Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
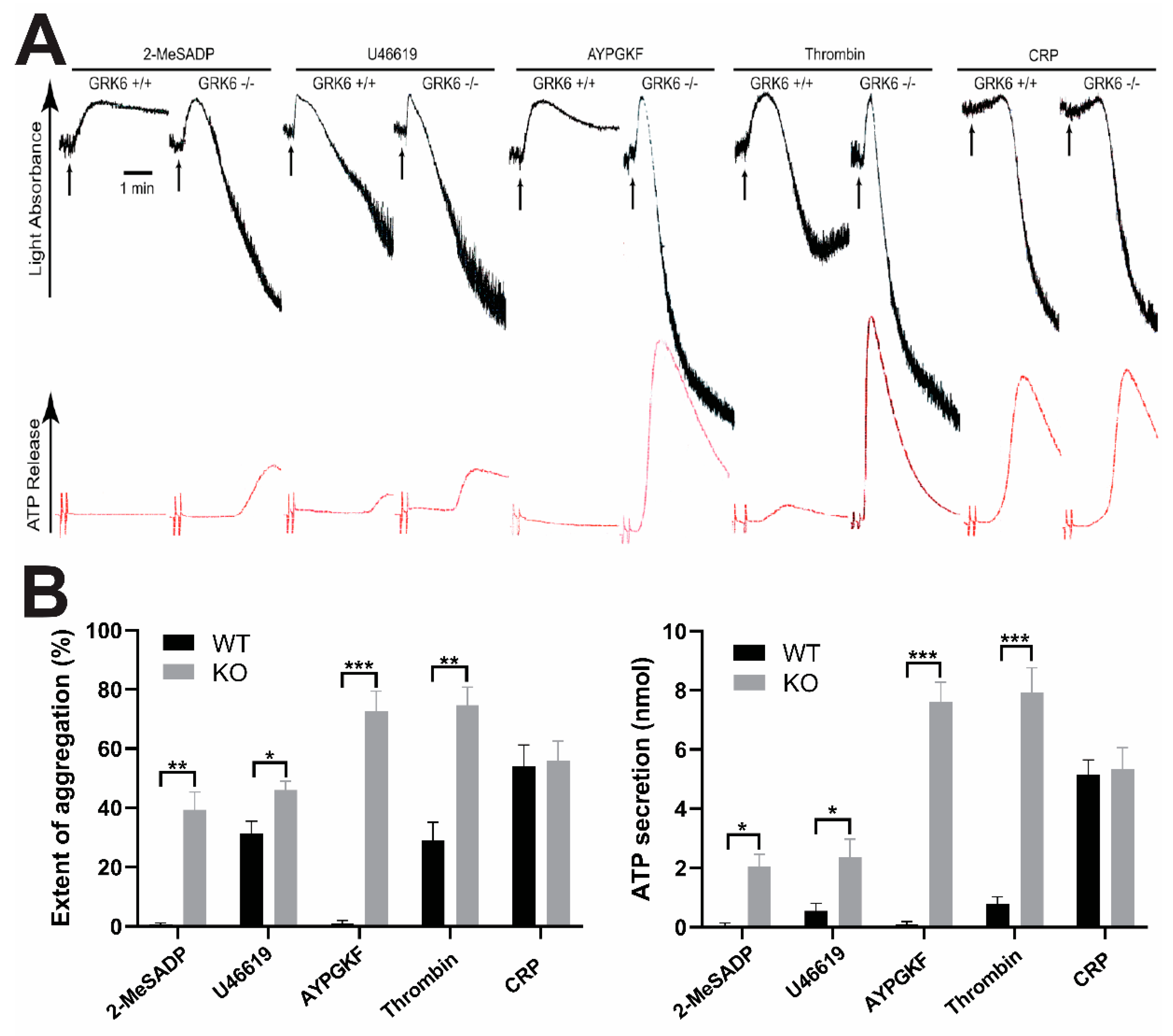
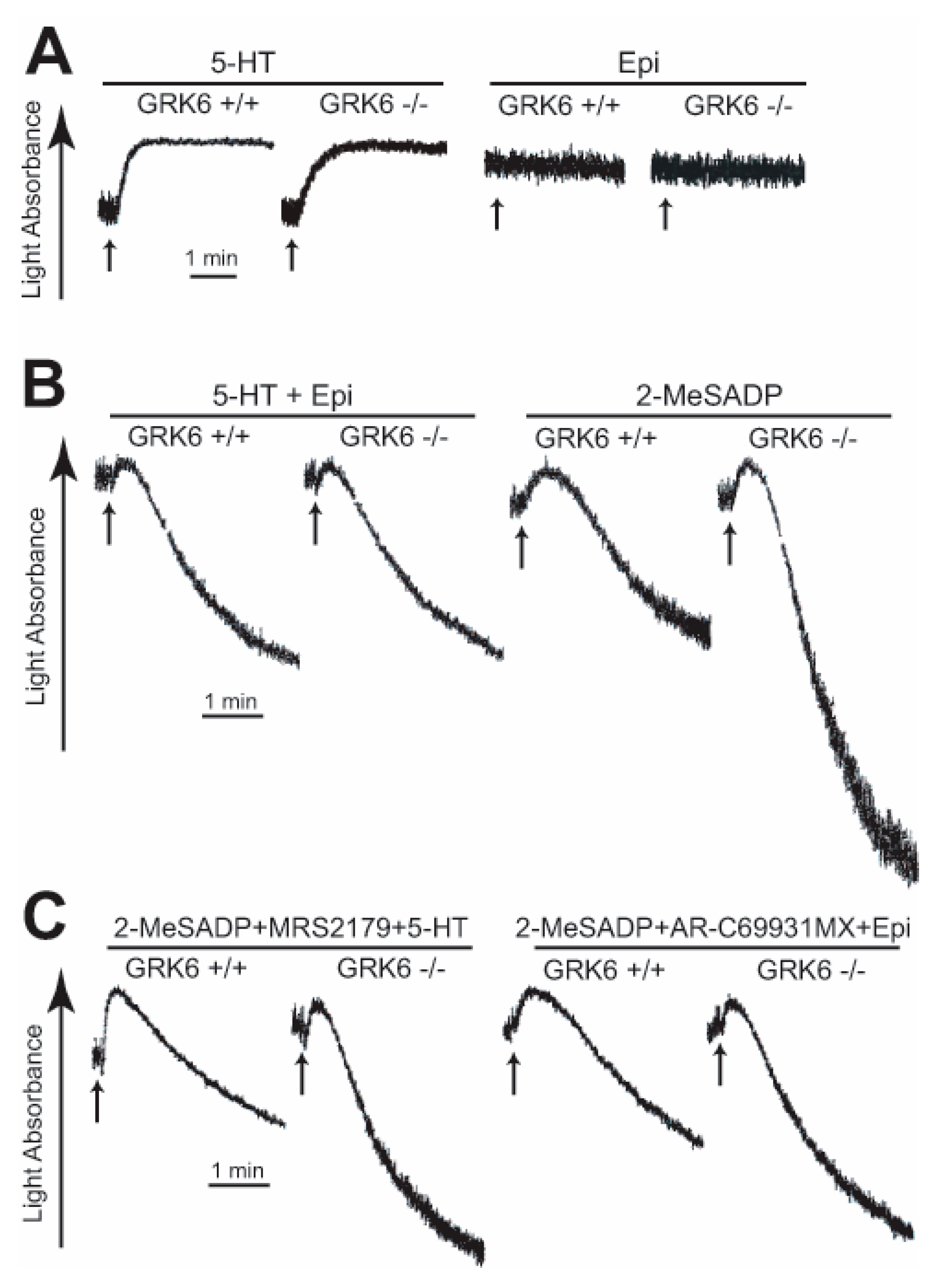
2.1. GRK6 Selectively Regulates GPCR-Mediated Platelet Functional Responses
2.2. GRK6 Regulates Specific GPCR-Mediated Platelet Aggregation
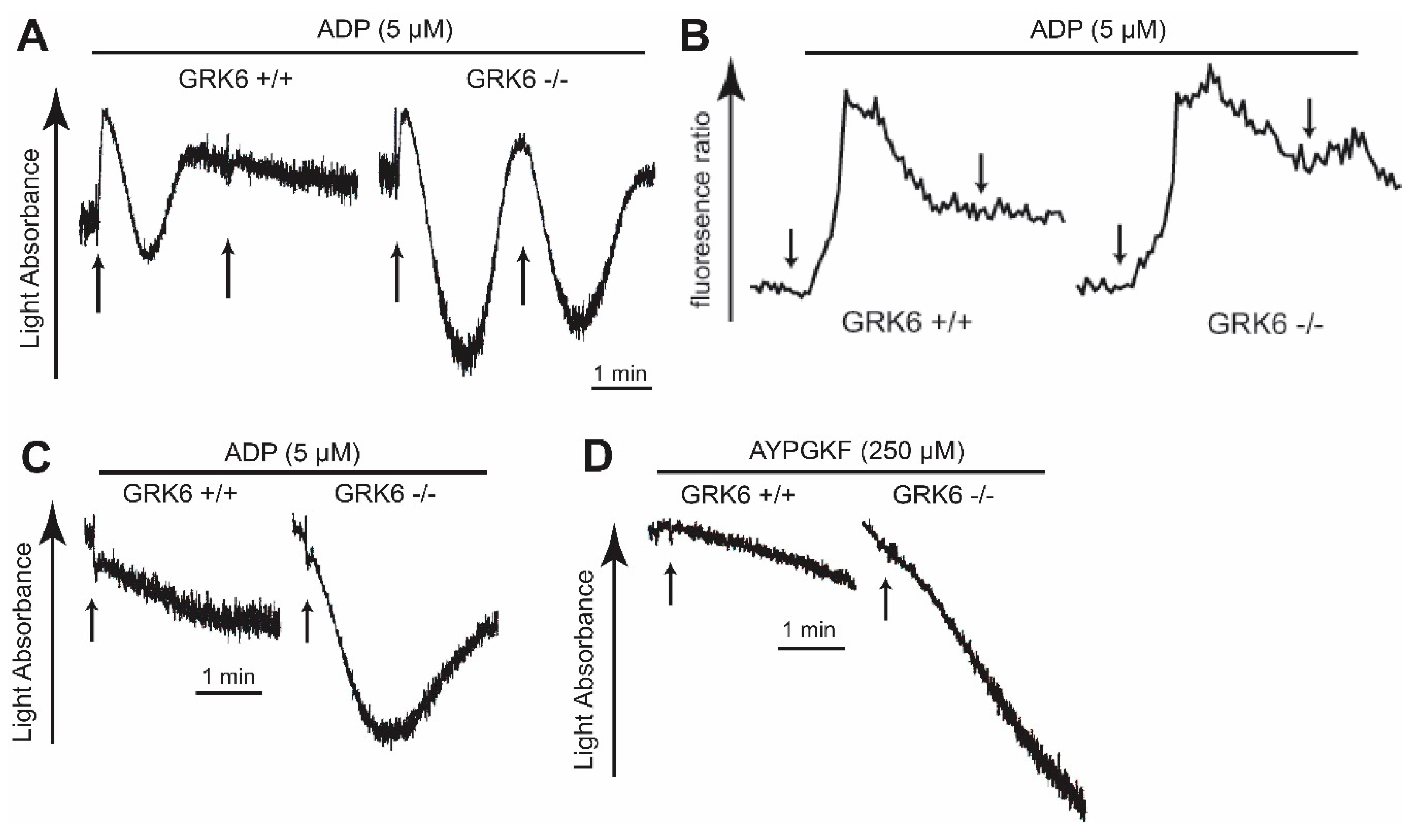
2.3. GRK6 Plays an Important Role in ADP and PAR4 Receptor Desensitization in Platelets
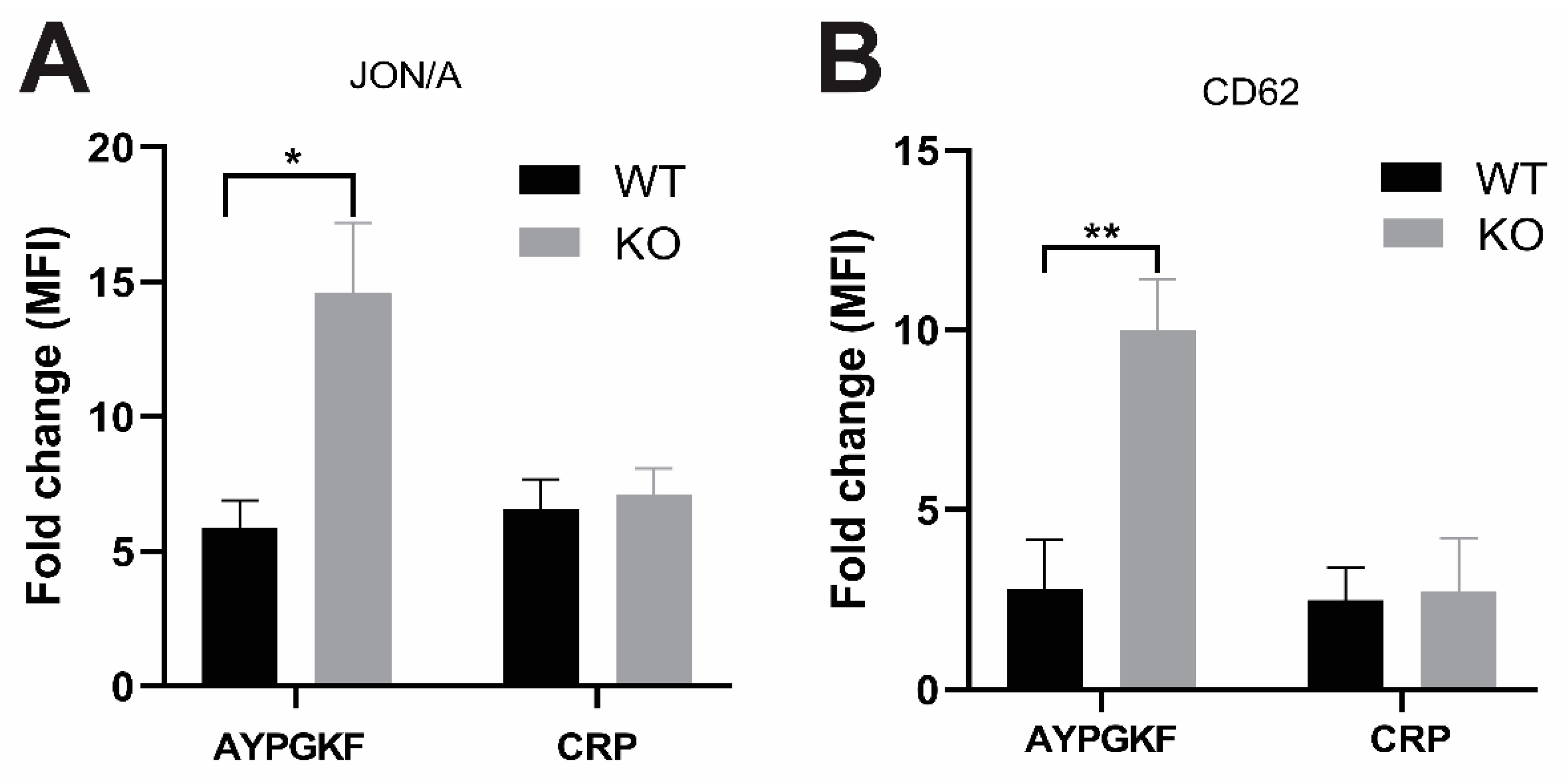
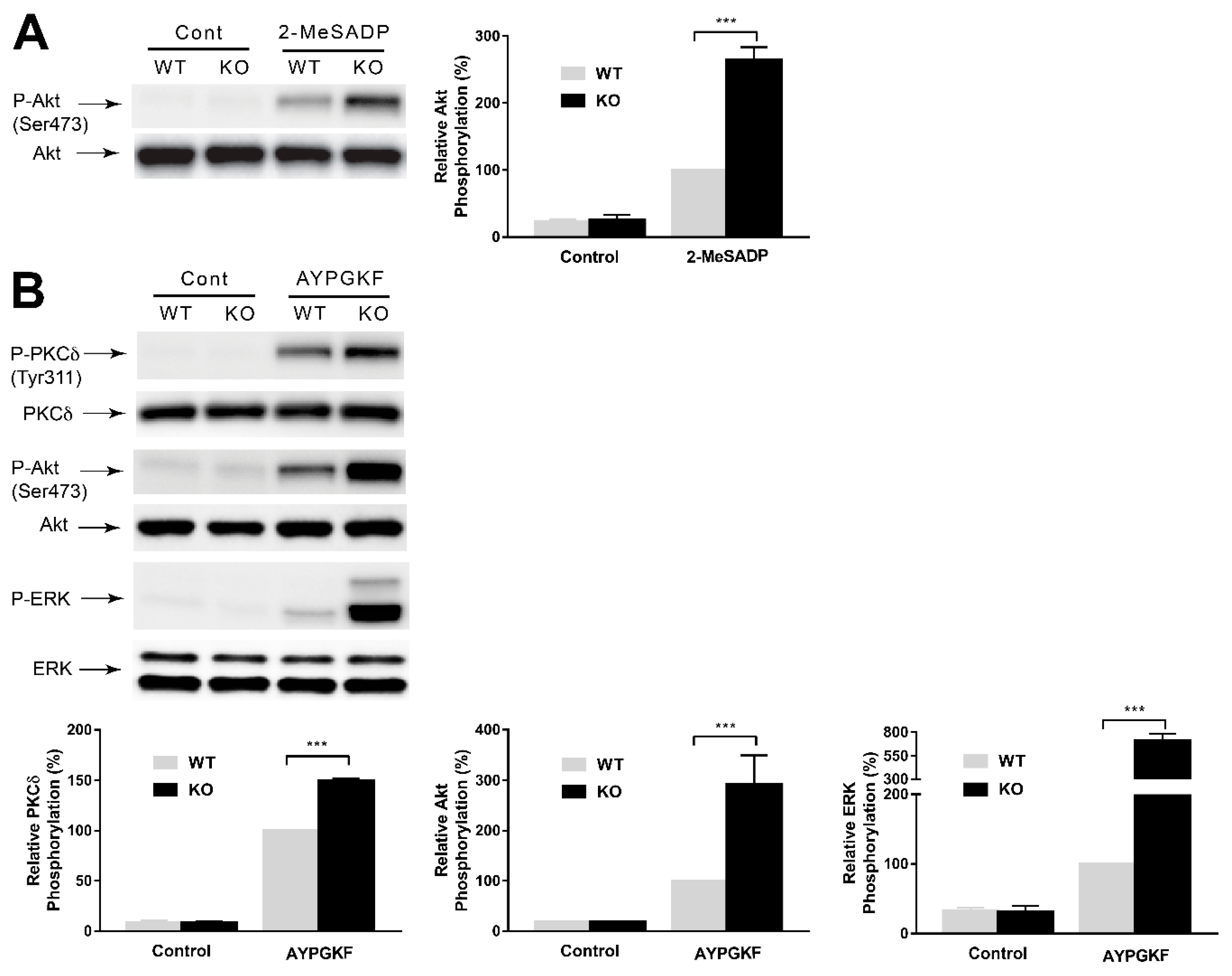
2.4. GPCR-Mediated Signaling Events are Potentiated in GRK6-Deficient Platelets
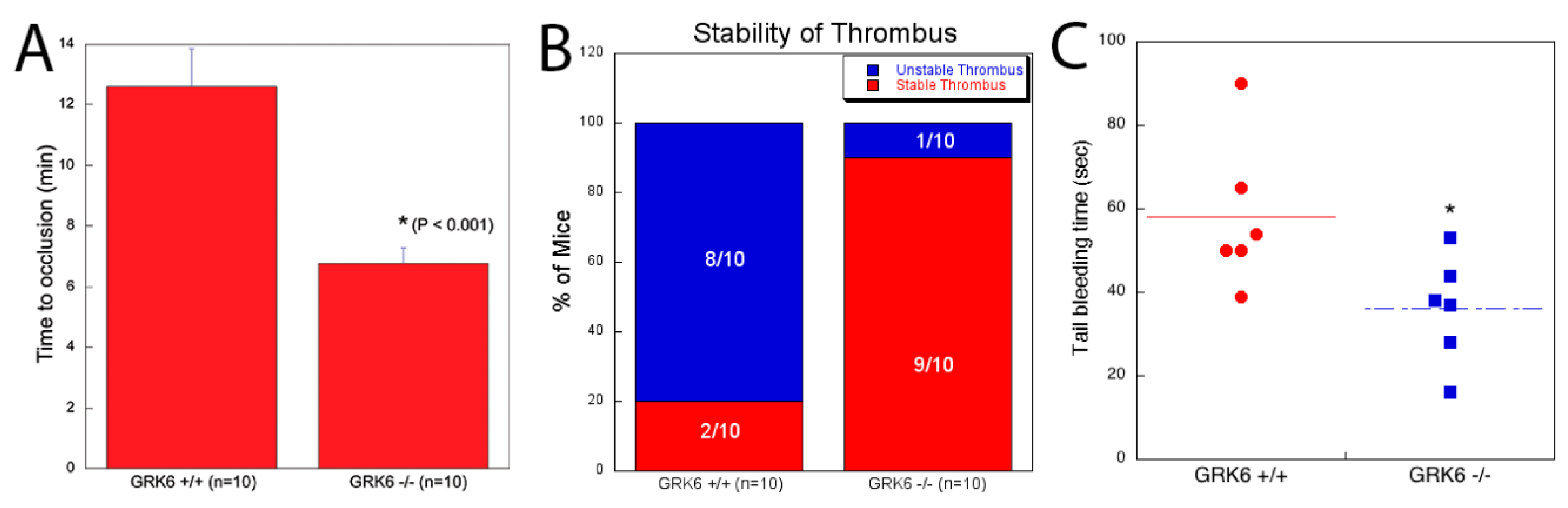
2.5. Contribution of GRK6 in the Platelet Function In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Preparation of Murine Platelets
4.4. Platelet Aggregation and Secretion
4.5. Flow Cytometry
4.6. Calcium Mobilization
4.7. Western Blotting
4.8. FeCl3-Induced In Vivo Thrombosis Model
4.9. Tail Bleeding Time Assay
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GPCR | G protein-coupled receptor |
| GRK | G protein-coupled receptor kinase |
| ADP | Adenosine diphosphate |
| CRP | Collagen-related peptide |
| PAR | Protease-activated receptor |
| ERK | Extracellular signal-related kinase |
| PKCδ | Protein Kinase Cδ |
| TP | Thromboxane receptor |
| PRP | Platelet-rich plasma |
References
- Jin, J.; Kunapuli, S.P. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc. Natl. Acad. Sci. USA 1998, 95, 8070–8074. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.Z.; Jin, J.; Kunapuli, S.P. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J. Biol. Chem. 1999, 274, 29108–29114. