases into reactive metabolites (thioacetamide-S-oxide to produce thioacetamide-S,S-dioxide and reactive oxygen species) [
24]. Our approach indicated cellular infiltration and fibrosis as the liver-injury endpoints most affected by thioacetamide [
19].
Recently, we found that our modular approach could predict in vivo injury endpoints from in vitro RNA sequence (RNA-seq) data with a strong correlation (
R2 > 0.6) [
25]. This study compared in vivo rat data with in vitro cellular data 24 h after treatment with thioacetamide-S-oxide, an intermediate metabolite of thioacetamide required to induce toxicity in vitro [
24]. The top ranked liver-injury modules identified by our in vitro studies using thioacetamide-S-oxide agreed with those identified in vivo using thioacetamide, indicating that in vitro cell injury was also associated with changes in the expression of fibrosis-related gene sets. These results suggest that, the predictions of our co-expressed gene module approach are more robust than those of gene signatures for specific pathologies, because they rely on groups of genes rather than individual genes.
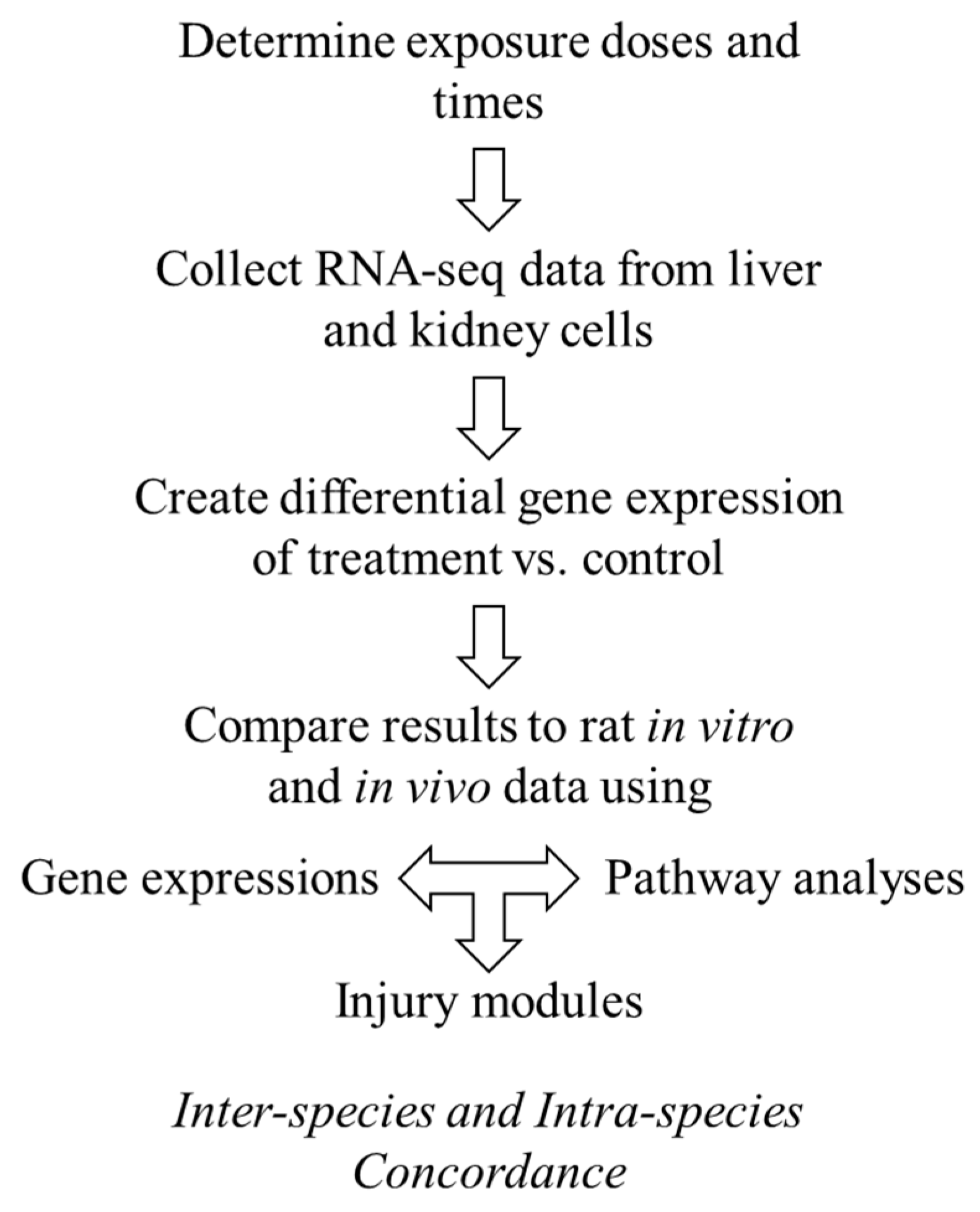
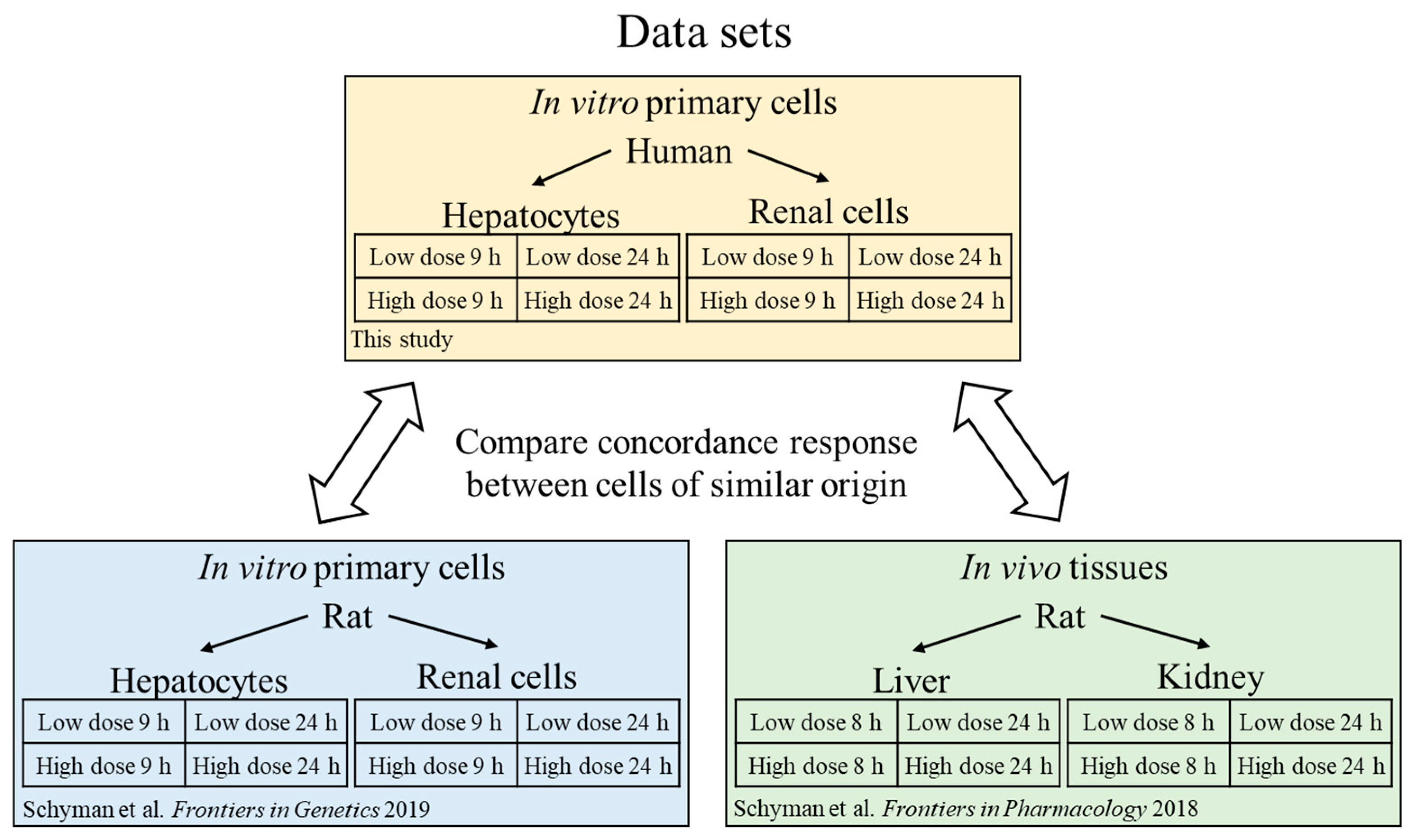
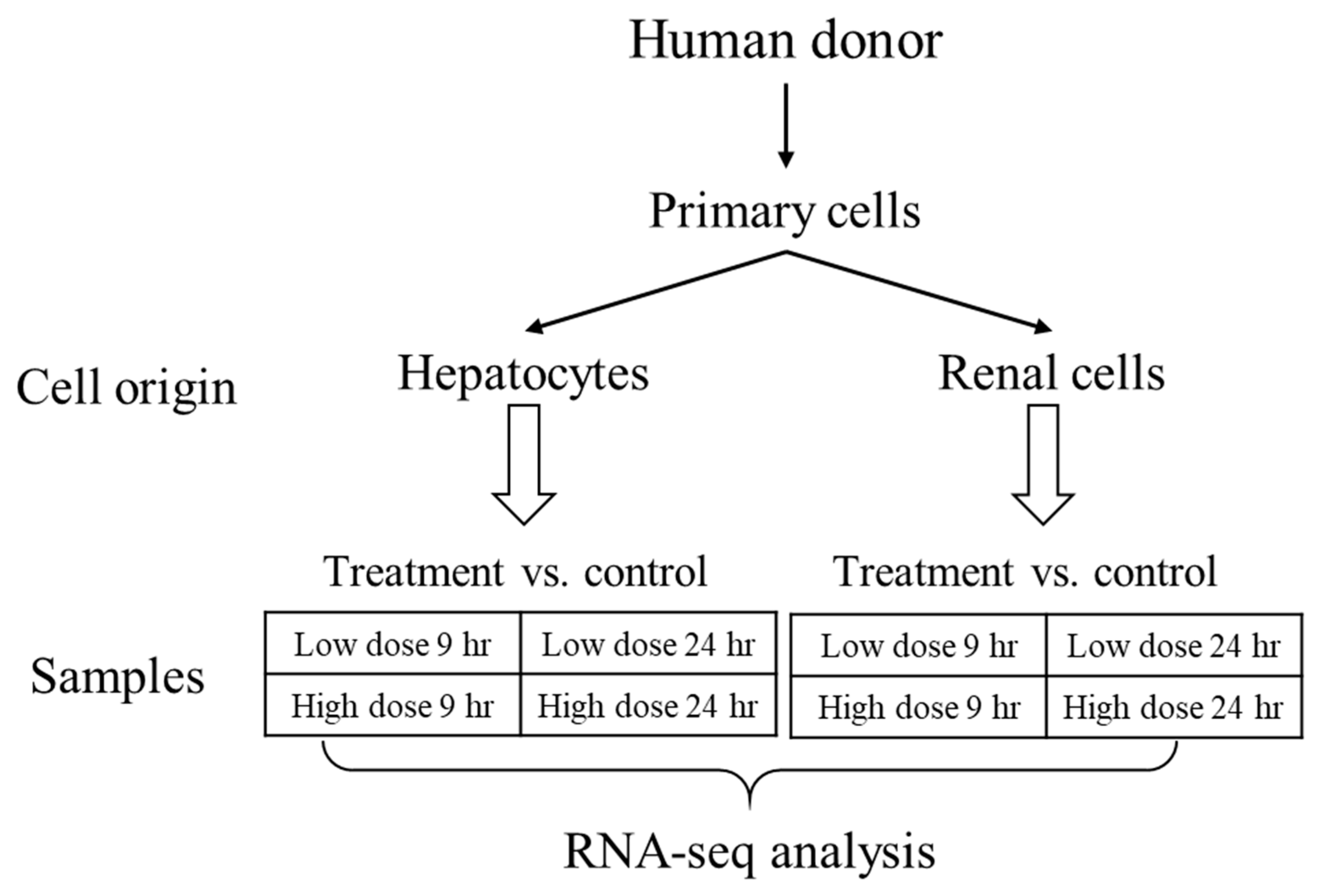
Here, we sought to explore the interspecies correlation using gene-expression responses in human cells exposed to thioacetamide-S-oxide in vitro with those induced by thioacetamide-S-oxide in cultured rat cells (in vitro) and by treating rats with thioacetamide (in vivo), following the diagram in
Figure 1. We chose thioacetamide as a model compound since it is known to cause liver diseases (fibrosis, cirrhosis, and carcinoma) in humans and is frequently used to study fibrosis/cirrhosis in animals [
26,
27,
28]. We used our injury modular approach to investigate interspecies correlations and compared it to gene-level and gene-pathway analyses. We found that the activation of our injury modules correlated well between humans and rats and between in vivo and in vitro studies, which was not the case at the individual gene or pathway level. To compare liver- and kidney-specific responses in vitro, we used two cell types from humans and rats—primary hepatocytes and renal tubule epithelial cells—and treated them with vehicle (control), a low dose of thioacetamide-S-oxide, or a high dose of the same compound.
