The Role of Pro-Inflammatory and Regulatory Signaling by IL-33 in the Brain and Liver: A Focused Systematic Review of Mouse and Human Data and Risk of Bias Assessment of the Literature



Abstract
1. Introduction
2. Methods
2.1. PRISMA Criteria
2.2. Protocol and Registration
2.3. Eligibility Criteria
2.4. Information Sources
2.5. Search
2.6. Study Selection
2.7. Data Collection Process
2.8. Data Items
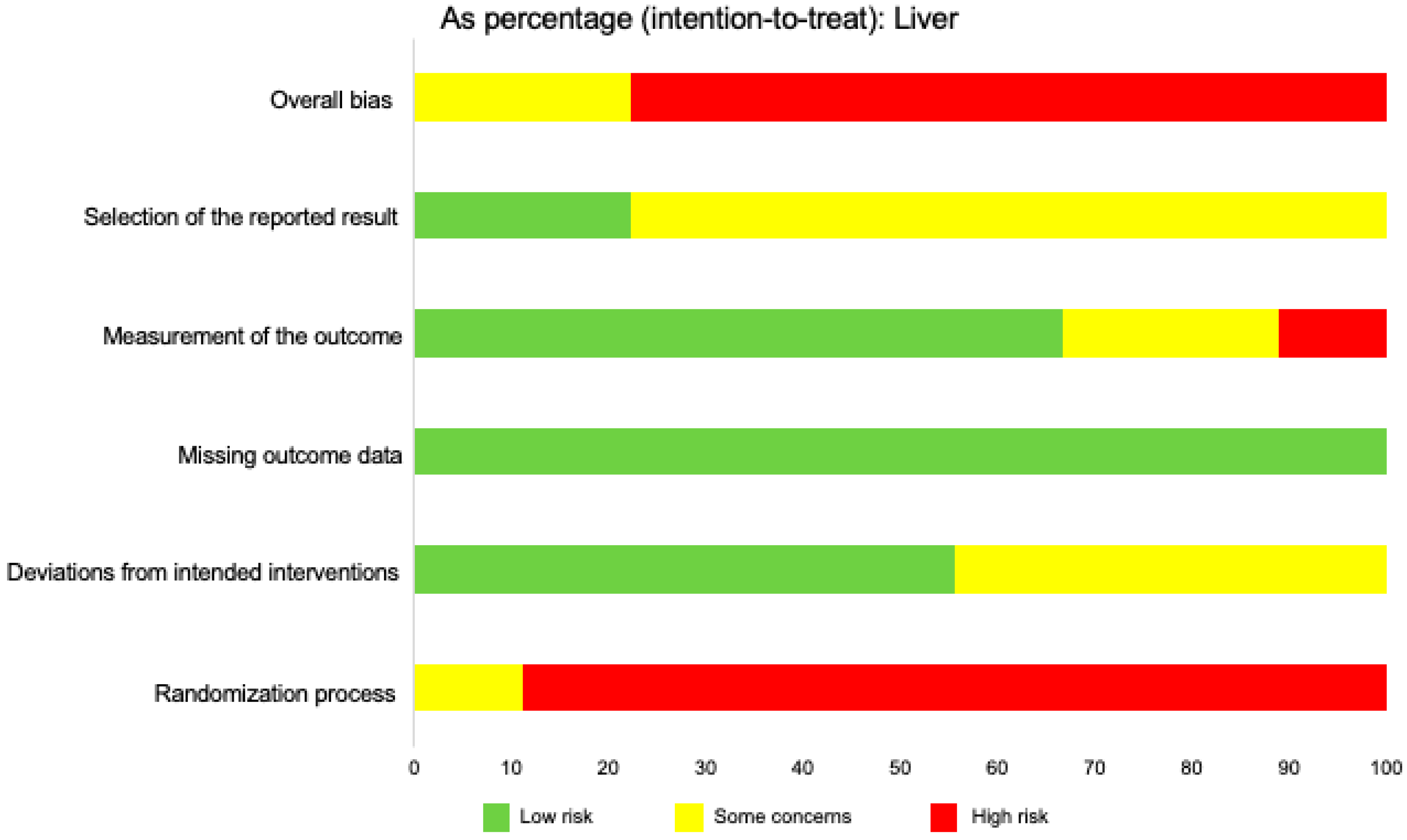
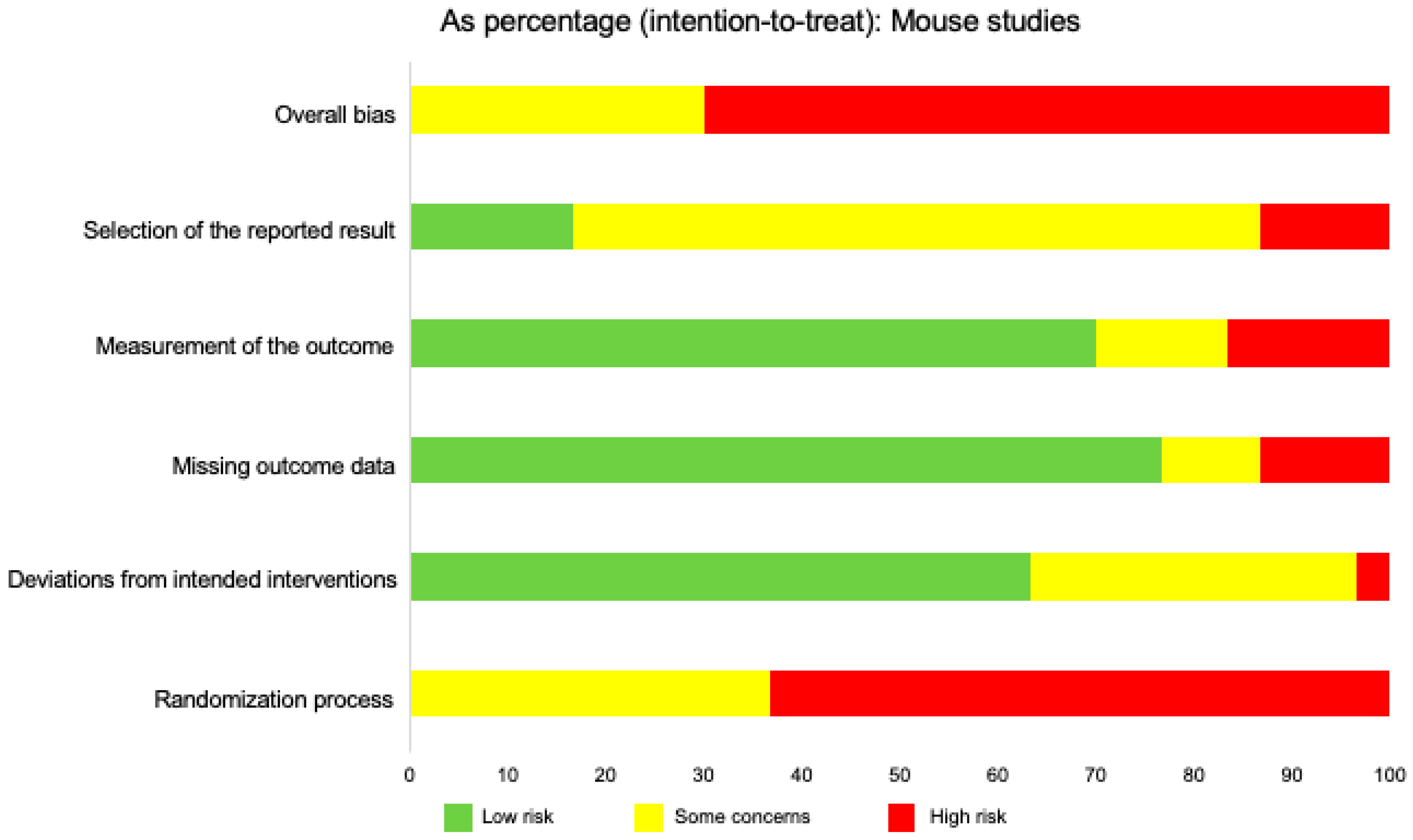
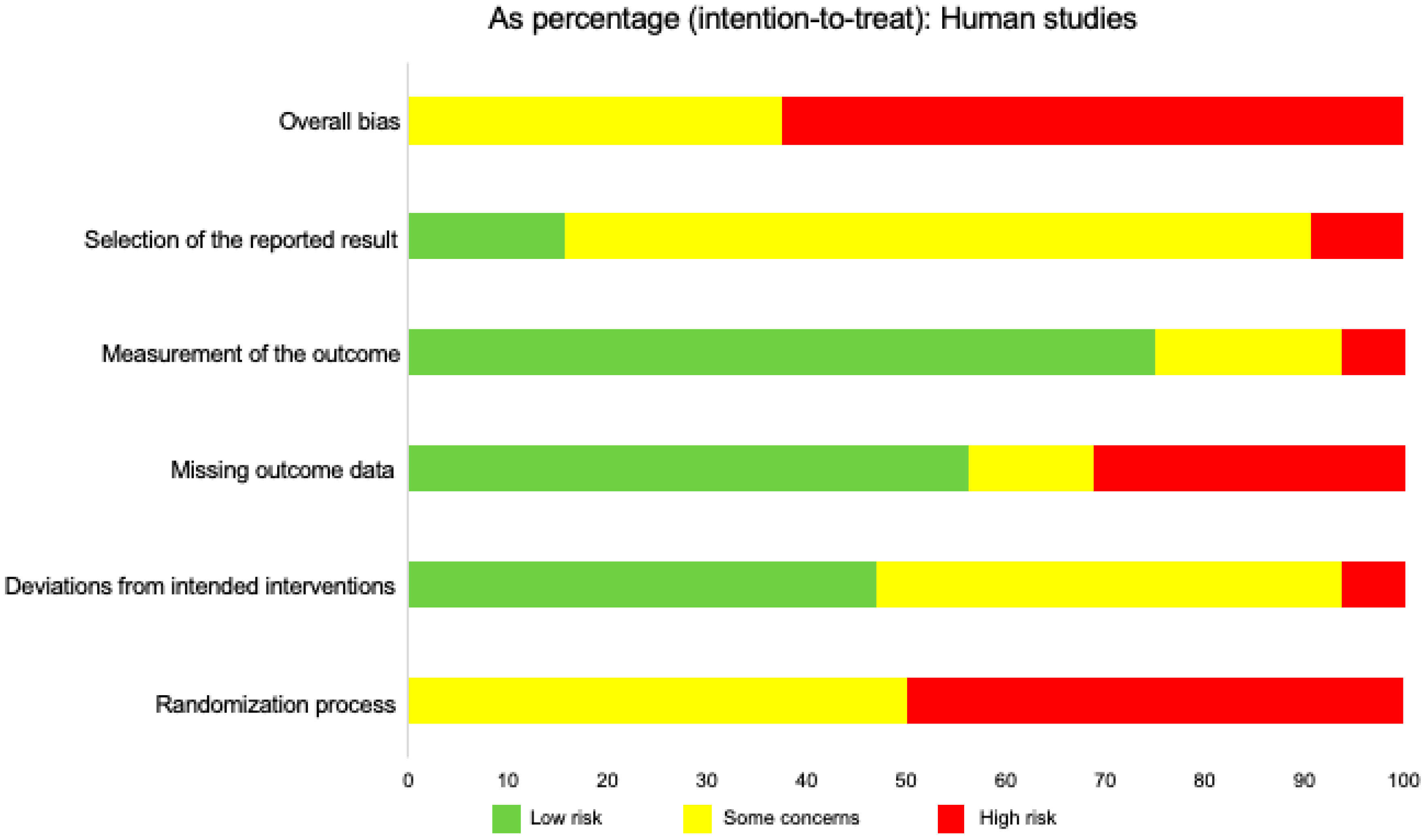
2.9. Risk of Bias in Individual Studies
2.10. Summary Measures
2.11. Synthesis of Results
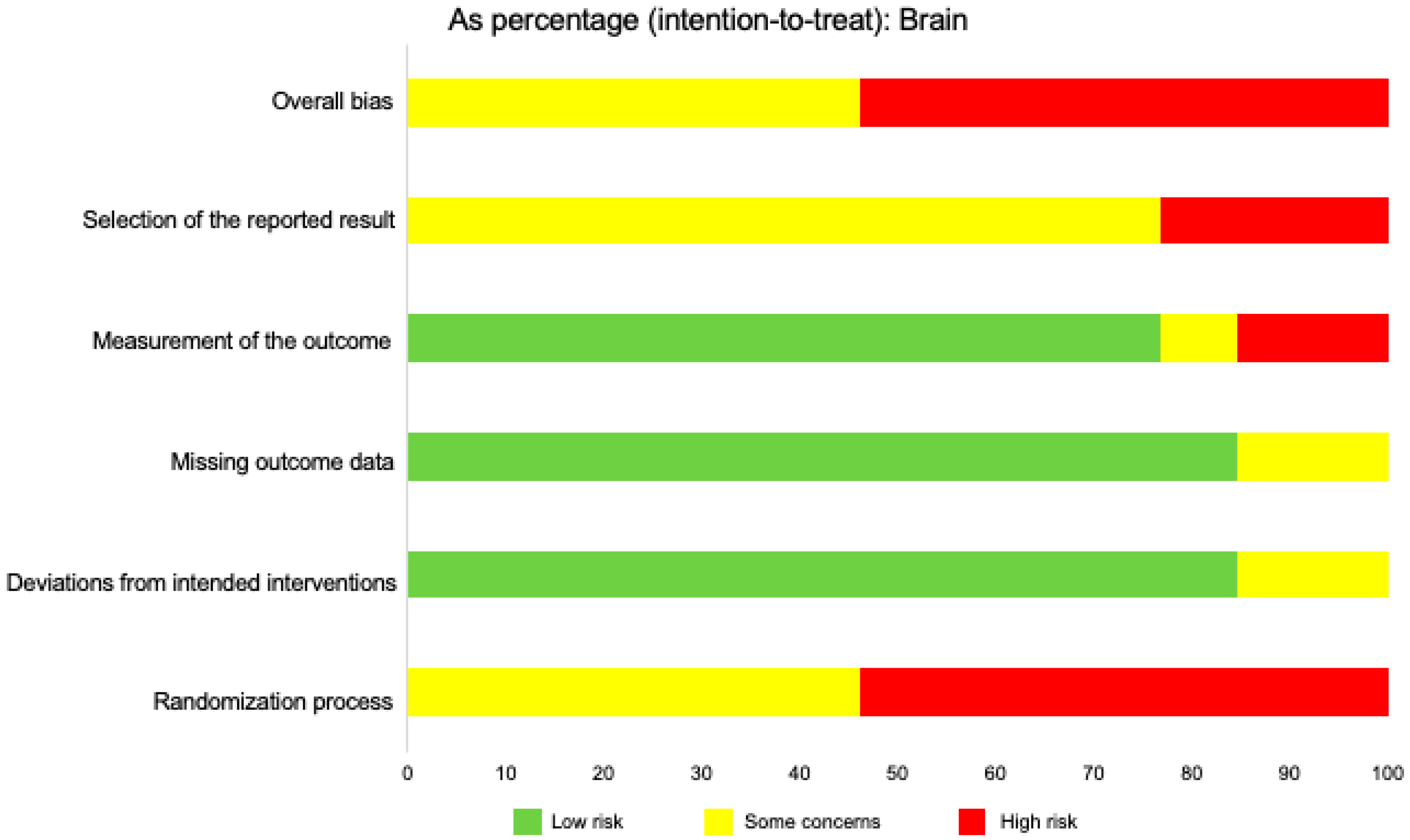
2.12. Risk of Bias across Studies
2.13. Additional Analyses
3. Results
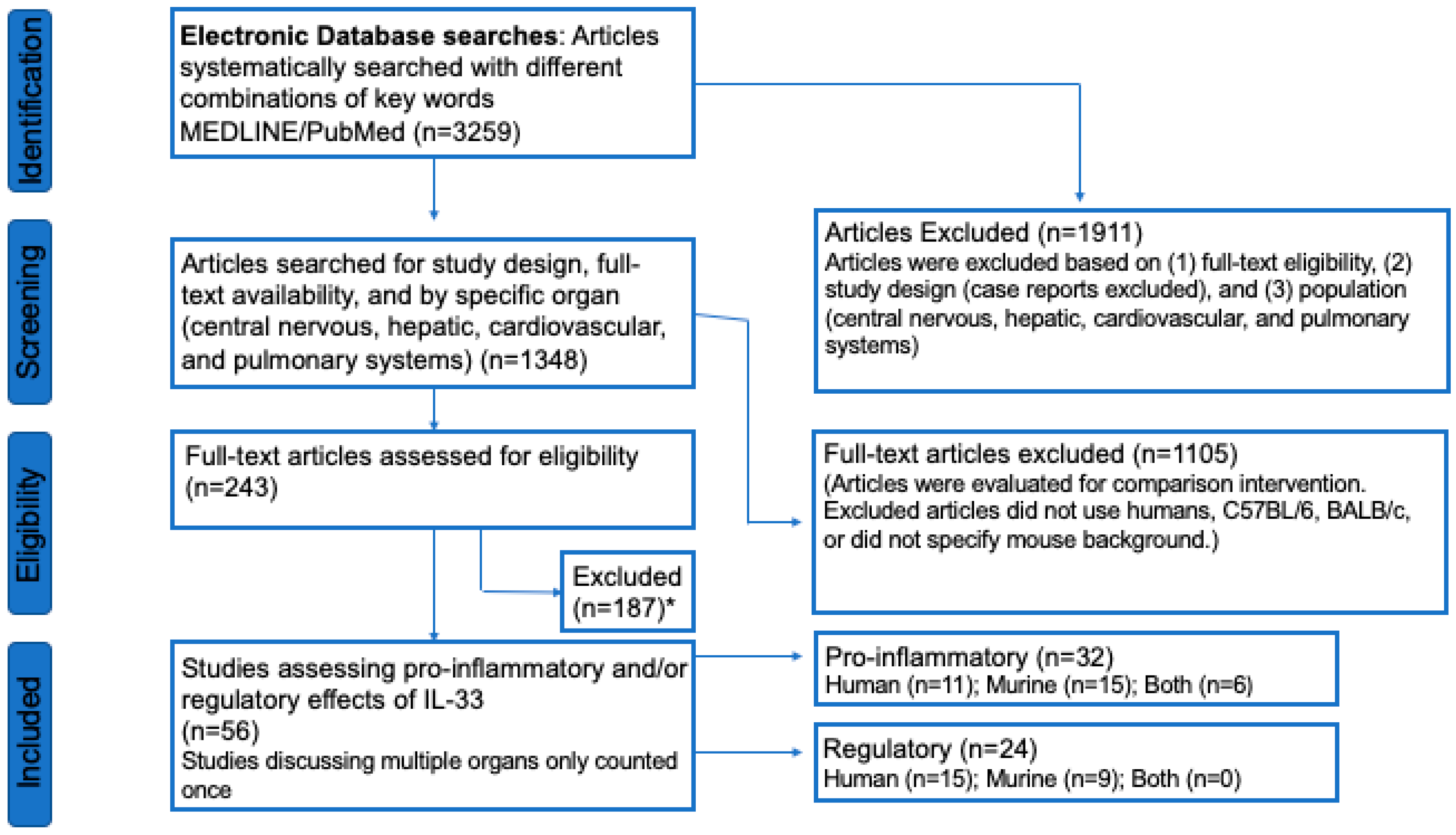
3.1. Study Selection
3.2. Effects of IL-33 on the Central Nervous System
3.3. Effects of IL-33 on the Liver
3.4. Effects of IL-33 on the Cardiovascular and Pulmonary Systems
4. Discussion
4.1. Summary of IL-33 Effects in Mice and Humans
4.2. Conclusions/Limitations
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Dohi, E.; Choi, E.Y.; Rose, I.; Murata, A.S.; Chow, S.; Niwa, M.; Kano, S.-I. Behavioral Changes in Mice Lacking Interleukin-33. Eneuro 2017, 4. [Google Scholar] [CrossRef]
- Marvie, P.; Lisbonne, M.; L’Helgoualc’H, A.; Rauch, M.; Turlin, B.; Preisser, L.; Bourd-Boittin, K.; Théret, N.; Gascan, H.; Piquet-Pellorce, C.; et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J. Cell. Mol. Med. 2009, 14, 1726–1739. [Google Scholar] [CrossRef]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrançais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 Is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33–LacZ Gene Trap Reporter Strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef]
- Molofsky, A.; Savage, A.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Iversen, L.; Johansen, C. Inflammasomes and inflammatory caspases in skin inflammation. Expert Rev. Mol. Diagn. 2008, 8, 697–705. [Google Scholar] [CrossRef]
- Takeishi, A.; Kuranaga, E.; Miura, M. Sensing and reacting to dangers by caspases: Caspase activation via inflammasomes. Drug Discov. Ther. 2008, 2, 14. [Google Scholar]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Miller, A.M. Role of IL-33 in inflammation and disease. J. Inflamm. 2011, 8, 22. [Google Scholar] [CrossRef]
