Role of Extracellular Matrix in Gastrointestinal Cancer-Associated Angiogenesis

 ,
,  , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. ECM and Angiogenesis: Highlights in GI Cancers
3. Impact of ECM on Endothelial Cells—Mechanisms of Action
3.1. Direct ECM-EC Interactions
3.2. Indirect Mechanisms
3.3. Mechanical Cues
4. Function of ECM Remodeling in GI Tumor-Associated Angiogenesis
5. ECM and Growth Factors in a Tangled Crosstalk
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Saluja, A.; Maitra, A. Pancreatitis and Pancreatic Cancer. Gastroenterology 2019, 156, 1937–1940. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Van, C.E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Marano, L.; Chiari, R.; Fabozzi, A.; De, V.F.; Boccardi, V.; Roviello, G.; Petrioli, R.; Marrelli, D.; Roviello, F.; Patriti, A. c-Met targeting in advanced gastric cancer: An open challenge. Cancer Lett. 2015, 365, 30–36. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Lievre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le, C.D.; Buc, E.; Ychou, M.; Bouche, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Allen, M.; Louise, J.J. Jekyll and Hyde: The role of the microenvironment on the progression of cancer. J. Pathol. 2011, 223, 162–176. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Leman, J.K.; Sandford, S.K.; Rhodes, J.L.; Kemp, R.A. Multiparametric analysis of colorectal cancer immune responses. World J. Gastroenterol. 2018, 24, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Canonica, K.; Dawson, H.; Sokol, L.; Karamitopoulou-Diamantis, E.; Lugli, A.; Zlobec, I. Phenotyping of tumor-associated macrophages in colorectal cancer: Impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunology 2016, 5, e1106677. [Google Scholar] [CrossRef]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Lichtenstern, C.R.; Ngu, R.K.; Shalapour, S.; Karin, M. Immunotherapy, Inflammation and Colorectal Cancer. Cells 2020, 9, 618. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.N.; Jung, S.A.; Joo, Y.H.; Yoo, K. Clinical significance of tissue levels of matrix metalloproteinases and tissue inhibitors of metalloproteinases in gastric cancer. J. Gastroenterol. 2007, 42, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lukaszewicz-Zajac, M.; Szmitkowski, M.; Litman-Zawadzka, A.; Mroczko, B. Matrix Metalloproteinases and Their Tissue Inhibitors in Comparison to Other Inflammatory Proteins in Gastric Cancer (GC). Cancer Investig. 2016, 34, 305–312. [Google Scholar] [CrossRef]
- Randles, M.J.; Humphries, M.J.; Lennon, R. Proteomic definitions of basement membrane composition in health and disease. Matrix Biol. 2017, 57–58, 12–28. [Google Scholar] [CrossRef]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular Matrix, a Hard Player in Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef]
- Mongiat, M.; Buraschi, S.; Andreuzzi, E.; Neill, T.; Iozzo, R.V. Extracellular matrix: The gatekeeper of tumor angiogenesis. Biochem. Soc. Trans. 2019, 47, 1543–1555. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Vallet, S.D. Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biol. 2019, 75–76, 170–189. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Neill, T.; Iozzo, R.V. Matrix modeling and remodeling: A biological interplay regulating tissue homeostasis and diseases. Matrix Biol. 2019, 75–76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nienhuser, H.; Schmidt, T. Angiogenesis and Anti-Angiogenic Therapy in Gastric Cancer. Int. J. Mol. Sci. 2017, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, M.; Miller, I.S.; Kang, G.; Hannis, A.N.; O’Farrell, A.C.; Barat, A.; Marston, G.; Coletta, P.L.; Byrne, A.T.; Prehn, J.H. Anti-angiogenic drug scheduling optimisation with application to colorectal cancer. Sci. Rep. 2018, 8, 11182. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Yurchenco, P.D.; Patton, B.L. Developmental and pathogenic mechanisms of basement membrane assembly. Curr. Pharm. Des. 2009, 15, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Colognato, H.; Yurchenco, P.D. Form and function: The laminin family of heterotrimers. Dev. Dyn. 2000, 218, 213–234. [Google Scholar] [CrossRef]
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adhes. Migr. 2013, 7, 56–63. [Google Scholar] [CrossRef]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef]
- Teller, I.C.; Beaulieu, J.F. Interactions between laminin and epithelial cells in intestinal health and disease. Expert Rev. Mol. Med. 2001, 3, 1–18. [Google Scholar] [CrossRef]
- Simon-Assmann, P.; Spenle, C.; Lefebvre, O.; Kedinger, M. The role of the basement membrane as a modulator of intestinal epithelial-mesenchymal interactions. Prog. Mol. Biol. Transl. Sci 2010, 96, 175–206. [Google Scholar] [CrossRef]
- Spenle, C.; Lefebvre, O.; Lacroute, J.; Mechine-Neuville, A.; Barreau, F.; Blottiere, H.M.; Duclos, B.; Arnold, C.; Hussenet, T.; Hemmerle, J.; et al. The laminin response in inflammatory bowel disease: Protection or malignancy? PLoS ONE 2014, 9, e111336. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Rupp, T.; Spenle, C.; Jivkov, I.; Shankaranarayanan, P.; Klein, A.; Pisarsky, L.; Mechine-Neuville, A.; Cremel, G.; Kedinger, M.; et al. Laminin a1 orchestrates VEGFA functions in the ecosystem of colorectal carcinoma. Biol Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Weeks, A.; Lim, S.Y.; Yuzhalin, A.; Lucotti, S.; Vermeer, J.A.F.; Jones, K.; Chen, J.; Muschel, R.J. Tumour-Derived Laminin alpha5 (LAMA5) Promotes Colorectal Liver Metastasis Growth, Branching Angiogenesis and Notch Pathway Inhibition. Cancers 2019, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- Maragoudakis, M.E.; Missirlis, E.; Karakiulakis, G.D.; Sarmonica, M.; Bastakis, M.; Tsopanoglou, N. Basement membrane biosynthesis as a target for developing inhibitors of angiogenesis with anti-tumor properties. Kidney Int. 1993, 43, 147–150. [Google Scholar] [CrossRef][Green Version]
- Sweeney, S.M.; DiLullo, G.; Slater, S.J.; Martinez, J.; Iozzo, R.V.; Lauer-Fields, J.L.; Fields, G.B.; San Antonio, J.D. Angiogenesis in collagen I requires alpha2beta1 ligation of a GFP*GER sequence and possibly p38 MAPK activation and focal adhesion disassembly. J. Biol. Chem. 2003, 278, 30516–30524. [Google Scholar] [CrossRef]
