The Structural Basis of the Binding of Various Aminopolycarboxylates by the Periplasmic EDTA-Binding Protein EppA from Chelativorans sp. BNC1

 , and
, and
Abstract
1. Introduction
2. Results
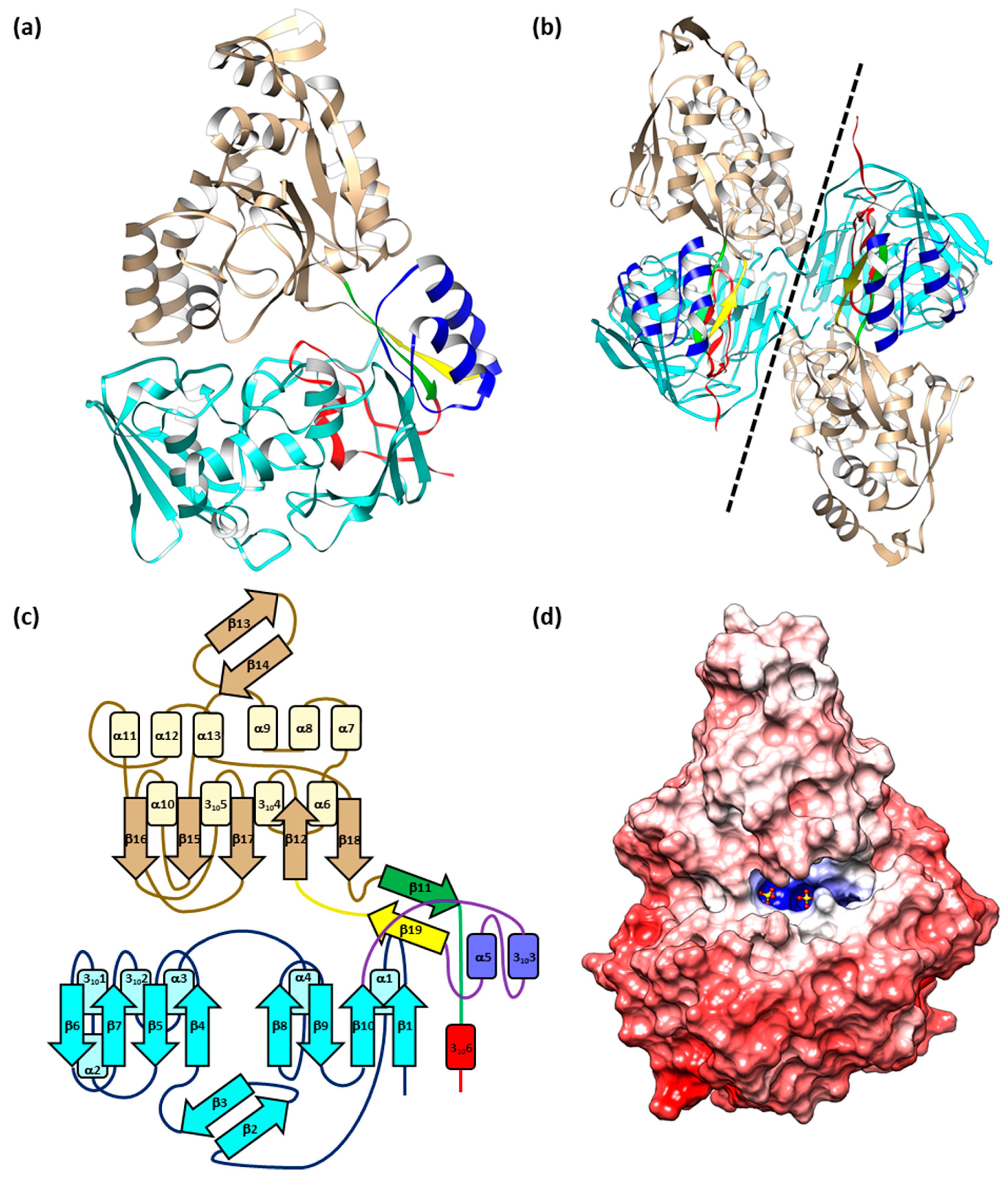
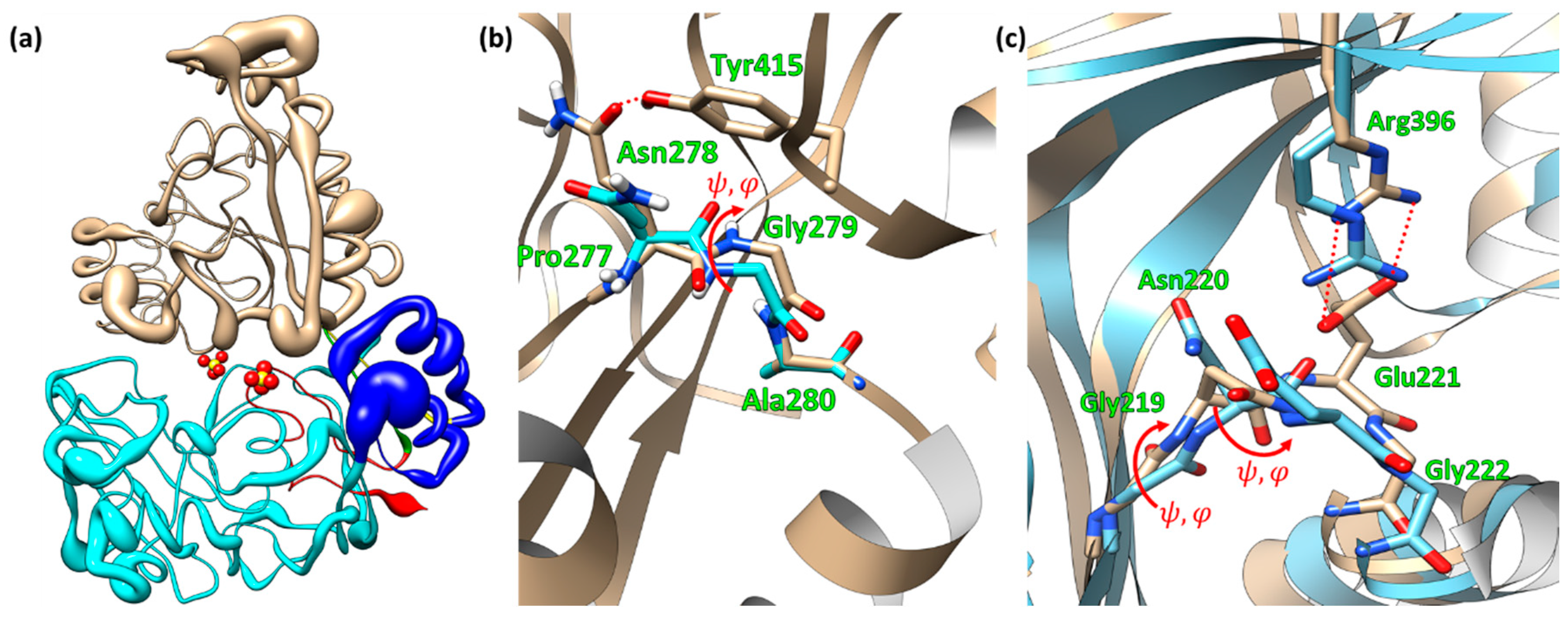
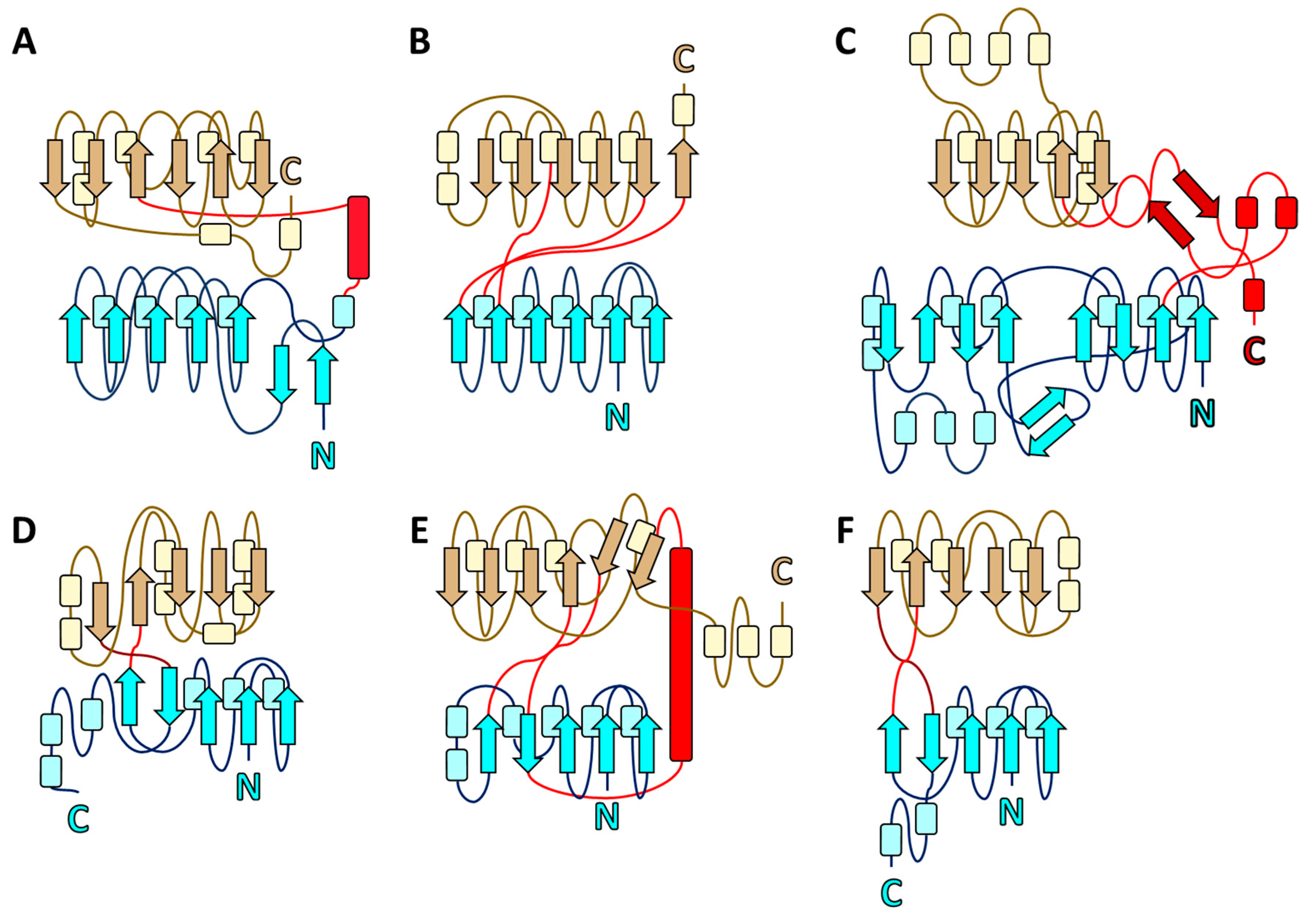
2.1. Structure of EppA
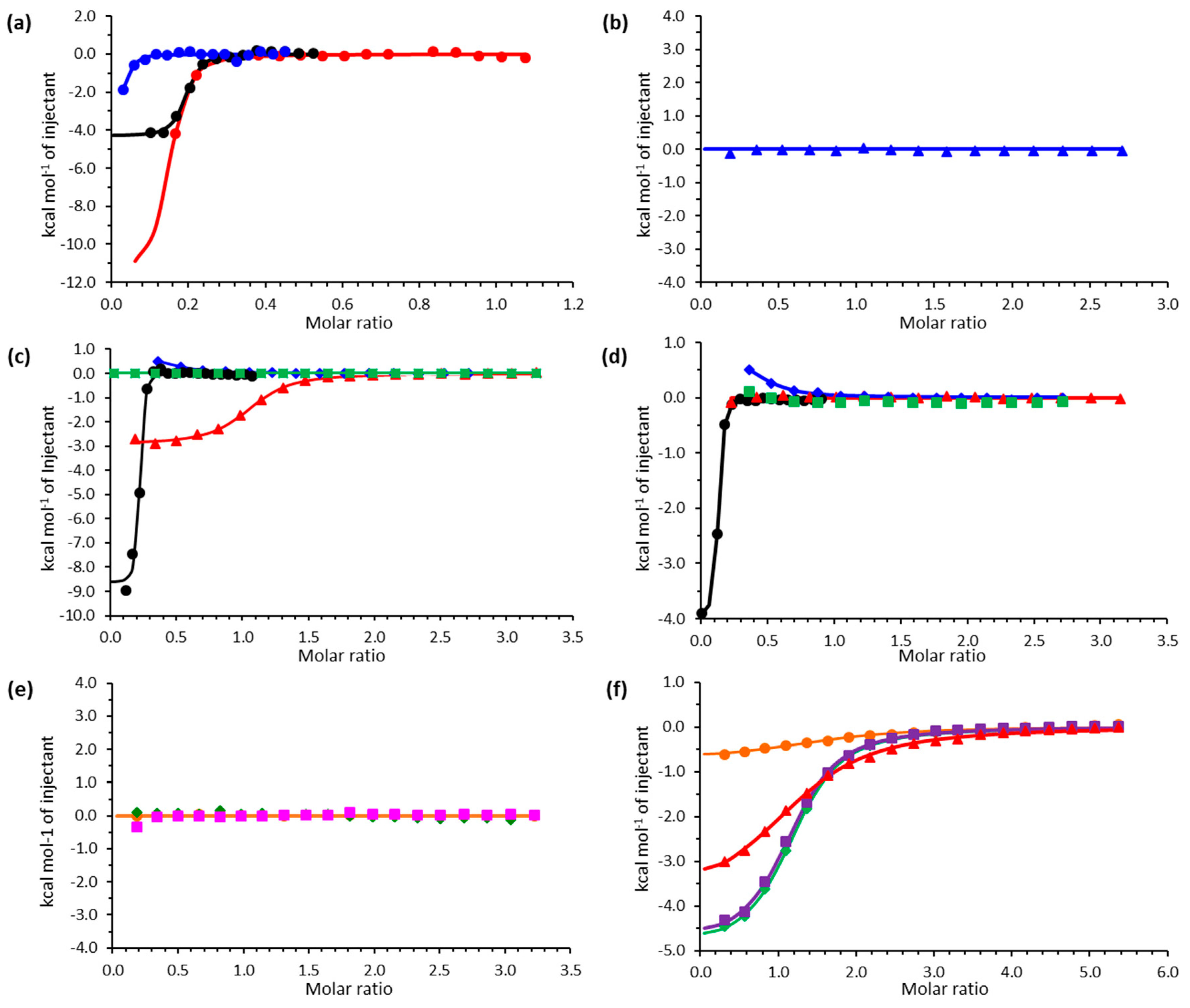
2.2. Isothermal Titration Calorimetry
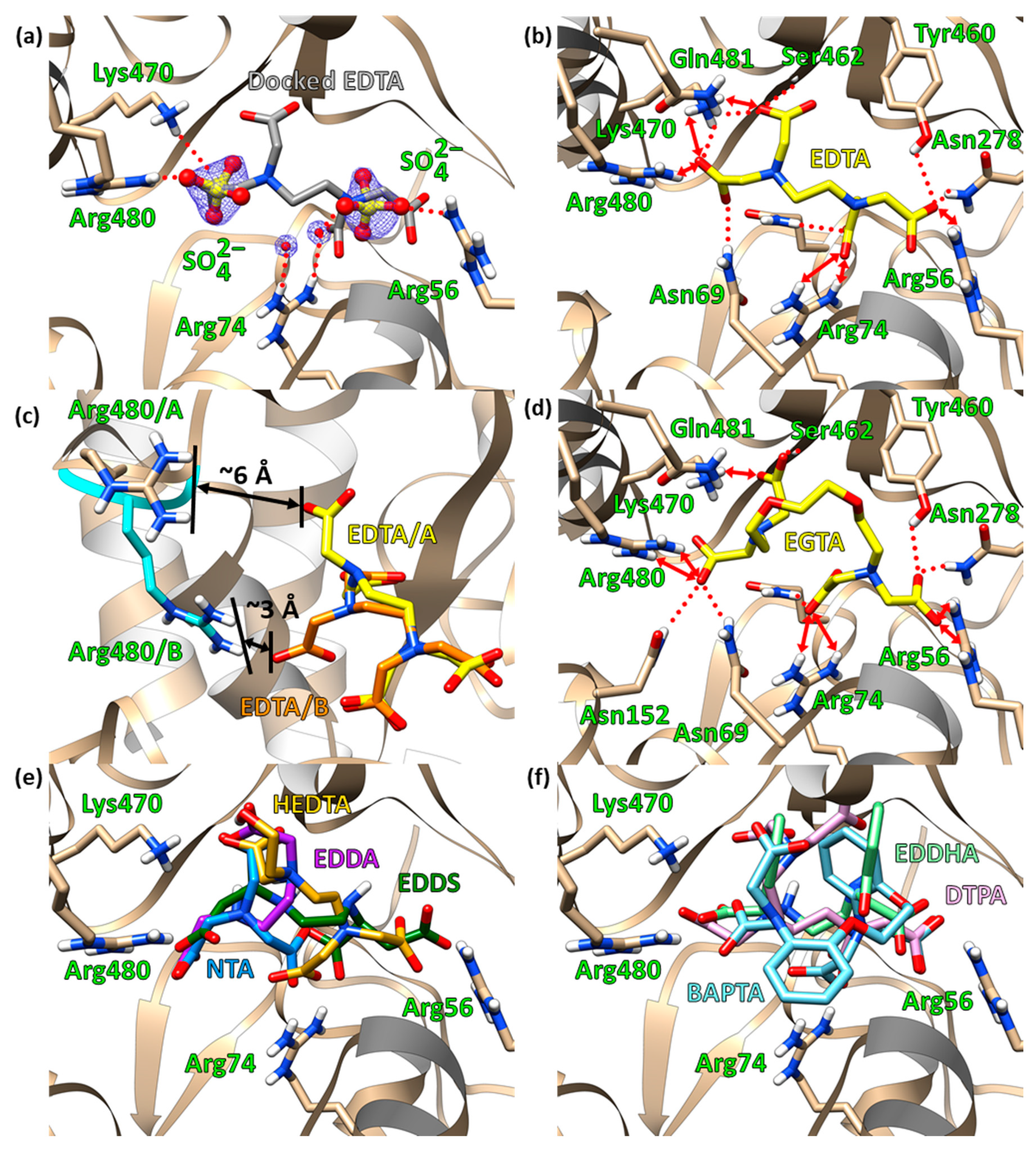
2.3. Molecular Docking
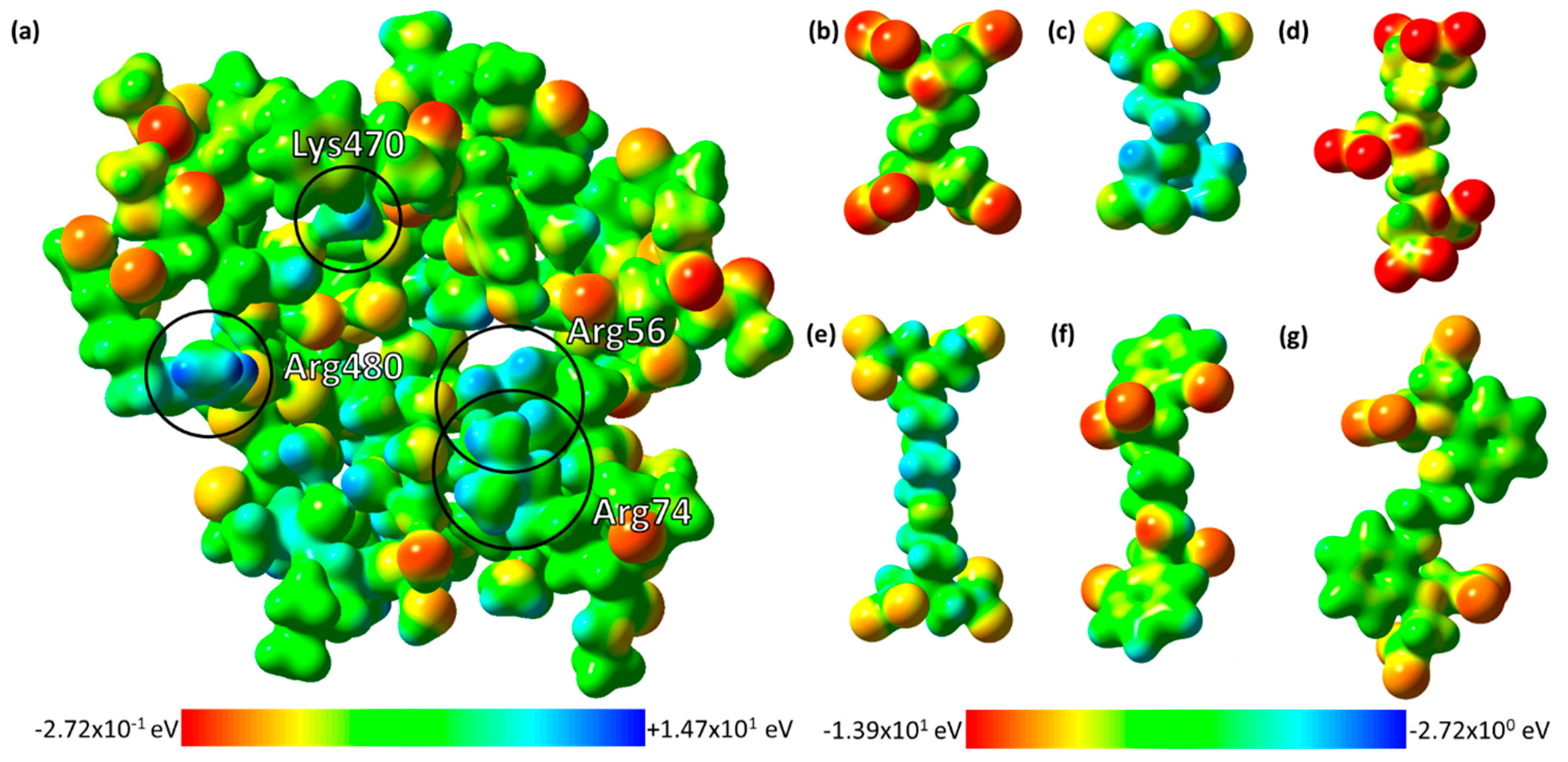
2.4. The Electrostatic Potentials of the Ligand-Binding Pocket
2.5. Site-Directed Mutagenetic Analysis of Key Binding Residues
2.6. Structural Homologs of EppA and Evolutionary Conservation
3. Discussion
4. Materials and Methods
4.1. Site-Directed Mutagenesis
4.2. Protein Expression and Purification
4.3. Molecular Mass Determination
4.4. Crystallization
4.5. Structure Determination
4.6. Structure Analysis
4.7. Isothermal Titration Calorimetry
4.8. Quantum Chemical Optimization and Electrostatics
4.9. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bucheli-Witschel, M.; Egli, T. Environmental fate and microbial degradation of aminopolycarboxylic acids. FEMS Microbiol Rev. 2001, 25, 69–106. [Google Scholar] [CrossRef] [PubMed]
- Toste, A.; Osborn, B.; Polach, K.; Lechner-Fish, T. Organic analyses of an actual and simulated mixed waste: Hanford’s organic complexant revisited. J. Radioanal. Nucl. Chem. 1995, 194, 25–34. [Google Scholar] [CrossRef]
- Barber, L.B.; Leenheer, J.A.; Pereira, W.E.; Noyes, T.I.; Brown, G.K.; Tabor, C.F.; Writer, J.H.E. Contaminants in the Mississippi River, 1987-92; U.S. Goverment Printing Office: Washington, DC, USA, 1995. [Google Scholar]
- Dirilgen, N. Effects of pH and chelator EDTA on Cr toxicity and accumulation in Lemma minor. Chemosphere 1998, 37, 771–783. [Google Scholar] [CrossRef]