- Talabot-Ayer, D.; Calo, N.; Vigne, S.; Lamacchia, C.; Gabay, C.; Palmer, G. The mouse interleukin (Il)33 gene is expressed in a cell type- and stimulus-dependent manner from two alternative promoters. J. Leukoc. Boil. 2011, 91, 119–125. [Google Scholar] [CrossRef]
- Kabata, H.; Moro, K.; Koyasu, S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol. Rev. 2018, 286, 37–52. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2017, 281, 154–168. [Google Scholar] [CrossRef]
- Drake, L.Y.; Kita, H. IL-33: Biological properties, functions, and roles in airway disease. Immunol. Rev. 2017, 278, 173–184. [Google Scholar] [CrossRef]
- Sterne, J.; Savović, J.; Page, M.J.; Elbers, R.G.; Blencowe, N.S.; Boutron, I.; Cates, C.J.; Cheng, H.-Y.; Corbett, M.S.; Eldridge, S.M.; et al. RoB 2: A revised tool for assessing risk of bias in randomised trials. BMJ 2019, 366, l4898. [Google Scholar] [CrossRef]
- Rachman, I.A.; Mohammad, F.; Chin, V.K.; Majid, R.A.; Lee, T.Y.; Abdullah, M.A.; Omenesa, R.B.; Ibraheem, Z.O.; Basir, R. Critical Roles of IL-33/ST2 Pathway in Neurological Disorders. Med. Inflamm. 2018, 5346413–5346419. [Google Scholar]
- Yndart, A.; Kaushik, A.; Agudelo, M.; Raymond, A.; Atluri, V.; Saxena, S.K.; Nair, M. Investigation of Neuropathogenesis in HIV-1 Clade B and C Infection Associated with IL-33 and ST2 Regulation. ACS Chem. Neurosci. 2015, 6, 1600–1612. [Google Scholar] [CrossRef]
- Yasuoka, S.; Kawanokuchi, J.; Parajuli, B.; Jin, S.; Doi, Y.; Noda, M.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Production and functions of IL-33 in the central nervous system. Brain Res. 2011, 1385, 8–17. [Google Scholar] [CrossRef]
- Priebe, K.; Brake, W.G.; Romeo, R.D.; Sisti, H.M.; Mueller, A.; McEwen, B.S.; Francis, D.D. Maternal influences on adult stress and anxiety-like behavior in C57BL/6J and BALB/cJ mice: A cross-fostering study. Dev. Psychobiol. 2005, 47, 398–407. [Google Scholar] [CrossRef]
- Miller, E.S.; Sakowicz, A.; Roy, A.; Yang, A.; Sullivan, J.T.; Grobman, W.A.; Wisner, K.L. Plasma, and cerebrospinal fluid inflammatory cytokines in perinatal depression. Am. J. Obstet. Gynecol. 2019, 220, 271. [Google Scholar] [CrossRef]
- Fairlie-Clarke, K.; Barbour, M.; Wilson, C.; Hridi, S.U.; Allan, D.; Jiang, H. Expression and Function of IL-33/ST2 Axis in the Central Nervous System Under Normal and Diseased Conditions. Front. Immunol. 2018, 9, 2596. [Google Scholar]