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S.; Laugwitz, K.-L.; Spicher, K.; Schultz, G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc. Natl. Acad. Sci. USA 1994, 91, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Shenker, A.; Goldsmith, P.; Unson, C.G.; Spiegel, A.M. The G protein coupled to the thromboxane A2 receptor in human platelets is a member of the novel Gq family. J. Biol. Chem. 1991, 266, 9309–9313. [Google Scholar]
- Kim, S.; Foster, C.; Lecchi, A.; Quinton, T.M.; Prosser, D.M.; Jin, J.; Cattaneo, M.; Kunapuli, S.P. Protease-activated receptors 1 and 4 do not stimulate Gi signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of Gisignaling. Blood 2002, 99, 3629–3636. [Google Scholar] [CrossRef]
- de Chaffoy de Courcelles, D.; Roevens, P.; van Belle, H.; de Clerck, F. The synergistic effect of serotonin and epinephrine on the human platelet at the level of signal transduction. FEBS Lett. 1987, 219, 283–288. [Google Scholar] [CrossRef][Green Version]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef]
- Benovic, J.L.; Gomez, J. Molecular cloning and expression of GRK6. A new member of the G protein-coupled receptor kinase family. J. Biol. Chem. 1993, 268, 19521–19527. [Google Scholar]
- Hisatomi, O.; Matsuda, S.; Satoh, T.; Kotaka, S.; Imanishi, Y.; Tokunaga, F. A novel subtype of G-protein-coupled receptor kinase, GRK7, in teleost cone photoreceptors. FEBS Lett. 1998, 424, 159–164. [Google Scholar] [CrossRef]
- Reiter, E.; Lefkowitz, R.J. GRKs and β-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef]