- Sillanpaa, M.; Oikari, A. Assessing the impact of complexation by EDTA and DTPA on heavy metal toxicity using Microtox bioassay. Chemosphere 1996, 32, 1485–1497. [Google Scholar] [CrossRef]
- Means, J.L.; Crerar, D.A.; Duguid, J.O. Migration of radioactive wastes: Radionuclide mobilization by complexing agents. Science 1978, 200, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, J.M.; Rees, T.F. Characterization of plutonium in maxey flats radioactive trench leachates. Science 1981, 212, 1506–1509. [Google Scholar] [CrossRef]
- Witschel, M.; Egli, T. Purification and characterization of a lyase from the EDTA-degrading bacterial strain DSM 9103 that catalyzes the splitting of [S,S]-ethylenediaminedisuccinate, a structural isomer of EDTA. Biodegradation 1997, 8, 419–428. [Google Scholar] [CrossRef]
- Pinto, I.S.; Neto, I.F.; Soares, H.M. Biodegradable chelating agents for industrial, domestic, and agricultural applications—A review. Environ. Sci Pollut. Res. Int. 2014, 21, 11893–11906. [Google Scholar] [CrossRef]
- Fang, H.Y.; Chen, S.C.; Chen, S.L. Degradation of ferric EDTA by Burkhol cepacia. Appl. Biochem. Biotechnol. 2003, 111, 81–92. [Google Scholar] [CrossRef]
- Lauff, J.J.; Steele, D.B.; Coogan, L.A.; Breitfeller, J.M. Degradation of the Ferric Chelate of EDTA by a Pure Culture of an Agrobacterium sp. Appl. Environ. Microbiol. 1990, 56, 3346–3353. [Google Scholar] [CrossRef]
- Nortemann, B. Biodegradation of EDTA. Appl. Microbiol. Biotechnol. 1999, 51, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Witschel, M.; Nagel, S.; Egli, T. Identification and characterization of the two-enzyme system catalyzing oxidation of EDTA in the EDTA-degrading bacterial strain DSM 9103. J. Bacteriol. 1997, 179, 6937–6943. [Google Scholar] [CrossRef] [PubMed]