- Wang, M.-M.; Miao, D.; Cao, X.-P.; Tan, L.; Tan, L. Innate immune activation in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 177. [Google Scholar] [CrossRef]
- Brown, M.A.; Weinberg, R.B. Mast Cells, and Innate Lymphoid Cells: Underappreciated Players in CNS Autoimmune Demyelinating Disease. Front. Immunol. 2018, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Weng, J.-F.; Luo, L.-F.; Cen, M.; Yu, W.-H.; Zheng, Y.-K.; Hu, W.; Pan, J.-W.; Dong, X.-Q. Serum ST2 as a potential prognostic biomarker for traumatic brain injury. Clin. Chim. Acta 2018, 487, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, A.; Yu, Y.; Zhang, L.; Yang, G.; Hu, H.; Luo, Y. Serum Soluble ST2 as a Novel Inflammatory Marker in Acute Ischemic Stroke. Clin. Lab. 2018, 64, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Ferhat, M.; Salamé, E.; Robin, A.; Herbelin, A.; Gombert, J.-M.; Silvain, C.; Barbarin, A. Interleukin-1 Family Cytokines: Keystones in Liver Inflammatory Diseases. Front. Immunol. 2019, 10, 2014. [Google Scholar] [CrossRef]
- Milovanovic, M.; Volarevic, V.; Radosavljevic, G.; Jovanovic, I.; Pejnovic, N.; Arsenijevic, N.; Lukic, M.L. IL-33/ST2 axis in inflammation and immunopathology. Immunol. Res. 2012, 52, 89–99. [Google Scholar]
- Cottagiri, M.; Nyandjo, M.; Stephens, M.; Mantilla, J.J.; Saito, H.; Mackay, I.R.; Rose, N.R.; Njoku, D.B. In drug-induced, immune-mediated hepatitis, interleukin-33 reduces hepatitis and improves survival independently and because of FoxP3+ T-cell activity. Cell. Mol. Immunol. 2018, 16, 706–717. [Google Scholar] [CrossRef]
- Jin, Y.; Kong, D.; Liu, C.; Gong, W. Role of IL-33 in transplant biology. Eur. Cytok. Netw. 2019, 30, 39–42. [Google Scholar]
- Sakai, N.; van Sweringen, H.L.; Quillin, R.C.; Schuster, R.; Blanchard, J.; Burns, J.M.; Tevar, A.D.; Edwards, M.J.; Lentsch, A.B. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology 2012, 56, 1468–1478. [Google Scholar] [CrossRef]
- Ferhat, M.H.; Robin, A.; Barbier, L.; Thierry, A.; Gombert, J.-M.; Barbarin, A.; Herbelin, A. The Impact of Invariant NKT Cells in Sterile Inflammation: The Possible Contribution of the Alarmin/Cytokine IL-33. Front. Immunol. 2018, 9, 2308. [Google Scholar] [CrossRef]