- Willets, J.M.; Challiss, R.A.; Nahorski, S.R. Non-visual GRKs: Are we seeing the whole picture? Trends Pharm. Sci. 2003, 24, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Trimarco, B.; Iaccarino, G. G-protein-coupled receptor kinase 2 and hypertension. High Blood Press. Cardiovasc. Prev. 2013, 20, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.J.; Lefkowitz, R.J. Desensitization of G protein-coupled receptors. Recent Prog. Horm. Res. 1996, 51, 319–351; discussion 352–313. [Google Scholar]
- Tiruppathi, C.; Yan, W.; Sandoval, R.; Naqvi, T.; Pronin, A.N.; Benovic, J.L.; Malik, A.B. G protein-coupled receptor kinase-5 regulates thrombin-activated signaling in endothelial cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7440–7445. [Google Scholar] [CrossRef]
- Ishii, K.; Chen, J.; Ishii, M.; Koch, W.J.; Freedman, N.J.; Lefkowitz, R.J.; Coughlin, S.R. Inhibition of thrombin receptor signaling by a G-protein coupled receptor kinase. Functional specificity among G-protein coupled receptor kinases. J. Biol. Chem. 1994, 269, 1125–1130. [Google Scholar]
- Hardy, A.R.; Conley, P.B.; Luo, J.; Benovic, J.L.; Poole, A.W.; Mundell, S.J. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase-dependent mechanisms. Blood 2005, 105, 3552–3560. [Google Scholar] [CrossRef]
- Willets, J.M.; Brighton, P.J.; Mistry, R.; Morris, G.E.; Konje, J.C.; Challiss, R.J. Regulation of oxytocin receptor responsiveness by G protein-coupled receptor kinase 6 in human myometrial smooth muscle. Mol. Endocrinol. 2009, 23, 1272–1280. [Google Scholar] [CrossRef]