- Weilenmann, H.U.; Engeli, B.; Bucheli-Witschel, M.; Egli, T. Isolation and growth characteristics of an EDTA-degrading member of the alpha-subclass of Proteobacteria. Biodegradation 2004, 15, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Doronina, N.V.; Kaparullina, E.N.; Trotsenko, Y.A.; Nortemann, B.; Bucheli-Witschel, M.; Weilenmann, H.U.; Egli, T. Chelativorans multitrophicus gen. nov., sp. nov. and Chelativorans oligotrophicus sp. nov., aerobic EDTA-degrading bacteria. Int. J. Syst. Evol. Microbiol. 2010, 60, 1044–1051. [Google Scholar] [CrossRef][Green Version]
- Bohuslavek, J.; Payne, J.W.; Liu, Y.; Bolton, H., Jr.; Xun, L. Cloning, sequencing, and characterization of a gene cluster involved in EDTA degradation from the bacterium BNC1. Appl. Environ. Microbiol. 2001, 67, 688–695. [Google Scholar] [CrossRef]
- Nortemann, B. Total degradation of EDTA by mixed cultures and a bacterial isolate. Appl. Environ. Microbiol. 1992, 58, 671–676. [Google Scholar] [CrossRef]
- Zhang, H.; Herman, J.P.; Bolton, H., Jr.; Zhang, Z.; Clark, S.; Xun, L. Evidence that bacterial ABC-type transporter imports free EDTA for metabolism. J. Bacteriol. 2007, 189, 7991–7997. [Google Scholar] [CrossRef][Green Version]
- Nissen, M.S.; Youn, B.; Knowles, B.D.; Ballinger, J.W.; Jun, S.Y.; Belchik, S.M.; Xun, L.; Kang, C. Crystal structures of NADH:FMN oxidoreductase (EmoB) at different stages of catalysis. J. Biol. Chem. 2008, 283, 28710–28720. [Google Scholar] [CrossRef]
- Jun, S.Y.; Lewis, K.M.; Youn, B.; Xun, L.; Kang, C. Structural and biochemical characterization of EDTA monooxygenase and its physical interaction with a partner flavin reductase. Mol. Microbiol. 2016, 100, 989–1003. [Google Scholar] [CrossRef]
- Liu, Y.; Louie, T.M.; Payne, J.; Bohuslavek, J.; Bolton, H., Jr.; Xun, L. Identification, purification, and characterization of iminodiacetate oxidase from the EDTA-degrading bacterium BNC1. Appl. Environ. Microbiol. 2001, 67, 696–701. [Google Scholar] [CrossRef]