- Kotsiou, O.S.; Gourgoulianis, K.I.; Zarogiannis, S.G. IL-33/ST2 Axis in Organ Fibrosis. Front. Immunol. 2018, 9, 2432. [Google Scholar]
- Neumann, K.; Schiller, B.; Tiegs, G. NLRP3 Inflammasome and IL-33: Novel Players in Sterile Liver Inflammation. Int. J. Mol. Sci. 2018, 19, 2732. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Liu, Q.; Jiang, R.; Lv, L.; Shoto, S.S.; Maillet, I.; Quesniaux, V.; Tang, J.; Zhang, W.; Sun, B.; et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell. Mol. Immunol. 2017, 15, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Jovicic, N.; Jeftic, I.; Jovanovic, I.; Radosavljevic, G.; Arsenijević, N.N.; Lukic, M.L.; Pejnovic, N. Differential Immunometabolic Phenotype in Th1 and Th2 Dominant Mouse Strains in Response to High-Fat Feeding. PLoS ONE 2015, 10, e0134089. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.; Zimmermann, M.; Lubsczyk, B.A.; Pilz, J.; Faybik, P.; Hetz, H.; Hacker, S.; Mangold, A.; Bacher, A.; Krenn, C.G.; et al. Up-Regulation of Interleukin 33 and Soluble ST2 Serum Levels in Liver Failure. J. Surg. Res. 2010, 163, 79–83. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, P.; Guo, H.; Sun, X.; Jiang, Z.; Xu, L.; Feng, J.; Niu, J.-Q.; Jiang, Y. Serum IL-33 Levels Are Associated with Liver Damage in Patients with Chronic Hepatitis C. Mediat. Inflamm. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Wang, J.; Cai, Y.; Ji, H.; Feng, J.; Ayana, D.A.; Niu, J.; Jiang, Y. Serum IL-33 Levels Are Associated with Liver Damage in Patients with Chronic Hepatitis B. J. Interf. Cytok. Res. 2012, 32, 248–253. [Google Scholar] [CrossRef]
- Polis, S.; Fernandez, R. Impact of physical and psychological factors on health-related quality of life in adult patients with liver cirrhosis: A systematic review protocol. JBI Database Syst. Rev. Implement. Rep. 2015, 13, 39–51. [Google Scholar] [CrossRef]
- Chen, J.; He, Y.; Xie, Z.; Wei, Y.; Duan, L. The Role of IL-33 in Experimental Heart Transplantation. Cardiol. Res. Pr. 2020, 6108362–6108367. [Google Scholar] [CrossRef]
- Ghali, R.; Altara, R.; Louch, W.E.; Cataliotti, A.; Mallat, Z.; Kaplan, A.; Zouein, F.A.; Booz, G.W. IL-33 (Interleukin-33)/sST2 Axis in Hypertension and Heart Failure. Hypertension 2018, 72, 818–828. [Google Scholar] [CrossRef]