3. Discussion
Extensive efforts have been made to understand the frequently poor correspondence of experimental results between different species or between in vitro and in vivo studies [
10,
29,
30,
31,
32]. We have previously used our toxicogenomic approach to predict rat in vivo toxicity endpoints from rat in vitro RNA-seq data with good concordance [
25]. A persisting challenge, however, has been to understand the often weak interspecies correlations. Here we sought to address this using human and rat data.
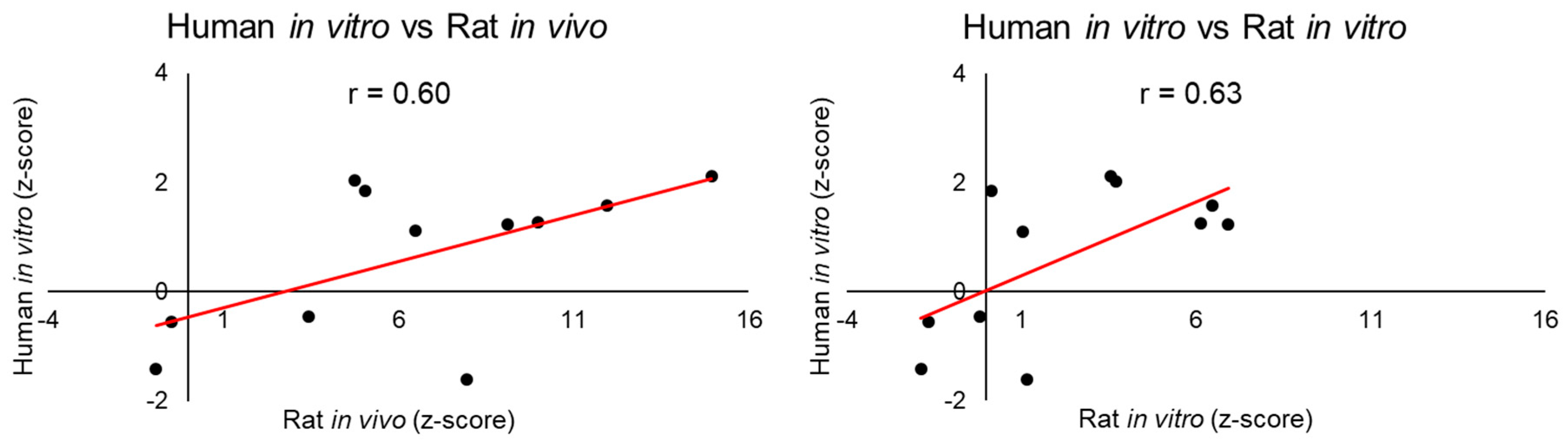
We examined the correlations between the injury modules activated in human-derived cells with those activated in rat tissues or cells. We found correlations between the liver-injury modules activated in human cells in vitro and those activated in the rat, both in vivo and in vitro, 24 h after high-dose treatment (
Figure 4). This is in accord with the observation that, a chemical-induced injury requires time to develop into an injury-specific phenotype, such as liver fibrosis. In contrast, no significant positive interspecies correlations were found among the kidney-injury modules. These findings demonstrate the sensitivity of the modular approach, in that injury modules show low activation scores when no injury is observed. The interspecies correlation result is encouraging given the relatively weak toxicity response in human primary cells. Another limitation of using commercially available human primary cells is that they are frequently derived from a single donor with potentially preexisting conditions, and whose cause of death is often unknown.