- Kang, C.; Jun, S.Y.; Bravo, A.G.; Vargas, E.M.; Liu, H.; Lewis, K.M.; Xun, L. Structural and Biochemical Characterization of Iminodiacetate Oxidase from Chelativorans sp. BNC1. Mol. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.B.; Graul, R.C.; Lee, A.Y.; Merkle, H.P.; Sadee, W. The Venus flytrap of periplasmic binding proteins: An ancient protein module present in multiple drug receptors. AAPS PharmSci 1999, 1, E2. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.H.; Pandit, J.; Kang, C.H.; Nikaido, K.; Gokcen, S.; Ames, G.F.; Kim, S.H. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J. Biol. Chem. 1993, 268, 11348–11355. [Google Scholar] [PubMed]
- Berntsson, R.P.; Smits, S.H.; Schmitt, L.; Slotboom, D.J.; Poolman, B. A structural classification of substrate-binding proteins. FEBS Lett. 2010, 584, 2606–2617. [Google Scholar] [CrossRef]
- Fukami-Kobayashi, K.; Tateno, Y.; Nishikawa, K. Domain dislocation: A change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 1999, 286, 279–290. [Google Scholar] [CrossRef]
- Painter, J.; Merritt, E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006, 39, 109–111. [Google Scholar] [CrossRef]
- Heddle, J.; Scott, D.J.; Unzai, S.; Park, S.Y.; Tame, J.R. Crystal structures of the liganded and unliganded nickel-binding protein NikA from Escherichia coli. J. Biol. Chem. 2003, 278, 50322–50329. [Google Scholar] [CrossRef]
- Rink, T.J.; Hallam, T.J. What turns platelets on? Trends Biochem. Sci. 1984, 9, 215–219. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993, 233, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using Evolutionary Data to Raise Testable Hypotheses about Protein Function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Scheepers, G.H.; Lycklama, A.N.J.A.; Poolman, B. An updated structural classification of substrate-binding proteins. FEBS Lett. 2016, 590, 4393–4401. [Google Scholar] [CrossRef]
- Braibant, M.; Gilot, P.; Content, J. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 2000, 24, 449–467. [Google Scholar] [CrossRef]