- Demyanets, S.; Kaun, C.; Pentz, R.; Krychtiuk, K.A.; Rauscher, S.; Pfaffenberger, S.; Zuckermann, A.; Aliabadi, A.; Gröger, M.; Maurer, G.; et al. Components of the interleukin-33/ST2 system are differentially expressed and regulated in human cardiac cells and in cells of the cardiac vasculature. J. Mol. Cell. Cardiol. 2013, 60, 16–26. [Google Scholar] [CrossRef]
- Vianello, E.; Dozio, E.; Tacchini, L.; Frati, L.; Corsi Romanelli, M.M. ST2/IL-33 signaling in cardiac fibrosis. Int. J. Biochem. Cell. Biol. 2019, 116. [Google Scholar] [CrossRef]
- Dhillon, O.S.; Narayan, H.K.; Quinn, P.A.; Squire, I.B.; Davies, J.E.; Ng, L.L. Interleukin 33 and ST2 in non–ST-elevation myocardial infarction: Comparison with Global Registry of Acute Coronary Events Risk Scoring and NT-proBNP. Am. Hear. J. 2011, 161, 1163–1170. [Google Scholar] [CrossRef]
- Pusceddu, I.; Dieplinger, B.; Mueller, T. ST2 and the ST2/IL-33 signalling pathway-biochemistry and pathophysiology in animal models and humans. Clin. Chim. Acta. 2019, 495, 493–500. [Google Scholar]
- Liu, C.L.; Shen, D.L.; Zhu, K.; Tang, J.N.; Wang, X.F.; Zhang, L.; Zhang, J.Y. Characterization of interleukin-33 and matrix metalloproteinase-28 in serum and their assocation with disease severity in patients with coronary heart disease. Coron. Artery Dis. 2014, 25, 498–504. [Google Scholar] [CrossRef]
- Krychtiuk, K.; Stojkovic, S.; Lenz, M.; Brekalo, M.; Huber, K.; Wojta, J.; Heinz, G.; Demyanets, S.; Speidl, W.S. Predictive value of low interleukin-33 in critically ill patients. Cytokine 2018, 103, 109–113. [Google Scholar] [CrossRef]
- Lyons, D.O.; Pullen, N.A. Beyond IgE: Alternative Mast Cell Activation Across Different Disease States. Int. J. Mol. Sci. 2020, 21, 1498. [Google Scholar] [CrossRef]
- Nechama, M.; Kwon, J.; Wei, S.; Kyi, A.T.; Welner, R.S.; Ben-Dov, I.Z.; Arredouani, M.S.; Asara, J.M.; Chen, C.-H.; Tsai, C.-Y.; et al. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity in IL-33-induced allergic airway inflammation. Nat. Commun. 2018, 9, 1603. [Google Scholar] [CrossRef]
- Yagami, A.; Orihara, K.; Morita, H.; Futamura, K.; Hashimoto, N.; Matsumoto, K.; Saito, H.; Matsuda, A. IL-33 Mediates Inflammatory Responses in Human Lung Tissue Cells. J. Immunol. 2010, 185, 5743–5750. [Google Scholar] [CrossRef]
- Lai, Y.; Altemeier, W.A.; Vandree, J.; Piliponsky, A.M.; Johnson, B.; Appel, C.L.; Frevert, C.W.; Hyde, D.M.; Ziegler, S.F.; Smith, D.E.; et al. Increased density of intraepithelial mast cells in exercise-induced bronchoconstriction regulated via epithelial-derived TSLP and IL-33. J. Allergy Clin. Immunol. 2013, 133, 1448–1455. [Google Scholar] [CrossRef]