- Li, Z.L.; Wang, Z.J.; Wei, G.H.; Yang, Y.; Wang, X.W. Changes in extracellular matrix in different stages of colorectal cancer and their effects on proliferation of cancer cells. World J Gastrointest. Oncol. 2020, 12, 267–275. [Google Scholar] [CrossRef]
- Zou, X.; Feng, B.; Dong, T.; Yan, G.; Tan, B.; Shen, H.; Huang, A.; Zhang, X.; Zhang, M.; Yang, P.; et al. Up-regulation of type I collagen during tumorigenesis of colorectal cancer revealed by quantitative proteomic analysis. J. Proteom. 2013, 94, 473–485. [Google Scholar] [CrossRef]
- Kehlet, S.N.; Sanz-Pamplona, R.; Brix, S.; Leeming, D.J.; Karsdal, M.A.; Moreno, V. Excessive collagen turnover products are released during colorectal cancer progression and elevated in serum from metastatic colorectal cancer patients. Sci. Rep. 2016, 6, 30599. [Google Scholar] [CrossRef]
- Mortensen, J.H.; Godskesen, L.E.; Jensen, M.D.; Van Haaften, W.T.; Klinge, L.G.; Olinga, P.; Dijkstra, G.; Kjeldsen, J.; Karsdal, M.A.; Bay-Jensen, A.C.; et al. Fragments of Citrullinated and MMP-degraded Vimentin and MMP-degraded Type III Collagen Are Novel Serological Biomarkers to Differentiate Crohn’s Disease from Ulcerative Colitis. J Crohns. Colitis 2015, 9, 863–872. [Google Scholar] [CrossRef]
- Willumsen, N.; Jorgensen, L.N.; Karsdal, M.A. Vastatin (the NC1 domain of human type VIII collagen a1 chain) is linked to stromal reactivity and elevated in serum from patients with colorectal cancer. Cancer Biol. Ther. 2019, 20, 692–699. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Zhou, J.F.; Bai, C.M.; Wang, Y.Z.; Li, X.Y.; Cheng, Y.J.; Chen, S.C. Endostar combined with chemotherapy for treatment of metastatic colorectal and gastric cancer: A pilot study. Chin. Med. J. 2011, 124, 4299–4303. [Google Scholar] [PubMed]
- Li, B.L.; Hu, X.L.; Zhao, X.H.; Sun, H.G.; Zhou, C.Y.; Zhang, Y. Endostar combined with irinotecan/calcium folinate/5-fluorouracil (FOLFIRI) for treating advanced colorectal cancer: A clinical study. J. Chemother. 2015, 27, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Liu, Q.; Li, P.; Han, Y.; Bian, X.; Tang, Y.; Kong, Q. Endostatin gene therapy delivered by attenuated Salmonella typhimurium in murine tumor models. Cancer Gene Ther. 2018, 25, 167–183. [Google Scholar] [CrossRef]
- Yang, H.; Sui, Y.; Guo, X.; Tan, X.; Li, Y.; Wang, M. Endostar continuous intravenous infusion combined with S-1 and oxaliplatin chemotherapy could be effective in treating liver metastasis from gastric cancer. J. Cancer Res. Ther. 2018, 14, S1148–S1151. [Google Scholar] [CrossRef]
- Lv, Y.; Zheng, J.P. The inhibitory effects of arresten protein on tumor formation. Chin. Med. Sci. J. 2012, 27, 11–17. [Google Scholar] [CrossRef]
- Hwang-Bo, J.; Yoo, K.H.; Park, J.H.; Jeong, H.S.; Chung, I.S. Recombinant canstatin inhibits angiopoietin-1-induced angiogenesis and lymphangiogenesis. Int. J. Cancer 2012, 131, 298–309. [Google Scholar] [CrossRef]
- Xing, Y.N.; Deng, P.; Xu, H.M. Canstatin induces apoptosis in gastric cancer xenograft growth in mice through the mitochondrial apoptotic pathway. Biosci. Rep. 2014, 34. [Google Scholar] [CrossRef]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; Hynes, R.O.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar] [CrossRef]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar] [CrossRef]
- Fernando, N.T.; Koch, M.; Rothrock, C.; Gollogly, L.K.; D’Amore, P.A.; Ryeom, S.; Yoon, S.S. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin. Cancer Res. 2008, 14, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Xun, A.Y.; Wei, X.X.; Yao, J.; Wang, J.Y.; Shi, R.Y.; Yang, G.H.; Li, Y.X.; Xu, Z.L.; Lai, M.G.; et al. Bifidobacteria Expressing Tumstatin Protein for Antitumor Therapy in Tumor-Bearing Mice. Technol. Cancer Res. Treat. 2016, 15, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Nicosia, R.F.; Bonanno, E.; Smith, M. Fibronectin promotes the elongation of microvessels during angiogenesis in vitro. J. Cell. Physiol. 1993, 154, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Milner, R. Fibronectin promotes brain capillary endothelial cell survival and proliferation through alpha5beta1 and alphavbeta3 integrins via MAP kinase signalling. J. Neurochem. 2006, 96, 148–159. [Google Scholar] [CrossRef]
- Zou, L.; Cao, S.; Kang, N.; Huebert, R.C.; Shah, V.H. Fibronectin induces endothelial cell migration through beta1 integrin and Src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J. Biol. Chem. 2012, 287, 7190–7202. [Google Scholar] [CrossRef]
- Smith, J.T.; Tomfohr, J.K.; Wells, M.C.; Beebe, T.P., Jr.; Kepler, T.B.; Reichert, W.M. Measurement of cell migration on surface-bound fibronectin gradients. Langmuir 2004, 20, 8279–8286. [Google Scholar] [CrossRef]
- Re, F.; Zanetti, A.; Sironi, M.; Polentarutti, N.; Lanfrancone, L.; Dejana, E.; Colotta, F. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured human endothelial cells. J. Cell. Biol. 1994, 127, 537–546. [Google Scholar] [CrossRef]
- Ilic, D.; Almeida, E.A.; Schlaepfer, D.D.; Dazin, P.; Aizawa, S.; Damsky, C.H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell. Biol. 1998, 143, 547–560. [Google Scholar] [CrossRef]
- Kim, S.; Bell, K.; Mousa, S.A.; Varner, J.A. Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am. J. Pathol. 2000, 156, 1345–1362. [Google Scholar] [CrossRef]
- Yi, W.; Xiao, E.; Ding, R.; Luo, P.; Yang, Y. High expression of fibronectin is associated with poor prognosis, cell proliferation and malignancy via the NF-kappaB/p53-apoptosis signaling pathway in colorectal cancer. Oncol. Rep. 2016, 36, 3145–3153. [Google Scholar] [CrossRef]
- Bootz, F.; Schmid, A.S.; Neri, D. Alternatively Spliced EDA Domain of Fibronectin Is a Target for Pharmacodelivery Applications in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1908–1917. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.N.; Roesli, C.; Kaspar, M.; Villa, A.; Neri, D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 2007, 67. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.J.; Wu, F.; Liang, H.J. Colorectal tumor derived fibronectin alternatively spliced EDA domain exserts lymphangiogenic effect on human lymphatic endothelial cells. Cancer Biol. Ther. 2010, 9, 186–191. [Google Scholar] [CrossRef][Green Version]