- Cherrier, M.V.; Martin, L.; Cavazza, C.; Jacquamet, L.; Lemaire, D.; Gaillard, J.; Fontecilla-Camps, J.C. Crystallographic and spectroscopic evidence for high affinity binding of FeEDTA(H2O)- to the periplasmic nickel transporter NikA. J. Am. Chem. Soc. 2005, 127, 10075–10082. [Google Scholar] [CrossRef]
- Cherrier, M.V.; Cavazza, C.; Bochot, C.; Lemaire, D.; Fontecilla-Camps, J.C. Structural characterization of a putative endogenous metal chelator in the periplasmic nickel transporter NikA. Biochemistry 2008, 47, 9937–9943. [Google Scholar] [CrossRef] [PubMed]
- Nishikiori, T.; Okuyama, A.; Naganawa, H.; Takita, T.; Hamada, M.; Takeuchi, T.; Aoyagi, T.; Umezawa, H. Production by actinomycetes of (S,S)-N,N’-ethylenediamine-disuccinic acid, an inhibitor of phospholipase C. J. Antibiot. (Tokyo) 1984, 37, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Paolieri, M. Ferdinand Münz: EDTA and 40 years of inventions. Bull. Hist. Chem. 2017, 42, 133–140. [Google Scholar]
- Payne, J.W.; Bolton, H., Jr.; Campbell, J.A.; Xun, L. Purification and characterization of EDTA monooxygenase from the EDTA-degrading bacterium BNC1. J. Bacteriol. 1998, 180, 3823–3827. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. 1), S162–S173. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D Biol. Cryst. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst. D Biol. Cryst. 2008, 64, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.M.; Greene, C.L.; Sattler, S.A.; Youn, B.; Xun, L.; Kang, C. Periplasmic EDTA-Binding Protein EppA, Tetragonal; Research Collaboratory for Structural Bioinformatics Protein Data Bank 2020, 6WM6 [dataset]. Available online: http://www.rcsb.org/ (accessed on 30 May 2020).
- Lewis, K.M.; Greene, C.L.; Sattler, S.A.; Youn, B.; Xun, L.; Kang, C. Periplasmic EDTA-Binding Protein EppA, Orthorhombic; Research Collaboratory for Structural Bioinformatics Protein Data Bank 2020, 6WM7 [dataset]. Available online: http://www.rcsb.org/ (accessed on 30 May 2020).