- Hirahara, K.; Mato, N.; Kagiwara, K.; Nakayama, T. The pathogenicity of IL-33 on steroid-resident eosinophilic inflammation via the activation of memory-type ST2(+) CD4(+) T cells. J. Leukoc. Biol. 2018, 104, 895–901. [Google Scholar]
- Li, E.; Knight, J.M.; Wu, Y.; Luong, A.; Rodriguez, A.; Kheradmand, F.; Corry, D.B. Airway mycosis in allergic airway disease. Adv. Immunol. 2019, 142, 85–140. [Google Scholar] [CrossRef]
- Luzina, I.G.; Pickering, E.M.; Kopach, P.; Kang, P.H.; Lockatell, V.; Todd, N.W.; Papadimitriou, J.C.; McKenzie, A.N.J.; Atamas, S.P. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J. Immunol. 2012, 189, 403–410. [Google Scholar] [CrossRef]
- Nelson, M.P.; Christmann, B.S.; Werner, J.L.; Metz, A.E.; Trevor, J.L.; Lowell, C.A.; Steele, C. IL-33 and M2a Alveolar Macrophages Promote Lung Defense against the Atypical Fungal Pathogen Pneumocystis murina. J. Immunol. 2011, 186, 2372–2381. [Google Scholar] [CrossRef]
- Watanabe, H.; Numata, K.; Ito, T.; Takagi, K.; Matsukawa, A. Innate Immune Response in TH1- and TH2-Dominant Mouse Strains. Shock 2004, 22, 460–466. [Google Scholar] [CrossRef]
- Hou, R.; Garner, M.; Holmes, C.; Osmond, C.; Teeling, J.; Lau, L.; Baldwin, D.S. Peripheral inflammatory cytokines, and immune balance in Generalised Anxiety Disorder: Case-controlled study. Brain Behav. Immun. 2017, 62, 212–218. [Google Scholar] [CrossRef]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Intervention | Comparison Intervention | Outcome Measures |
|---|---|---|---|
| C57BL/6 mice BALB/c mice Humans Murine model Central Nervous System Brain Liver Hepatic System Cardiovascular System Heart Cardiac Pulmonary System Lungs | IL-33 Interleukin-33 IL-1 Cytokine Family IL-10 IL-3 IL-5 Microglia Chemokines Cytokines Astrocytes Oligodendrocytes Neurons Sinusoidal endothelial cells Macrophages M2 Macrophages Hepatic stellate cells Human liver hepatocytes Cardiomyocytes Fibroblasts FoxP3+ Tregs Regulatory T Cells Endothelial cells Pulmonary endothelial cells ST2 receptor sST2 Th2 Cells ILC2s | C57BL/6 mice BALB/c mice Humans Murine model Central Nervous System Brain Liver Hepatic System Cardiovascular System Heart Cardiac | Pro-inflammatory Inflammatory Anti-inflammatory Regulatory Suppression |