- Zhou, H.; Yan, F.; Tai, H.-H. Phosphorylation and desensitization of the human thromboxane receptor-α by G protein-coupled receptor kinases. J. Pharmacol. Exp. Ther. 2001, 298, 1243–1251. [Google Scholar]
- Parent, J.-L.; Labrecque, P.; Orsini, M.J.; Benovic, J.L. Internalization of the TXA2 Receptor α and β Isoforms Role of the differentially spliced COOH terminus in agonist-promoted receptor internalization. J. Biol. Chem. 1999, 274, 8941–8948. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, S.; Ren, X.-R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jin, J.; Kunapuli, S.P. Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood 2006, 107, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; De, S.; Damron, D.S.; Chen, W.S.; Hay, N.; Byzova, T.V. Impaired platelet responses to thrombin and collagen in AKT-1–deficient mice. Blood 2004, 104, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jin, J.; Kunapuli, S.P. Akt activation in platelets depends on Gi signaling pathways. J. Biol. Chem. 2004, 279, 4186–4195. [Google Scholar] [CrossRef] [PubMed]
- Murugappan, S.; Tuluc, F.; Dorsam, R.T.; Shankar, H.; Kunapuli, S.P. Differential role of protein kinase Cδ isoform in agonist-induced dense granule secretion in human platelets. J. Biol. Chem. 2004, 279, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Shankar, H.; Garcia, A.; Prabhakar, J.; Kim, S.; Kunapuli, S. P2Y12 receptor-mediated potentiation of thrombin-induced thromboxane A2 generation in platelets occurs through regulation of Erk1/2 activation. J. Thromb. Haemost. 2006, 4, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, D.; Jiang, H.; Morgans, A.; Monks, R.; Birnbaum, M.; Brass, L.F. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J. Clin. Investig. 2004, 113, 441–450. [Google Scholar] [CrossRef]