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Schrödinger, L. Schrödinger Suite 2016-2 Protein Preparation Wizard; Maestro, Version 10.2; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 2797–2803. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5.0. 9; Semichem, Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Moriarty, N.W.; Grosse-Kunstleve, R.W.; Adams, P.D. Electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Cryst. D Biol. Cryst. 2009, 65, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Dunning Jr, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kd (μM) | ∆H (kcal mol−1) | ∆S (cal mol−1 K−1) | |
|---|---|---|---|
| Asp10 b | n.b. c | n.b. | n.b. |
| EDDA d | 0.340 ± 0.118 | −0.851 ± 0.893 | 26.7 |
| EDTA | 0.0095 ± 0.006 | −2.283 ± 0.07 | 29 |
| EGTA | 0.169 ± 0.71 | −4.34 ± 0.114 | 16.4 |
| MgEDTA | 0.315 ± 3.26 | −4.028 ± 0.037 | 16.2 |
| CaEDTA | 2.320 ± 20.41 | −2.917 ± 0.038 | 16.0 |
| SrEDTA | 1.69 ± 7.69 | −4.392 ± 0.118 | 27.1 |
| BaEDTA | n.b. | n.b. | n.b. |
| CrEDTA | 0.019 ± 0.002 | −2.573 ± 0.145 | 30.5 |
| MnEDTA | n.b. | n.b. | n.b. |
| FeEDTA | n.b. | n.b. | n.b. |
| CoEDTA | n.b. | n.b. | n.b. |
| NiEDTA | n.b. | n.b. | n.b. |
| CuEDTA | n.b. | n.b. | n.b. |
| ZnEDTA | n.b. | n.b. | n.b. |
| CdEDTA | 0.28 ± 0.49 | −0.555 ± 0.002 | 28.1 |
| PrEDTA | 6.17 ± 55.25 | −5.366 ± 0.133 | 5.83 |
| NdEDTA | 8.85 ± 141.2 | −4.889 ± 0.063 | 6.73 |
| EuEDTA | 26.45 ± 363.6 | −3.943 ± 0.105 | 7.72 |
| TbEDTA | 31.25 ± 116.4 | −0.751 ± 0.069 | 18.1 |
| MgEGTA | 0.315 ± 3.26 | −4.028 ± 0.037 | 16.2 |
| CaEGTA | n.b. | n.b. | n.b. |
| SrEGTA | 4.50 ± 10.5 | 0.794 ± 0.164 | 27.1 |
| BaEGTA | n.b. | n.b. | n.b. |
| Ligand * | ΔGbinding,A (kcal mol−1) | ΔGbinding,B (kcal mol−1) |
|---|---|---|
| EDTA | −6.4 | −6.6 |
| EGTA | −6.0 | −6.1 |