| Location | Action/Mechanism |
|---|---|
| Astrocytes | Constitutive IL-33 expression |
| Microglia | High expression of IL-33; pro-inflammatory response (TNF-α, IL-10, IL-1β, oxidative stress molecules) |
| Oligodendrocytes | Constitutive IL-33 expression |
| Neurons | Constitutive IL-33 expression |
| Brain Region(s) | IL-33 Expression |
|---|---|
| Corpus callosum & secondary motor cortex | Oligodendrocytes (Olig2+) |
| Medial prefrontal cortex | Oligodendrocytes and astrocytes (Olig2+ and S100β+) |
| Periventricular hypothalamic nucleus, basolateral amygdala, cortical amygdala | Astrocytes (S100β+) |
| Neurons & microglia | No expression |
| Location | Effect (Pro-inflammatory or Regulatory) |
|---|---|
| Central Nervous System | Pro-inflammatory roles: Microglia (via release of pro-inflammatory cytokines, chemokines, and oxidative stress molecules) |
| Regulatory roles: Astrocytes, oligodendrocytes, neurons | |
| Hepatic System | Pro-inflammatory roles: Sinusoidal endothelial cells (HSECs), macrophages, hepatic stellate cells (HSCs) |
| Regulatory roles: FoxP3+ Tregs | |
| Cardiovascular System | Pro-inflammatory roles: Endothelial cells |
| Regulatory roles: Unknown | |
| Pulmonary System | Pro-inflammatory roles: Pulmonary endothelial cells, macrophages, epithelial cells |
| Regulatory roles: ST2 receptor, M2 macrophages |
| Location | Effect |
|---|---|
| Central Nervous System | Constitutive expression in glial cells (oligodendrocytes and astrocytes) |
| Hepatic System | Nuclear factor and inflammatory mediator; expressed in human liver hepatocytes (cytoplasm and nucleus) |
| Cardiovascular System | Constitutive expression in endothelial cells, cardiomyocytes, and fibroblasts |
| Pulmonary System | Pro-inflammatory role in lung epithelial cells |
| Organ/Tissue | GTEx | Human Protein Atlas | ProteomicsDB |
|---|---|---|---|
| Adipose tissue | Y | Y | N |
| Adrenal gland | Y | N | N |
| Amygdala | Y | N | N |
| Appendix | N | Y | N |
| Basal ganglia | Y | N | N |
| Bone marrow | N | Y | N |
| Breast | Y | Y | N |
| Bronchus | N | Y | N |
| Caudate | N | Y | N |
| Cerebellum | Y | Y | N |
| Cerebral cortex | Y | Y | N |
| Cervix, uterine | Y | Y | Y |
| Colon | Y | Y | Y |
| Duodenum | N | N | N |
| Endometrium | Y | Y | N |