- Xiang, L.; Xie, G.; Ou, J.; Wei, X.; Pan, F.; Liang, H. The extra domain A of fibronectin increases VEGF-C expression in colorectal carcinoma involving the PI3K/AKT signaling pathway. PLoS ONE 2012, 7, e35378. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Deng, J.; Wei, X.; Xie, G.; Zhou, R.; Yu, L.; Liang, H. Fibronectin extra domain A (EDA) sustains CD133(+)/CD44(+) subpopulation of colorectal cancer cells. Stem Cell Res. 2013, 11, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Peng, Y.; Deng, J.; Miao, H.; Zhou, J.; Zha, L.; Zhou, R.; Yu, L.; Shi, H.; Liang, H. Endothelial cell-derived fibronectin extra domain A promotes colorectal cancer metastasis via inducing epithelial-mesenchymal transition. Carcinogenesis 2014, 35, 1661–1670. [Google Scholar] [CrossRef]
- Mongiat, M.; Sweeney, S.M.; San Antonio, J.D.; Fu, J.; Iozzo, R.V. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 2003, 278, 4238–4249. [Google Scholar] [CrossRef]
- Gubbiotti, M.A.; Buraschi, S.; Kapoor, A.; Iozzo, R.V. Proteoglycan signaling in tumor angiogenesis and endothelial cell autophagy. Semin. Cancer Biol. 2020, 62, 1–8. [Google Scholar] [CrossRef]
- Zoeller, J.J.; Whitelock, J.M.; Iozzo, R.V. Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. 2009, 28, 284–291. [Google Scholar] [CrossRef]
- Douglass, S.; Goyal, A.; Iozzo, R.V. The role of perlecan and endorepellin in the control of tumor angiogenesis and endothelial cell autophagy. Connect. Tissue Res. 2015, 56, 381–391. [Google Scholar] [CrossRef]
- Sharma, B.; Handler, M.; Eichstetter, I.; Whitelock, J.M.; Nugent, M.A.; Iozzo, R.V. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J. Clin. Investig. 1998, 102, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Iozzo, R.V. Transcriptional silencing of perlecan gene expression by interferon- gamma. J. Biol. Chem. 1998, 273, 4642–4646. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Ji, H.; Kapp, E.A.; Simpson, R.J. Sulindac modulates secreted protein expression from LIM1215 colon carcinoma cells prior to apoptosis. Biochim. Biophys. Acta 2013, 1834, 2293–2307. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.M.; Reed, C.C.; Bix, G.; Fu, J.; Zhang, Y.; Gopalakrishnan, B.; Greenspan, D.S.; Iozzo, R.V. BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J. Biol. Chem. 2005, 280, 7080–7087. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ma, Y.; Xiao, J.; Zheng, H.; Song, C.; Gong, Y.; Xing, X. Up-regulated biglycan expression correlates with the malignancy in human colorectal cancers. Clin. Exp. Med. 2012, 12, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, G.X.; Zhang, S.G.; Wang, Q.; Wen, Y.G.; Tang, H.M.; Zhou, C.Z.; Xing, A.Y.; Fan, J.W.; Yan, D.W.; et al. Biglycan expression correlates with aggressiveness and poor prognosis of gastric cancer. Exp. Biol. Med. 2011, 236, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Gu, X.; Ma, T.; Ye, H. Biglycan up-regulated vascular endothelial growth factor (VEGF) expression and promoted angiogenesis in colon cancer. Tumour. Biol. 2015, 36, 1773–1780. [Google Scholar] [CrossRef]
- Caon, I.; Bartolini, B.; Parnigoni, A.; Carava, E.; Moretto, P.; Viola, M.; Karousou, E.; Vigetti, D.; Passi, A. Revisiting the hallmarks of cancer: The role of hyaluronan. Semin. Cancer Biol. 2020, 62, 9–19. [Google Scholar] [CrossRef]
- Filpa, V.; Bistoletti, M.; Caon, I.; Moro, E.; Grimaldi, A.; Moretto, P.; Baj, A.; Giron, M.C.; Karousou, E.; Viola, M.; et al. Changes in hyaluronan deposition in the rat myenteric plexus after experimentally-induced colitis. Sci. Rep. 2017, 7, 17644. [Google Scholar] [CrossRef]
- Viola, M.; Karousou, E.; D’Angelo, M.L.; Caon, I.; De, L.G.; Passi, A.; Vigetti, D. Regulated Hyaluronan Synthesis by Vascular Cells. Int. J. Cell Biol. 2015, 2015, 208303. [Google Scholar] [CrossRef]
- Yamada, Y.; Itano, N.; Narimatsu, H.; Kudo, T.; Morozumi, K.; Hirohashi, S.; Ochiai, A.; Ueda, M.; Kimata, K. Elevated transcript level of hyaluronan synthase1 gene correlates with poor prognosis of human colon cancer. Clin. Exp. Metastasis 2004, 21, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Passi, A.; Vigetti, D.; Buraschi, S.; Iozzo, R.V. Dissecting the role of hyaluronan synthases in the tumor microenvironment. FEBS J. 2019, 286, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Makkar, S.; Riehl, T.E.; Chen, B.; Yan, Y.; Alvarado, D.M.; Ciorba, M.A.; Stenson, W.F. Hyaluronic Acid Binding to TLR4 Promotes Proliferation and Blocks Apoptosis in Colon Cancer. Mol. Cancer Ther. 2019, 18, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Nishiguchi, Y.; Kang, S.M.; Yashiro, M.; Onoda, N.; Sawada, T.; Ishikawa, T.; Hirakawa, K. Expression of thrombospondin-1 inversely correlated with tumor vascularity and hematogenous metastasis in colon cancer. Oncol. Rep. 2001, 8, 763–766. [Google Scholar] [CrossRef]
- Kaio, E.; Tanaka, S.; Oka, S.; Hiyama, T.; Kitadai, Y.; Haruma, K.; Chayama, K. Clinical significance of thrombospondin-1 expression in relation to vascular endothelial growth factor and interleukin-10 expression at the deepest invasive tumor site of advanced colorectal carcinoma. Int. J. Oncol. 2003, 23, 901–911. [Google Scholar]
- Tsuchida, T.; Kijima, H.; Tokunaga, T.; Oshika, Y.; Hatanaka, H.; Fukushima, Y.; Abe, Y.; Kawai, K.; Yoshida, Y.; Miura, S.; et al. Expression of the thrombospondin 1 receptor CD36 is correlated with decreased stromal vascularisation in colon cancer. Int. J. Oncol. 1999, 14, 47–51. [Google Scholar] [CrossRef]
- Jo, W.S.; Mizukami, Y.; Duerr, E.M.; Zukerberg, L.R.; Chung, D.C. Wnt signaling can repress thrombospondin-1 expression in colonic tumorigenesis. Cancer Biol. Ther. 2005, 4, 1361–1366. [Google Scholar] [CrossRef]
- Li, Q.; Ahuja, N.; Burger, P.C.; Issa, J.P. Methylation and silencing of the Thrombospondin-1 promoter in human cancer. Oncogene 1999, 18, 3284–3289. [Google Scholar] [CrossRef]
- Kanai, Y.; Ushijima, S.; Kondo, Y.; Nakanishi, Y.; Hirohashi, S. DNA methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int. J. Cancer 2001, 91, 205–212. [Google Scholar] [CrossRef]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar] [CrossRef]