- Nagy, B.; Bhavaraju, K.; Getz, T.; Bynagari, Y.S.; Kim, S.; Kunapuli, S.P. Impaired activation of platelets lacking protein kinase C-θ isoform. Blood 2009, 113, 2557–2567. [Google Scholar] [CrossRef]
- Bynagari-Settipalli, Y.S.; Lakhani, P.; Jin, J.; Bhavaraju, K.; Rico, M.C.; Kim, S.; Woulfe, D.; Kunapuli, S.P. Protein kinase C isoform epsilon negatively regulates ADP-induced calcium mobilization and thromboxane generation in platelets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1211–1219. [Google Scholar] [CrossRef]
- O’Brien, J.R.; Etherington, M.; Jamieson, S. Refractory state of platelet aggregation with major operations. Lancet 1971, 2, 741–743. [Google Scholar] [CrossRef]
- Shapiro, M.J.; Weiss, E.J.; Faruqi, T.R.; Coughlin, S.R. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J. Biol. Chem. 2000, 275, 25216–25221. [Google Scholar] [CrossRef] [PubMed]
- Molino, M.; Bainton, D.F.; Hoxie, J.A.; Coughlin, S.R.; Brass, L.F. Thrombin receptors on human platelets initial localization and subsequent redistribution during platelet activation. J. Biol. Chem. 1997, 272, 6011–6017. [Google Scholar] [CrossRef] [PubMed]
- Raychowdhury, M.K.; Yukawa, M.; Collins, L.J.; McGrail, S.H.; Kent, K.C.; Ware, J.A. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J. Biol. Chem. 1994, 269, 19256–19261. [Google Scholar] [PubMed]