| NTA | −5.2 | −6.5 |
| EDDA | −5.2 | −4.9 |
| HEDTA | −6.0 | −6.1 |
| EDDS | −6.1 | −6.7 |
| DTPA | −6.6 | −6.7 |
| EDDHA | −7.0 | −7.0 |
| BAPTA | −6.9 | −6.9 |
| Kd (nM) | ∆H (kcal mol−1) | ∆S (cal mol−1 K−1) | |
|---|---|---|---|
| Wild-type | 9.52 ± 6.25 | −2.283 ± 0.070 | 29 |
| R56A | 161 ± 272 | −1.802 ± 0.147 | 25 |
| R74A | 249 ± 382 | −1.877 ± 0.195 | 23.9 |
| K470A | 27.1 ± 20.2 | −1.872 ± 0.117 | 28.3 |
| R480A | 40.8 ± 26.5 | −1.138 ± 0.089 | 30 |
| Protein | PDB ID | Source | % Identity | % Similarity | Z-Score |
|---|---|---|---|---|---|
| LpqW | 2GRV | Mycobacterium smegmatis | 22.5 | 38.8 | 29.0 |
| CBP | 2O7I | Thermotoga maritima | 19.6 | 33.5 | 24.4 |
| AgaA | 6HLX | Rhizobium radiobacter | 18.8 | 35.7 | 31.4 |
| OppA | 3DRF | Lactococcus lactis | 18.7 | 35.2 | 27.6 |
| OppA2 | 2WOK | Streptomyces clavuligerus | 18.3 | 32.3 | 29.1 |
| MnBP3 | 4PFT | Thermotoga maritima | 18.3 | 35.3 | 27.6 |
| MoaA | 6TFX | Rhizobium radiobacter | 18.2 | 33.5 | 30.3 |
| NikZ | 4OET | Campylobacter jejuni | 18.1 | 36.8 | 34.4 |
| CtaP | 5ISU | Listeria monocytogenes | 18.0 | 33.9 | 34.2 |
| CosBP | 1ZTY | Vibrio cholerae | 17.9 | 33.3 | 21.0 |
| EppA (apo) | EppA (EDTA Soak) | |
|---|---|---|
| Data collection | ||
| Space group | P43212 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 87.81, 87.81, 164.48 | 84.34, 90.61, 165.27 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 |
| Resolution (Å) | 49.46—1.42 (1.47—1.42) | 47.07—1.56 (1.62—1.56) |
| Rmerge | 0.16 (0.860) | 0.125 (1.59) |
| Wavelength (Å) | 0.9793 | 1.000 |
| Unique reflections | 122,095 (11,723) | 179,416 (17,354) |
| Completeness (%) | 97.40 (89.55) | 94.44 (77.22) |
| <I>/σI | 10.57 (4.77) | 12.65 (1.22) |
| CC1/2 | 0.996 (0.887) | 0.999 (0.375) |
| CC * | 0.999 (0.97) | 1 (0.738) |
| Multiplicity | 20.5 (16.6) | 13.5 (6.2) |
| Refinement | ||
| Rwork/Rfree | 0.145 (0.187)/0.153 (0.210) | 0.153 (0.310)/0.169 (0.317) |