- Punekar, S.; Zak, S.; Kalter, V.G.; Dobransky, L.; Punekar, I.; Lawler, J.W.; Gutierrez, L.S. Thrombospondin 1 and its mimetic peptide ABT-510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiology 2008, 75, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Zak, S.; Treven, J.; Nash, N.; Gutierrez, L.S. Lack of thrombospondin-1 increases angiogenesis in a model of chronic inflammatory bowel disease. Int. J. Colorectal Dis. 2008, 23, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.S.; Ling, J.; Nye, D.; Papathomas, K.; Dickinson, C. Thrombospondin peptide ABT-898 inhibits inflammation and angiogenesis in a colitis model. World J. Gastroenterol. 2015, 21, 6157–6166. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Tokunaga, T.; Hatanaka, H.; Tsuchida, T.; Tomii, Y.; Osada, H.; Onoda, N.; Morino, F.; Nagata, J.; Kijima, H.; et al. Interleukin 10 expression is correlated with thrombospondin expression and decreased vascular involvement in colon cancer. Int. J. Oncol. 2001, 18, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Wang, Q.; Huska, M.R.; Andrade-Navarro, M.A.; Lemm, M.; Fichtner, I.; Dahlmann, M.; Kobelt, D.; Walther, W.; Smith, J.; et al. SPON2, a newly identified target gene of MACC1, drives colorectal cancer metastasis in mice and is prognostic for colorectal cancer patient survival. Oncogene 2016, 35, 5942–5952. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.H.; Huang, L.; Zhang, X.; Zhang, P.; Zhang, S.M.; Guan, H.; Zhang, Y.; Zhu, X.Y.; Tian, S.; Deng, K.; et al. Mindin regulates vascular smooth muscle cell phenotype and prevents neointima formation. Clin. Sci. 2015, 129, 129–145. [Google Scholar] [CrossRef]
- Wang, L.F.; Liu, Y.S.; Yang, B.; Li, P.; Cheng, X.S.; Xiao, C.X.; Liu, J.J.; Li, S.; Ren, J.L.; Guleng, B. The extracellular matrix protein mindin attenuates colon cancer progression by blocking angiogenesis via Egr-1-mediated regulation. Oncogene 2018, 37, 601–615. [Google Scholar] [CrossRef]
- Guleng, B.; Lian, Y.M.; Ren, J.L. Mindin is upregulated during colitis and may activate NF-kappaB in a TLR-9 mediated manner. World J. Gastroenterol. 2010, 16, 1070–1075. [Google Scholar] [CrossRef]
- Lu, H.; Feng, Y.; Hu, Y.; Guo, Y.; Liu, Y.; Mao, Q.; Xue, W. Spondin 2 promotes the proliferation, migration and invasion of gastric cancer cells. J. Cell Mol. Med. 2020, 24, 98–113. [Google Scholar] [CrossRef]
- Doliana, R.; Canton, A.; Bucciotti, F.; Mongiat, M.; Bonaldo, P.; Colombatti, A. Structure, chromosomal localization, and promoter analysis of the human elastin microfibril interfase located proteIN (EMILIN) gene. J. Biol. Chem. 2000, 275, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Mungiguerra, G.; Bot, S.; Mucignat, M.T.; Giacomello, E.; Doliana, R.; Colombatti, A. Self-assembly and supramolecular organization of EMILIN. J. Biol. Chem. 2000, 275, 25471–25480. [Google Scholar] [CrossRef] [PubMed]
- Colombatti, A.; Spessotto, P.; Doliana, R.; Mongiat, M.; Bressan, G.M.; Esposito, G. The EMILIN/Multimerin family. Front. Immunol. 2011, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Bot, S.; Andreuzzi, E.; Capuano, A.; Schiavinato, A.; Colombatti, A.; Doliana, R. Multiple-interactions among EMILIN1 and EMILI. Matrix Biol. 2015, 41, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moncasi, M.P.; Garin-Chesa, P.; Stockert, E.; Jaffe, E.A.; Old, L.J.; Rettig, W.J. Identification of a high molecular weight endothelial cell surface glycoprotein, endoGlyx-1, in normal and tumor blood vessels. Lab. Investig. 1994, 71, 366–373. [Google Scholar] [PubMed]
- Lorenzon, E.; Colladel, R.; Andreuzzi, E.; Marastoni, S.; Todaro, F.; Schiappacassi, M.; Ligresti, G.; Colombatti, A.; Mongiat, M. MULTIMERIN2 impairs tumor angiogenesis and growth by interfering with VEGF-A/VEGFR2 pathway. Oncogene 2012, 31, 3136–3147. [Google Scholar] [CrossRef]
- Colladel, R.; Pellicani, R.; Andreuzzi, E.; Paulitti, A.; Tarticchio, G.; Todaro, F.; Colombatti, A.; Mongiat, M. MULTIMERIN2 binds VEGF-A primarily via the carbohydrate chains exerting an angiostatic function and impairing tumor growth. Oncotarget 2016, 7, 2022–2037. [Google Scholar] [CrossRef]
- Andreuzzi, E.; Colladel, R.; Pellicani, R.; Tarticchio, G.; Cannizzaro, R.; Spessotto, P.; Bussolati, B.; Brossa, A.; De, P.P.; Canzonieri, V.; et al. The angiostatic molecule Multimerin 2 is processed by MMP-9 to allow sprouting angiogenesis. Matrix Biol. 2017, 64, 40–53. [Google Scholar] [CrossRef]
- Andreuzzi, E.; Capuano, A.; Pellicani, R.; Poletto, E.; Doliana, R.; Maiero, S.; Fornasarig, M.; Magris, R.; Colombatti, A.; Cannizzaro, R.; et al. Loss of Multimerin-2 and EMILIN-2 Expression in Gastric Cancer Associate with Altered Angiogenesis. Int. J. Mol. Sci. 2018, 19, 3983. [Google Scholar] [CrossRef]
- Handsley, M.M.; Edwards, D.R. Metalloproteinases and their inhibitors in tumor angiogenesis. Int. J. Cancer 2005, 115, 849–860. [Google Scholar] [CrossRef]
- Lee, S.; Rho, S.S.; Park, H.; Park, J.A.; Kim, J.; Lee, I.K.; Koh, G.Y.; Mochizuki, N.; Kim, Y.M.; Kwon, Y.G. Carbohydrate-binding protein CLEC14A regulates VEGFR-2- and VEGFR-3-dependent signals during angiogenesis and lymphangiogenesis. J. Clin. Investig. 2017, 127, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Dobashi, Y.; Hatakeyama, Y.; Tajiri, R.; Fujimura, T.; Heldin, C.H.; Ooi, A. Clinicopathological significance of platelet-derived growth factor (PDGF)-B and vascular endothelial growth factor-A expression, PDGF receptor-beta phosphorylation, and microvessel density in gastric cancer. BMC Cancer 2010, 10, 659. [Google Scholar] [CrossRef] [PubMed]
- Pellicani, R.; Poletto, E.; Andreuzzi, E.; Paulitti, A.; Doliana, R.; Bizzotto, D.; Braghetta, P.; Colladel, R.; Tarticchio, G.; Sabatelli, P.; et al. Multimerin-2 maintains vascular stability and permeability. Matrix Biol. 2020, 87, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Marastoni, S.; Ligresti, G.; Lorenzon, E.; Colombatti, A.; Mongiat, M. Extracellular matrix: A matter of life and death. Connect. Tissue Res. 2008, 49, 203–206. [Google Scholar] [CrossRef]
- Mongiat, M.; Ligresti, G.; Marastoni, S.; Lorenzon, E.; Doliana, R.; Colombatti, A. Regulation of the extrinsic apoptotic pathway by the extracellular matrix glycoprotein EMILIN2. Mol. Cell. Biol. 2007, 27, 7176–7187. [Google Scholar] [CrossRef]
- Marastoni, S.; Andreuzzi, E.; Paulitti, A.; Colladel, R.; Pellicani, R.; Todaro, F.; Schiavinato, A.; Bonaldo, P.; Colombatti, A.; Mongiat, M. EMILIN2 down-modulates the Wnt signalling pathway and suppresses breast cancer cell growth and migration. J. Pathol. 2014, 232, 391–404. [Google Scholar] [CrossRef]