| Epididymis | N | N | N |
| Esophagus | Y | Y | Y |
| Fallopian tube | Y | N | N |
| Gallbladder | N | N | Y |
| Heart muscle | Y | Y | Y |
| Hippocampus | Y | Y | N |
| Hypothalamus | Y | N | N |
| Kidney | Y | N | N |
| Liver | Y | N | Y |
| Lung | Y | Y | Y |
| Lymph node | N | Y | Y |
| Midbrain | Y | N | N |
| Myometrium | N | N | Y |
| Nasopharynx | N | Y | N |
| Oral Mucosa | N | N | N |
| Ovary | Y | N | N |
| Pancreas | Y | Y | Y |
| Parathyroid gland | N | N | N |
| Pituitary gland | Y | N | N |
| Placenta | N | N | Y |
| Prostate | Y | Y | Y |
| Rectum | N | N | N |
| Salivary gland | Y | Y | N |
| Seminal vesicle | N | N | N |
| Skeletal muscle | Y | Y | N |
| Skin | Y | Y | Y |
| Small Intestine | Y | N | N |
| Smooth muscle | N | Y | N |
| Soft tissue | N | Y | N |
| Spinal cord | Y | N | N |
| Spleen | Y | Y | N |
| Stomach | Y | Y | N |
| Testis | Y | Y | Y |
| Thyroid gland | Y | N | N |
| Tonsil | N | Y | N |
| Urinary bladder | Y | Y | Y |
| Uterus | N | N | Y |
| Vagina | Y | Y | N |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zharichenko, N.; Njoku, D.B. The Role of Pro-Inflammatory and Regulatory Signaling by IL-33 in the Brain and Liver: A Focused Systematic Review of Mouse and Human Data and Risk of Bias Assessment of the Literature. Int. J. Mol. Sci. 2020, 21, 3933. https://doi.org/10.3390/ijms21113933
Zharichenko N, Njoku DB. The Role of Pro-Inflammatory and Regulatory Signaling by IL-33 in the Brain and Liver: A Focused Systematic Review of Mouse and Human Data and Risk of Bias Assessment of the Literature. International Journal of Molecular Sciences. 2020; 21(11):3933. https://doi.org/10.3390/ijms21113933
Chicago/Turabian StyleZharichenko, Nika, and Dolores B. Njoku. 2020. "The Role of Pro-Inflammatory and Regulatory Signaling by IL-33 in the Brain and Liver: A Focused Systematic Review of Mouse and Human Data and Risk of Bias Assessment of the Literature" International Journal of Molecular Sciences 21, no. 11: 3933. https://doi.org/10.3390/ijms21113933
APA StyleZharichenko, N., & Njoku, D. B. (2020). The Role of Pro-Inflammatory and Regulatory Signaling by IL-33 in the Brain and Liver: A Focused Systematic Review of Mouse and Human Data and Risk of Bias Assessment of the Literature. International Journal of Molecular Sciences, 21(11), 3933. https://doi.org/10.3390/ijms21113933