We examined the DEGs in the liver 24 h after high-dose treatment, where we observed interspecies correlations (
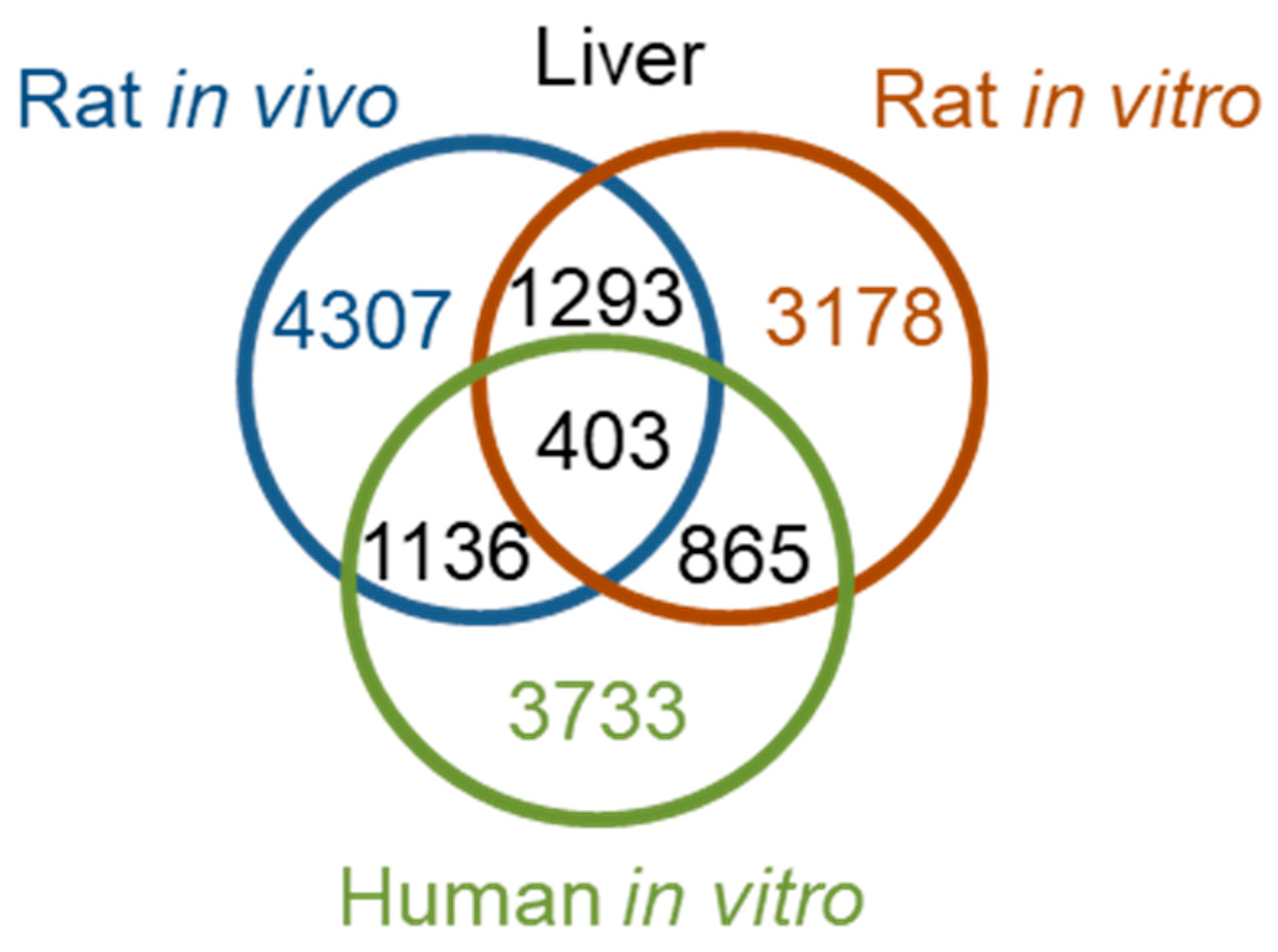
Table 3). Notably, 24 h after thioacetamide/thioacetamide-S-oxide treatment, the numbers of DEGs in human primary hepatocytes, rat hepatocytes
in vivo, and rat primary hepatocytes were comparable, amounting to 3733 (the number of rat genes orthologous to the 4267 human DEGs, identified using the
Ensemble website [
33]), 4307, and 3178, respectively.