- Mongiat, M.; Marastoni, S.; Ligresti, G.; Lorenzon, E.; Schiappacassi, M.; Perris, R.; Frustaci, S.; Colombatti, A. The extracellular matrix glycoprotein elastin microfibril interface located protein 2: A dual role in the tumor microenvironment. Neoplasia 2010, 12, 294–304. [Google Scholar] [CrossRef]
- Paulitti, A.; Andreuzzi, E.; Bizzotto, D.; Pellicani, R.; Tarticchio, G.; Marastoni, S.; Pastrello, C.; Jurisica, I.; Ligresti, G.; Bucciotti, F.; et al. The ablation of the matricellular protein EMILIN2 causes defective vascularization due to impaired EGFR-dependent IL-8 production affecting tumor growth. Oncogene 2018, 37, 3399–3414. [Google Scholar] [CrossRef]
- Andreuzzi, E.; Fejza, A.; Capuano, A.; Poletto, E.; Pivetta, E.; Doliana, R.; Pellicani, R.; Favero, A.; Maiero, S.; Fornasarig, M.; et al. Deregulated expression of Elastin Microfibril Interfacer 2 (EMILIN2) in gastric cancer affects tumor growth and angiogenesis. Matrix Biol. Plus 2020. [Google Scholar] [CrossRef]
- Shi, J.; Wei, P.K. Interleukin-8: A potent promoter of angiogenesis in gastric cancer. Oncol. Lett. 2016, 11, 1043–1050. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Velazquez-Quesada, I.; Murdamoothoo, D.; Ahowesso, C.; Yilmaz, A.; Spenle, C.; Averous, G.; Erne, W.; Oberndorfer, F.; Oszwald, A.; et al. Tenascin-C increases lung metastasis by impacting blood vessel invasions. Matrix Biol. 2019, 83, 26–47. [Google Scholar] [CrossRef] [PubMed]
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Hussenet, T.; Langlois, B.; Orend, G. Advances in tenascin-C biology. Cell. Mol. Life Sci. 2011, 68, 3175–3199. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Mishra, D.P. Matrix reloaded: CCN, tenascin and SIBLING group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol. Ther. 2016, 168, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Brant, S.R.; Okou, D.T.; Simpson, C.L.; Cutler, D.J.; Haritunians, T.; Bradfield, J.P.; Chopra, P.; Prince, J.; Begum, F.; Kumar, A.; et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans With Inflammatory Bowel Disease. Gastroenterology 2017, 152, 206–217. [Google Scholar] [CrossRef]
- Riedl, S.; Tandara, A.; Reinshagen, M.; Hinz, U.; Faissner, A.; Bodenmuller, H.; Buhr, H.J.; Herfarth, C.; Moller, P. Serum tenascin-C is an indicator of inflammatory bowel disease activity. Int. J. Colorectal. Dis. 2001, 16, 285–291. [Google Scholar] [CrossRef]
- Islam, M.S.; Kusakabe, M.; Horiguchi, K.; Iino, S.; Nakamura, T.; Iwanaga, K.; Hashimoto, H.; Matsumoto, S.; Murata, T.; Hori, M.; et al. PDGF and TGF-beta promote tenascin-C expression in subepithelial myofibroblasts and contribute to intestinal mucosal protection in mice. Br. J. Pharmacol. 2014, 171, 375–388. [Google Scholar] [CrossRef]
- Hanamura, N.; Yoshida, T.; Matsumoto, E.; Kawarada, Y.; Sakakura, T. Expression of fibronectin and tenascin-C mRNA by myofibroblasts, vascular cells and epithelial cells in human colon adenomas and carcinomas. Int. J. Cancer 1997, 73, 10–15. [Google Scholar] [CrossRef]
- Murakami, T.; Kikuchi, H.; Ishimatsu, H.; Iino, I.; Hirotsu, A.; Matsumoto, T.; Ozaki, Y.; Kawabata, T.; Hiramatsu, Y.; Ohta, M.; et al. Tenascin C in colorectal cancer stroma is a predictive marker for liver metastasis and is a potent target of miR-198 as identified by microRNA analysis. Br. J. Cancer 2017, 117, 1360–1370. [Google Scholar] [CrossRef]
- Kawamura, T.; Yamamoto, M.; Suzuki, K.; Suzuki, Y.; Kamishima, M.; Sakata, M.; Kurachi, K.; Setoh, M.; Konno, H.; Takeuchi, H. Tenascin-C Produced by Intestinal Myofibroblasts Promotes Colitis- associated Cancer Development Through Angiogenesis. Inflamm. Bowel. Dis. 2019, 25, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.D. Diverse biological functions of the SPARC family of proteins. Int. J. Biochem. Cell Biol. 2012, 44, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhang, H.; Ge, W.; Liu, X.; Loera, S.; Chu, P.; Chen, H.; Peng, J.; Zhou, L.; Yu, S.; et al. Secreted protein acidic and rich in cysteines-like 1 suppresses aggressiveness and predicts better survival in colorectal cancers. Clin. Cancer Res. 2012, 18, 5438–5448. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.P.; Springer, T.A. Cloning from purified high endothelial venule cells of hevin, a close relative of the antiadhesive extracellular matrix protein SPARC. Immunity 1995, 2, 113–123. [Google Scholar] [CrossRef]
- Girard, J.P.; Springer, T.A. Modulation of endothelial cell adhesion by hevin, an acidic protein associated with high endothelial venules. J. Biol. Chem. 1996, 271, 4511–4517. [Google Scholar] [CrossRef] [PubMed]
- Cydzik, M.; Abdul-Wahid, A.; Park, S.; Bourdeau, A.; Bowden, K.; Prodeus, A.; Kollara, A.; Brown, T.J.; Ringuette, M.J.; Gariepy, J. Slow binding kinetics of secreted protein, acidic, rich in cysteine-VEGF interaction limit VEGF activation of VEGF receptor 2 and attenuate angiogenesis. FASEB J. 2015, 29, 3493–3505. [Google Scholar] [CrossRef]
- Naschberger, E.; Liebl, A.; Schellerer, V.S.; Schutz, M.; Britzen-Laurent, N.; Kolbel, P.; Schaal, U.; Haep, L.; Regensburger, D.; Wittmann, T.; et al. Matricellular protein SPARCL1 regulates tumor microenvironment-dependent endothelial cell heterogeneity in colorectal carcinoma. J. Clin. Investig. 2016, 126, 4187–4204. [Google Scholar] [CrossRef]
- Eichmann, A.; Makinen, T.; Alitalo, K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev. 2005, 19, 1013–1021. [Google Scholar] [CrossRef]
- Larrivee, B.; Freitas, C.; Trombe, M.; Lv, X.; Delafarge, B.; Yuan, L.; Bouvree, K.; Breant, C.; Del, T.R.; Brechot, N.; et al. Activation of the UNC5B receptor by Netrin-1 inhibits sprouting angiogenesis. Genes Dev. 2007, 21, 2433–2447. [Google Scholar] [CrossRef]
- Lu, X.; Le, N.F.; Yuan, L.; Jiang, Q.; De, L.B.; Sugiyama, D.; Breant, C.; Claes, F.; De, S.F.; Thomas, J.L.; et al. The netrin receptor UNC5B mediates guidance events controlling morphogenesis of the vascular system. Nature 2004, 432, 179–186. [Google Scholar] [CrossRef]
- Wilson, B.D.; Ii, M.; Park, K.W.; Suli, A.; Sorensen, L.K.; Larrieu-Lahargue, F.; Urness, L.D.; Suh, W.; Asai, J.; Kock, G.A.; et al. Netrins promote developmental and therapeutic angiogenesis. Science 2006, 313, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Cai, H. Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed- forward mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 6530–6535. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Crouse, D.; Lee, M.; Karnik, S.K.; Sorensen, L.K.; Murphy, K.J.; Kuo, C.J.; Li, D.Y. The axonal attractant Netrin-1 is an angiogenic factor. Proc. Natl. Acad. Sci. USA 2004, 101, 16210–16215. [Google Scholar] [CrossRef]