| CCwork/CCfree | 0.971 (0.919)/0.966 (0.915) | 0.968 (0.709)/0.966 (0.691) |
| Number of atoms | ||
| Protein | 4209 | 8310 |
| Sulfate | 20 | 15 |
| Ethylene glycol | 63 | 80 |
| Water | 604 | 1285 |
| ADP (Å2) | ||
| All atoms | 26.05 | 21.33 |
| Protein | 24.45 | 19.57 |
| Ligands | 34.5 | 30.2 |
| Solvent | 36.58 | 32.04 |
| R.m.s deviations | ||
| Bonds (Å) | 0.008 | 0.008 |
| Angles (°) | 1.31 | 1.27 |
| Ramachandrans | ||
| % Favored | 98.31 | 98.12 |
| % Outliers | 0 | 0 |
| Rotamer outliers | 0.45 | 0.23 |
| Clashscore | 1.79 | 2.29 |
| TLS groups | 3 | 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, K.M.; Greene, C.L.; Sattler, S.A.; Youn, B.; Xun, L.; Kang, C. The Structural Basis of the Binding of Various Aminopolycarboxylates by the Periplasmic EDTA-Binding Protein EppA from Chelativorans sp. BNC1. Int. J. Mol. Sci. 2020, 21, 3940. https://doi.org/10.3390/ijms21113940
Lewis KM, Greene CL, Sattler SA, Youn B, Xun L, Kang C. The Structural Basis of the Binding of Various Aminopolycarboxylates by the Periplasmic EDTA-Binding Protein EppA from Chelativorans sp. BNC1. International Journal of Molecular Sciences. 2020; 21(11):3940. https://doi.org/10.3390/ijms21113940
Chicago/Turabian StyleLewis, Kevin M., Chelsie L. Greene, Steven A. Sattler, Buhyun Youn, Luying Xun, and ChulHee Kang. 2020. "The Structural Basis of the Binding of Various Aminopolycarboxylates by the Periplasmic EDTA-Binding Protein EppA from Chelativorans sp. BNC1" International Journal of Molecular Sciences 21, no. 11: 3940. https://doi.org/10.3390/ijms21113940
APA StyleLewis, K. M., Greene, C. L., Sattler, S. A., Youn, B., Xun, L., & Kang, C. (2020). The Structural Basis of the Binding of Various Aminopolycarboxylates by the Periplasmic EDTA-Binding Protein EppA from Chelativorans sp. BNC1. International Journal of Molecular Sciences, 21(11), 3940. https://doi.org/10.3390/ijms21113940