Figure 5 shows a Venn diagram of the interspecies overlap of DEGs. There were 403 DEGs common to all three datasets—about 10% of the total DEGs identified in human primary hepatocytes, rat liver in vivo, and rat primary hepatocytes. Although the pair-wise overlaps were roughly 2 to 3 times higher, many DEGs were not shared between species or between in vitro and in vivo studies. In fact, the numbers of overlapping genes were close to the numbers expected to overlap by chance and would result in poor interspecies correlation. To analyze the KEGG pathways in which the 403 overlapping DEGs are involved, we used the David Gene Functional Classification Tool [
34,
35].
Table 5 shows the 10 most enriched KEGG pathways. The most significantly enriched pathway was the complement and coagulation cascade pathway, which plays an important role in fibrosis. However, it is frequently involved in immune responses, and is therefore not injury specific. Therefore, any mechanistic interpretation across species based on KEGG pathways would be difficult.
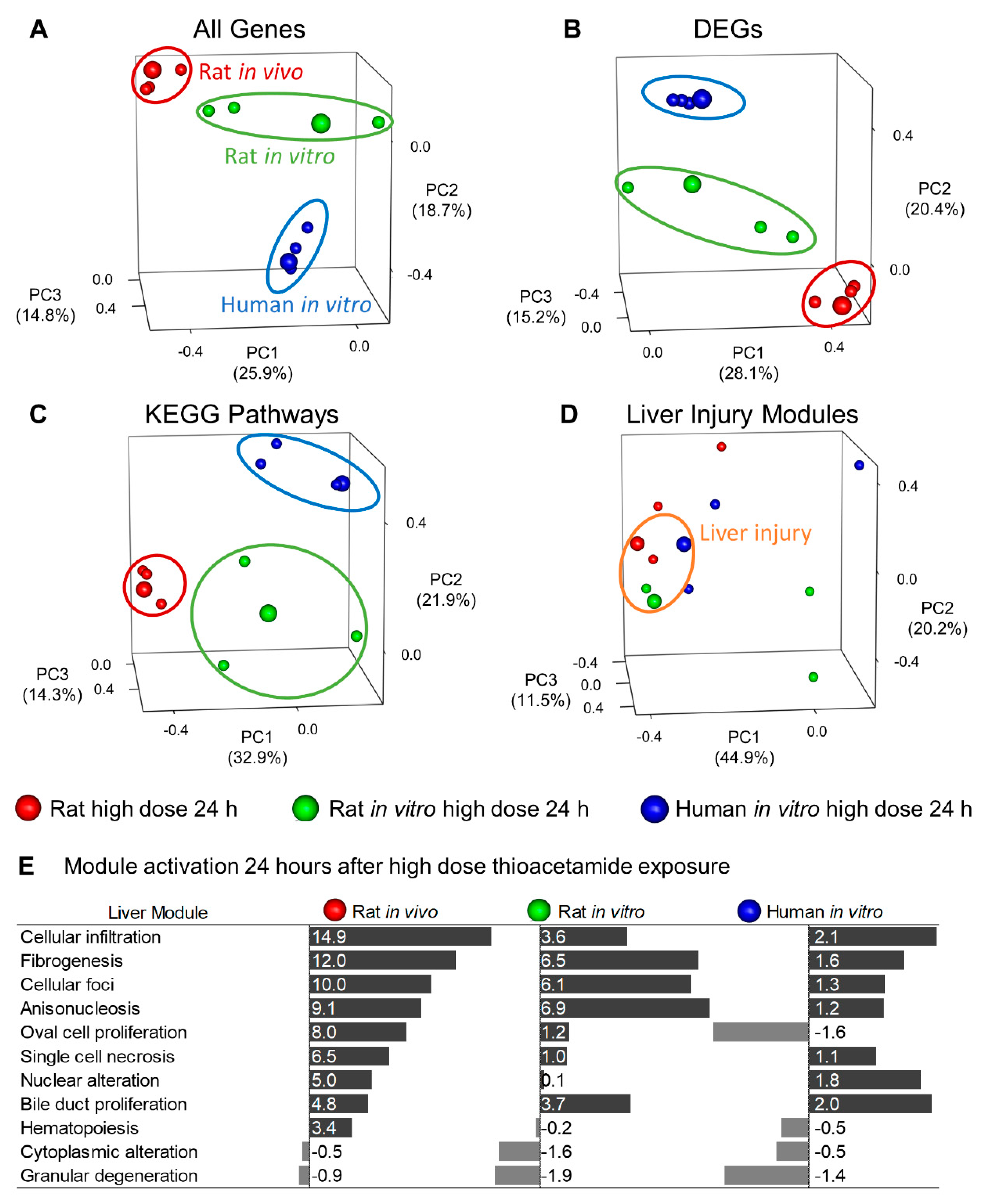
We performed principal component analysis (PCA) to qualitatively compare commonly used approaches (e.g., DEG and KEGG pathway approaches) for analyzing in vitro, in vivo, and interspecies correlations. To facilitate interpretation of the PCA plots, we have enlarged the spheres corresponding to the high-dose 24-h condition, where we expect the highest toxicity. The PCA plots in
Figure 6A–C show clustering of the rat in vivo, rat in vitro, and human in vitro data by systems rather than by conditions, as the three enlarged spheres were distinctly separated for each analysis approach.
Although we may have expected system-specific separation in the PCA plot for the individual-gene approach, we expected to see greater condition-specific separation in the plot for the KEGG pathway approach, which involves sets of genes. The KEGG Pathway plot illustrates that different KEGG pathways drive the principal components in different systems, making it difficult to mechanistically interpret an interspecies or in vitro–in vivo comparison. However, the injury module approach clustered the data points by conditions (enlarged spheres) associated with liver injury (orange ellipse). The small red and green spheres within the orange ellipse represent the low-dose 24-h conditions for the rat in vivo and in vitro data, respectively. The injury module approach properly clustered these two conditions with the large spheres, as our injury modules also predicted liver injury for both conditions. The PCA in