- Cirulli, V.; Yebra, M. Netrins: Beyond the brain. Nat. Rev. Mol. Cell Biol. 2007, 8, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Eveno, C.; Broqueres-You, D.; Feron, J.G.; Rampanou, A.; Tijeras-Raballand, A.; Ropert, S.; Leconte, L.; Levy, B.I.; Pocard, M. Netrin-4 delays colorectal cancer carcinomatosis by inhibiting tumor angiogenesis. Am. J. Pathol. 2011, 178, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Eveno, C.; Contreres, J.O.; Hainaud, P.; Nemeth, J.; Dupuy, E.; Pocard, M. Netrin-4 overexpression suppresses primary and metastatic colorectal tumor progression. Oncol. Rep. 2013, 29, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Minor, A.J.; Coulombe, K.L.K. Engineering a collagen matrix for cell-instructive regenerative angiogenesis. J. Biomed. Mater. Res. B Appl. Biomater. 2020. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Lu, X.; Lu, D.; Scully, M.; Kakkar, V. The role of integrins in cancer and the development of anti-integrin therapeutic agents for cancer therapy. Perspect. Med. Chem. 2008, 2, 57–73. [Google Scholar] [CrossRef]
- Perruzzi, C.A.; de Fougerolles, A.R.; Koteliansky, V.E.; Whelan, M.C.; Westlin, W.F.; Senger, D.R. Functional overlap and cooperativity among alphav and beta1 integrin subfamilies during skin angiogenesis. J. Investig. Dermatol. 2003, 120, 1100–1109. [Google Scholar] [CrossRef]
- Le, G.M.; Chambard, J.C.; Breittmayer, J.P.; Grall, D.; Pouyssegur, J.; Van Obberghen-Schilling, E. The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol. Biol. Cell 2000, 11, 1103–1112. [Google Scholar] [CrossRef]
- Stewart, J.A., Jr.; West, T.A.; Lucchesi, P.A. Nitric oxide-induced collagen IV expression and angiogenesis: FAK or fiction? Focus on “Collagen IV contributes to nitric oxide-induced angiogenesis of lung endothelial cells”. Am. J. Physiol. Cell Physiol. 2011, 300, C968–C969. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Colorado, P.C.; Torre, A.; Kamphaus, G.; Maeshima, Y.; Hopfer, H.; Takahashi, K.; Volk, R.; Zamborsky, E.D.; Herman, S.; Sarkar, P.K.; et al. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000, 60, 2520–2526. [Google Scholar]
- Sudhakar, A.; Nyberg, P.; Keshamouni, V.G.; Mannam, A.P.; Li, J.; Sugimoto, H.; Cosgrove, D.; Kalluri, R. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J. Clin. Investig. 2005, 115, 2801–2810. [Google Scholar] [CrossRef]
- Magnon, C.; Opolon, P.; Ricard, M.; Connault, E.; Ardouin, P.; Galaup, A.; Metivier, D.; Bidart, J.M.; Germain, S.; Perricaudet, M.; et al. Radiation and inhibition of angiogenesis by canstatin synergize to induce HIF-1alpha-mediated tumor apoptotic switch. J. Clin. Investig. 2007, 117, 1844–1855. [Google Scholar] [CrossRef]
- Boosani, C.S.; Mannam, A.P.; Cosgrove, D.; Silva, R.; Hodivala-Dilke, K.M.; Keshamouni, V.G.; Sudhakar, A. Regulation of COX-2 mediated signaling by alpha3 type IV noncollagenous domain in tumor angiogenesis. Blood 2007, 110, 1168–1177. [Google Scholar] [CrossRef]
- Maeshima, Y.; Colorado, P.C.; Torre, A.; Holthaus, K.A.; Grunkemeyer, J.A.; Ericksen, M.B.; Hopfer, H.; Xiao, Y.; Stillman, I.E.; Kalluri, R. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J. Biol. Chem. 2000, 275, 21340–21348. [Google Scholar] [CrossRef]
- Li, Y.J.; Sun, L.C.; He, Y.; Liu, X.H.; Liu, M.; Wang, Q.M.; Jin, X.M. The anti-tumor properties of two tumstatin peptide fragments in human gastric carcinoma. Acta Pharmacol. Sin. 2009, 30, 1307–1315. [Google Scholar] [CrossRef]
- Goyal, A.; Pal, N.; Concannon, M.; Paul, M.; Doran, M.; Poluzzi, C.; Sekiguchi, K.; Whitelock, J.M.; Neill, T.; Iozzo, R.V. Endorepellin, the angiostatic module of perlecan, interacts with both the alpha2beta1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2): A dual receptor antagonism. J. Biol. Chem. 2011, 286, 25947–25962. [Google Scholar] [CrossRef]
- Bix, G.; Fu, J.; Gonzalez, E.M.; Macro, L.; Barker, A.; Campbell, S.; Zutter, M.M.; Santoro, S.A.; Kim, J.K.; Hook, M.; et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J. Cell Biol. 2004, 166, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Poluzzi, C.; Willis, C.D.; Smythies, J.; Shellard, A.; Neill, T.; Iozzo, R.V. Endorepellin affects angiogenesis by antagonizing diverse vascular endothelial growth factor receptor 2 (VEGFR2)-evoked signaling pathways: Transcriptional repression of hypoxia-inducible factor 1alpha and VEGFA and concurrent inhibition of nuclear factor of activated T cell 1 (NFAT1) activation. J. Biol. Chem. 2012, 287, 43543–43556. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Andreuzzi, E.; Wang, Z.X.; Peiper, S.C.; Mongiat, M.; Iozzo, R.V. Endorepellin remodels the endothelial transcriptome toward a pro-autophagic and pro-mitophagic gene signature. J. Biol. Chem. 2018, 293, 12137–12148. [Google Scholar] [CrossRef] [PubMed]
- Bix, G.; Castello, R.; Burrows, M.; Zoeller, J.J.; Weech, M.; Iozzo, R.A.; Cardi, C.; Thakur, M.L.; Barker, C.A.; Camphausen, K.; et al. Endorepellin in vivo: Targeting the tumor vasculature and retarding cancer growth and metabolism. J. Natl. Cancer Inst. 2006, 98, 1634–1646. [Google Scholar] [CrossRef]
- Resovi, A.; Pinessi, D.; Chiorino, G.; Taraboletti, G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014, 37, 83–91. [Google Scholar] [CrossRef]
- Calzada, M.J.; Sipes, J.M.; Krutzsch, H.C.; Yurchenco, P.D.; Annis, D.S.; Mosher, D.F.; Roberts, D.D. Recognition of the N-terminal modules of thrombospondin-1 and thrombospondin-2 by alpha6beta1 integrin. J. Biol. Chem. 2003, 278, 40679–40687. [Google Scholar] [CrossRef]
- Yee, K.O.; Connolly, C.M.; Duquette, M.; Kazerounian, S.; Washington, R.; Lawler, J. The effect of thrombospondin-1 on breast cancer metastasis. Breast Cancer Res. Treat. 2009, 114, 85–96. [Google Scholar] [CrossRef]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Jimenez, B.; Volpert, O.V.; Reiher, F.; Chang, L.; Munoz, A.; Karin, M.; Bouck, N. c-Jun N-terminal kinase activation is required for the inhibition of neovascularization by thrombospondin-1. Oncogene 2001, 20, 3443–3448. [Google Scholar] [CrossRef]