- Kelley-Hickie, L.P.; O’Keeffe, M.B.; Reid, H.M.; Kinsella, B.T. Homologous desensitization of signalling by the alpha (α) isoform of the human thromboxane A2 receptor: A specific role for nitric oxide signalling. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 970–989. [Google Scholar] [CrossRef]
- Habib, A.; Vezza, R.; Créminon, C.; Maclouf, J.; FitzGerald, G.A. Rapid, Agonist-dependent Phosphorylation in Vivo of Human Thromboxane Receptor Isoforms Minimal Involvement of Protein Kinase C. J. Biol. Chem. 1997, 272, 7191–7200. [Google Scholar] [CrossRef]
- Hechler, B.; Leon, C.; Vial, C.; Vigne, P.; Frelin, C.; Cazenave, J.P.; Gachet, C. The P2Y1 receptor is necessary for adenosine 5′-diphosphate-induced platelet aggregation. Blood 1998, 92, 152–159. [Google Scholar] [CrossRef]
- Smith, S.K.; Limbird, L.E. Solubilization of human platelet alpha-adrenergic receptors: Evidence that agonist occupancy of the receptor stabilizes receptor—Effector interactions. Proc. Natl. Acad. Sci. USA 1981, 78, 4026–4030. [Google Scholar] [CrossRef]
- Yang, J.; Wu, J.; Kowalska, M.A.; Dalvi, A.; Prevost, N.; O’Brien, P.J.; Manning, D.; Poncz, M.; Lucki, I.; Blendy, J.A. Loss of signaling through the G protein, Gz, results in abnormal platelet activation and altered responses to psychoactive drugs. Proc. Natl. Acad. Sci. USA 2000, 97, 9984–9989. [Google Scholar] [CrossRef]
- Cerrito, F.; Lazzaro, M.P.; Gaudio, E.; Arminio, P.; Aloisi, G. 5HT2-receptors and serotonin release: Their role in human platelet aggregation. Life Sci. 1993, 53, 209–215. [Google Scholar] [CrossRef]
- Daniel, J.L.; Dangelmaier, C.; Jin, J.; Kim, Y.B.; Kunapuli, S.P. Role of intracellular signaling events in ADP-induced platelet aggregation. Thromb. Haemost. 1999, 82, 1322–1326. [Google Scholar] [CrossRef]