Figure 6D illustrates that our modular approach to track injury was conserved across species and between in vitro and in vivo systems.
The primary liver-injury endpoint of thioacetamide is fibrosis. Although the fibrogenesis module was not the top-ranked injury module in the human in vitro assay (
Figure 6E), other injury modules linked to known histopathological endpoints, such as those of bile duct proliferation and cellular infiltration, were activated [
17]. Notably, 24 h after high-dose treatment in the human in vitro assay with thioacetamide-S-oxide, we observed bile duct proliferation and cellular infiltration (
Figure 6E and
Table 1), which are associated with fibrosis [
36].
In our previous studies [
19,
25], we found that activation of the fibrogenesis module increased over time in the liver, an effect which we also observed here when comparing fibrogenesis module activation at 9 and 24 h in
Table 1. The fibrogenesis module activation score would likely have continued to increase after 24 h, consistent with the observation that this phenotype takes longer than 24 h to become histologically visible. The delay in the fibrosis response in the human study relative to the fibrosis response in the rat studies may also reflect a species difference.
In human renal cells, 24 h of high-dose thioacetamide-S-oxide treatment activated the fibrogenesis and degeneration modules (
Table 2). It is likely that these kidney-injury modules were activated within 24 h of thioacetamide-S-oxide exposure because a relatively high dose (1 mM) was required to detect clear indications of injury in human renal cells. In fact, the dose for the low-dose thioacetamide-S-oxide treatment of human renal cells (0.25 mM) was equivalent to that of the high-dose treatment of human primary hepatocytes, and the only low-dose activated kidney-injury module was cellular infiltration—a common response to chemical toxicity.
In summary, we show that using human in vitro cell responses based on RNA-seq data with an injury-module approach, rather than the commonly used DEGs/pathway approach, yields significant correlations with injury-module activation of rat in vivo and in vitro responses to a similar exposure of the toxicant, thioacetamide or its metabolite. Consequently, our injury-module approach could potentially be used to screen large numbers of chemicals in vitro and predict liver and kidney injuries, and thereby improve the efficiency of toxicity assessments by reducing the number of animals needed in experiments.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}