- Rupp, T.; Langlois, B.; Koczorowska, M.M.; Radwanska, A.; Sun, Z.; Hussenet, T.; Lefebvre, O.; Murdamoothoo, D.; Arnold, C.; Klein, A.; et al. Tenascin-C Orchestrates Glioblastoma Angiogenesis by Modulation of Pro- and Anti-angiogenic Signaling. Cell Rep. 2016, 17, 2607–2619. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [PubMed]
- Castets, M.; Coissieux, M.M.; Delloye-Bourgeois, C.; Bernard, L.; Delcros, J.G.; Bernet, A.; Laudet, V.; Mehlen, P. Inhibition of endothelial cell apoptosis by netrin-1 during angiogenesis. Dev. Cell 2009, 16, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Navankasattusas, S.; Whitehead, K.J.; Suli, A.; Sorensen, L.K.; Lim, A.H.; Zhao, J.; Park, K.W.; Wythe, J.D.; Thomas, K.R.; Chien, C.B.; et al. The netrin receptor UNC5B promotes angiogenesis in specific vascular beds. Development 2008, 135, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Wink, D.A.; Roberts, D.D. Thrombospondin-1 antagonizes nitric oxide-stimulated vascular smooth muscle cell responses. Cardiovasc. Res 2006, 71, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Priya, M.K.; Sahu, G.; Soto-Pantoja, D.R.; Goldy, N.; Sundaresan, A.M.; Jadhav, V.; Barathkumar, T.R.; Saran, U.; Jaffar Ali, B.M.; Roberts, D.D.; et al. Tipping off endothelial tubes: Nitric oxide drives tip cells. Angiogenesis 2015, 18, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Gaffney, J.; Matou, S.; West, D.; Delisser, H.; Savani, R.C.; Kumar, S. Hyaluronan-mediated angiogenesis in vascular disease: Uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol. 2007, 26, 58–68. [Google Scholar] [CrossRef]
- Slevin, M.; Kumar, S.; Gaffney, J. Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses. J. Biol. Chem. 2002, 277, 41046–41059. [Google Scholar] [CrossRef]
- Galvagni, F.; Nardi, F.; Spiga, O.; Trezza, A.; Tarticchio, G.; Pellicani, R.; Andreuzzi, E.; Caldi, E.; Toti, P.; Tosi, G.M.; et al. Dissecting the CD93-Multimerin 2 interaction involved in cell adhesion and migration of the activated endothelium. Matrix Biol. 2017, 64, 112–127. [Google Scholar] [CrossRef]
- Khan, K.A.; Naylor, A.J.; Khan, A.; Noy, P.J.; Mambretti, M.; Lodhia, P.; Athwal, J.; Korzystka, A.; Buckley, C.D.; Willcox, B.E.; et al. Multimerin-2 is a ligand for group 14 family C-type lectins CLEC14A, CD93 and CD248 spanning the endothelial pericyte interface. Oncogene 2017, 36, 6097–6108. [Google Scholar] [CrossRef]
- Orlandini, M.; Galvagni, F.; Bardelli, M.; Rocchigiani, M.; Lentucci, C.; Anselmi, F.; Zippo, A.; Bini, L.; Oliviero, S. The characterization of a novel monoclonal antibody against CD93 unveils a new antiangiogenic target. Oncotarget 2014, 5, 2750–2760. [Google Scholar] [CrossRef]
- Lugano, R.; Vemuri, K.; Yu, D.; Bergqvist, M.; Smits, A.; Essand, M.; Johansson, S.; Dejana, E.; Dimberg, A. CD93 promotes beta1 integrin activation and fibronectin fibrillogenesis during tumor angiogenesis. J. Clin. Investig. 2018, 128, 3280–3297. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef] [PubMed]
- De, P.M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Luo, J.; Chen, X.Q.; Li, P. The Role of TGF-beta and Its Receptors in Gastrointestinal Cancers. Transl. Oncol. 2019, 12, 475–484. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Ten, D.P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Ten, D.P.; Arthur, H.M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef]
- Agah, A.; Kyriakides, T.R.; Lawler, J.; Bornstein, P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am. J. Pathol. 2002, 161, 831–839. [Google Scholar] [CrossRef]
- Kuroda, K.; Yashiro, M.; Sera, T.; Yamamoto, Y.; Kushitani, Y.; Sugimoto, A.; Kushiyama, S.; Nishimura, S.; Togano, S.; Okuno, T.; et al. The clinicopathological significance of Thrombospondin-4 expression in the tumor microenvironment of gastric cancer. PLoS ONE 2019, 14, e0224727. [Google Scholar] [CrossRef]
- Stenina-Adognravi, O.; Plow, E.F. Thrombospondin-4 in tissue remodeling. Matrix Biol. 2019, 75–76, 300–313. [Google Scholar] [CrossRef]
- Martino, M.M.; Briquez, P.S.; Guc, E.; Tortelli, F.; Kilarski, W.W.; Metzger, S.; Rice, J.J.; Kuhn, G.A.; Muller, R.; Swartz, M.A.; et al. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science 2014, 343, 885–888. [Google Scholar] [CrossRef]
- Hynes, R.O. Extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Bastos, B.M.; Jiang, W.G.; Cai, J. Tumour-Endothelial Cell Communications: Important and Indispensable Mediators of Tumour Angiogenesis. Anticancer Res. 2016, 36, 1119–1126. [Google Scholar] [PubMed]
- Parsons-Wingerter, P.; Chandrasekharan, U.M.; McKay, T.L.; Radhakrishnan, K.; DiCorleto, P.E.; Albarran, B.; Farr, A.G. A VEGF165-induced phenotypic switch from increased vessel density to increased vessel diameter and increased endothelial NOS activity. Microvasc. Res. 2006, 72, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Marisi, G.; Scarpi, E.; Passardi, A.; Nanni, O.; Ragazzini, A.; Valgiusti, M.; Casadei, G.A.; Neri, L.M.; Frassineti, G.L.; Amadori, D.; et al. Circulating VEGF and eNOS variations as predictors of outcome in metastatic colorectal cancer patients receiving bevacizumab. Sci. Rep. 2017, 7, 1293. [Google Scholar] [CrossRef]
- Liu, W.; Dong, Z.; Hu, R.; Wang, C. Association of Vascular Endothelial Growth Factor (VEGF) Gene Polymorphisms with Gastric Cancer and Its Development, Prognosis, and Survival. Technol. Cancer Res. Treat. 2018, 17, 1533034617753810. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Longatto-Filho, A.; Martins, S.F. Gastric Cancer and Angiogenesis: Is VEGF a Useful Biomarker to Assess Progression and Remission? J. Gastric. Cancer 2017, 17, 1–10. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Levin, E.R. Extracellular signal-regulated protein kinase/Jun kinase cross-talk underlies vascular endothelial cell growth factor-induced endothelial cell proliferation. J. Biol. Chem. 1998, 273, 26722–26728. [Google Scholar] [CrossRef]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol 2009, 21, 154–165. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, X.B.; Wang, J.S.; Wang, H.C.; Li, L.P. Function of fibroblast growth factor 2 in gastric cancer occurrence and prognosis. Mol. Med. Rep. 2020, 21, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. FGFR2-related pathogenesis and FGFR2-targeted therapeutics (Review). Int. J. Mol. Med. 2009, 23, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.M.; Hubbell, J.A. The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010, 24, 4711–4721. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley. Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Belotti, D.; Capelli, C.; Resovi, A.; Introna, M.; Taraboletti, G. Thrombospondin-1 promotes mesenchymal stromal cell functions via TGFbeta and in cooperation with PDGF. Matrix Biol. 2016, 55, 106–116. [Google Scholar] [CrossRef]