- Kim, S.; Kunapuli, S.P. P2Y12 receptor in platelet activation. Platelets 2011, 22, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Aiyar, N.; Disa, J.; Dang, K.; Pronin, A.N.; Benovic, J.L.; Nambi, P. Involvement of G protein-coupled receptor kinase-6 in desensitization of CGRP receptors. Eur. J. Pharmacol. 2000, 403, 1–7. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Bohn, L.M.; Sotnikova, T.D.; Cyr, M.; Laakso, A.; Macrae, A.D.; Torres, G.E.; Kim, K.-M.; Lefkowitz, R.J.; Caron, M.G. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron 2003, 38, 291–303. [Google Scholar] [CrossRef]
- Baurand, A.; Eckly, A.; Bari, N.; Léon, C.; Hechler, B.; Cazenave, J.-P.; Gachet, C. Desensitization of the platelet aggregation response to ADP: Differential down-regulation of the P2Y1 and P2cyc receptors. Thromb. Haemost. 2000, 84, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Kahner, B.N.; Shankar, H.; Murugappan, S.; Prasad, G.L.; Kunapuli, S.P. Nucleotide receptor signaling in platelets. J. Thromb. Haemost. 2006, 4, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gupta, S.; Cooper, M.; de Helian, D.; Zhao, X.; Naik, M.U.; Wurtzel, J.G.; Stalker, T.J.; Goldfinger, L.E.; Benovic, J. GRK6 regulates the hemostatic response to injury through its rate-limiting effects on GPCR signaling in platelets. Blood Adv. 2020, 4, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Cipolla, L.; Guidetti, G.; Okigaki, M.; Jin, J.; Torti, M.; Kunapuli, S.P. Distinct role of Pyk2 in mediating thromboxane generation downstream of both G12/13 and integrin αIIbβ3 in platelets. J. Biol. Chem. 2013, 288, 18194–18203. [Google Scholar] [CrossRef]
- Daniel, J.L.; Dangelmaier, C.A.; Mada, S.; Buitrago, L.; Jin, J.; Langdon, W.Y.; Tsygankov, A.Y.; Kunapuli, S.P.; Sanjay, A. Cbl-b is a novel physiologic regulator of glycoprotein VI-dependent platelet activation. J. Biol. Chem. 2010, 285, 17282–17291. [Google Scholar] [CrossRef]
- Kim, S.; Kunapuli, S.P. Negative regulation of Gq-mediated pathways in platelets by G12/13 pathways through Fyn kinase. J. Biol. Chem. 2011, 286, 24170–24179. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, P.K.; Kim, S.; Jee, Y.; Lee, S.-H.; Park, K.-M.; Kim, S. Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization. Int. J. Mol. Sci. 2020, 21, 3932. https://doi.org/10.3390/ijms21113932
Chaudhary PK, Kim S, Jee Y, Lee S-H, Park K-M, Kim S. Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization. International Journal of Molecular Sciences. 2020; 21(11):3932. https://doi.org/10.3390/ijms21113932
Chicago/Turabian StyleChaudhary, Preeti Kumari, Sanggu Kim, Youngheun Jee, Seung-Hun Lee, Kyung-Mee Park, and Soochong Kim. 2020. "Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization" International Journal of Molecular Sciences 21, no. 11: 3932. https://doi.org/10.3390/ijms21113932
APA StyleChaudhary, P. K., Kim, S., Jee, Y., Lee, S.-H., Park, K.-M., & Kim, S. (2020). Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization. International Journal of Molecular Sciences, 21(11), 3932. https://doi.org/10.3390/ijms21113932