- Murakami, M.; Nguyen, L.T.; Zhuang, Z.W.; Moodie, K.L.; Carmeliet, P.; Stan, R.V.; Simons, M. The FGF system has a key role in regulating vascular integrity. J. Clin. Investig. 2008, 118, 3355–3366. [Google Scholar] [CrossRef]
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57–58, 1–11. [Google Scholar] [CrossRef]
- Li, S.; Shimono, C.; Norioka, N.; Nakano, I.; Okubo, T.; Yagi, Y.; Hayashi, M.; Sato, Y.; Fujisaki, H.; Hattori, S.; et al. Activin A binds to perlecan through its pro-region that has heparin/heparan sulfate binding activity. J. Biol. Chem. 2010, 285, 36645–36655. [Google Scholar] [CrossRef]
- Poluzzi, C.; Iozzo, R.V.; Schaefer, L. Endostatin and endorepellin: A common route of action for similar angiostatic cancer avengers. Adv. Drug Deliv. Rev 2016, 97, 156–173. [Google Scholar] [CrossRef]
- Whitelock, J.M.; Melrose, J.; Iozzo, R.V. Diverse cell signaling events modulated by perlecan. Biochemistry 2008, 47, 11174–11183. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Fu, J.; Oldershaw, R.; Greenhalgh, R.; Gown, A.M.; Iozzo, R.V. Perlecan protein core interacts with extracellular matrix protein 1 (ECM1), a glycoprotein involved in bone formation and angiogenesis. J. Biol. Chem. 2003, 278, 17491–17499. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Otto, J.; Oldershaw, R.; Ferrer, F.; Sato, J.D.; Iozzo, R.V. Fibroblast growth factor-binding protein is a novel partner for perlecan protein core. J. Biol. Chem. 2001, 276, 10263–10271. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Taylor, K.; Otto, J.; Aho, S.; Uitto, J.; Whitelock, J.M.; Iozzo, R.V. The protein core of the proteoglycan perlecan binds specifically to fibroblast growth factor-7. J. Biol. Chem. 2000, 275, 7095–7100. [Google Scholar] [CrossRef]
- Gonzalez, E.M.; Mongiat, M.; Slater, S.J.; Baffa, R.; Iozzo, R.V. A novel interaction between perlecan protein core and progranulin: Potential effects on tumor growth. J. Biol. Chem. 2003, 278, 38113–38116. [Google Scholar] [CrossRef]
- Chuang, C.Y.; Lord, M.S.; Melrose, J.; Rees, M.D.; Knox, S.M.; Freeman, C.; Iozzo, R.V.; Whitelock, J.M. Heparan sulfate-dependent signaling of fibroblast growth factor 18 by chondrocyte-derived perlecan. Biochemistry 2010, 49, 5524–5532. [Google Scholar] [CrossRef]
- Lord, M.S.; Chuang, C.Y.; Melrose, J.; Davies, M.J.; Iozzo, R.V.; Whitelock, J.M. The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol. 2014, 35, 112–122. [Google Scholar] [CrossRef]
- Oladipupo, S.S.; Kabir, A.U.; Smith, C.; Choi, K.; Ornitz, D.M. Impaired tumor growth and angiogenesis in mice heterozygous for Vegfr2 (Flk1). Sci. Rep. 2018, 8, 14724. [Google Scholar] [CrossRef]
- Briquez, P.S.; Hubbell, J.A.; Martino, M.M. Extracellular Matrix-Inspired Growth Factor Delivery Systems for Skin Wound Healing. Adv. Wound Care 2015, 4, 479–489. [Google Scholar] [CrossRef]
- van, C.H.; Giaccone, G.; Hoekman, K. Epidermal growth factor receptor and angiogenesis: Opportunities for combined anticancer strategies. Int. J. Cancer 2005, 117, 883–888. [Google Scholar] [CrossRef]
- Minder, P.; Zajac, E.; Quigley, J.P.; Deryugina, E.I. EGFR regulates the development and microarchitecture of intratumoral angiogenic vasculature capable of sustaining cancer cell intravasation. Neoplasia 2015, 17, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Hwang, S.; Kim, Y.M.; Pyun, B.J.; Kim, T.Y.; Lee, S.T.; Gho, Y.S.; Kwon, Y.G. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J. Biol. Chem. 2002, 277, 27872–27879. [Google Scholar] [CrossRef] [PubMed]
- Zanotelli, M.R.; Reinhart-King, C.A. Mechanical Forces in Tumor Angiogenesis. Adv. Exp. Med. Biol. 2018, 1092, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef]
- Giverso, C.; Ciarletta, P. Tumour angiogenesis as a chemo-mechanical surface instability. Sci. Rep. 2016, 6, 22610. [Google Scholar] [CrossRef]
- Vaeyens, M.M.; Jorge-Penas, A.; Barrasa-Fano, J.; Steuwe, C.; Heck, T.; Carmeliet, P.; Roeffaers, M.; Van, O.H. Matrix deformations around angiogenic sprouts correlate to sprout dynamics and suggest pulling activity. Angiogenesis 2020. [Google Scholar] [CrossRef]
- Rivron, N.C.; Vrij, E.J.; Rouwkema, J.; Le, G.S.; van den Berg, A.; Truckenmuller, R.K.; van Blitterswijk, C.A. Tissue deformation spatially modulates VEGF signaling and angiogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 6886–6891. [Google Scholar] [CrossRef]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Mysliwiec, A.G.; Ornstein, D.L. Matrix metalloproteinases in colorectal cancer. Clin. Colorectal Cancer 2002, 1, 208–219. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Zuidwijk, K.; Verspaget, H.W.; de Jonge-Muller, E.S.; van, D.W.; Ferreira, V.; Fontijn, R.D.; David, G.; Hommes, D.W.; Lamers, C.B.; et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer 2008, 44, 1904–1913. [Google Scholar] [CrossRef]
- Zheng, H.; Takahashi, H.; Murai, Y.; Cui, Z.; Nomoto, K.; Niwa, H.; Tsuneyama, K.; Takano, Y. Expressions of MMP-2, MMP-9 and VEGF are closely linked to growth, invasion, metastasis and angiogenesis of gastric carcinoma. Anticancer Res. 2006, 26, 3579–3583. [Google Scholar] [PubMed]
- Maeshima, Y.; Sudhakar, A.; Lively, J.C.; Ueki, K.; Kharbanda, S.; Kahn, C.R.; Sonenberg, N.; Hynes, R.O.; Kalluri, R. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science 2002, 295, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, Y.; Manfredi, M.; Reimer, C.; Holthaus, K.A.; Hopfer, H.; Chandamuri, B.R.; Kharbanda, S.; Kalluri, R. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J. Biol. Chem. 2001, 276, 15240–15248. [Google Scholar] [CrossRef] [PubMed]
- Di, D.T.; Orlandi, P.; Fioravanti, A.; Ali, G.; Cremolini, C.; Loupakis, F.; Gentile, D.; Banchi, M.; Cucchiara, F.; Antoniotti, C.; et al. Chemotherapeutic and antiangiogenic drugs beyond tumor progression in colon cancer: Evaluation of the effects of switched schedules and related pharmacodynamics. Biochem. Pharmacol. 2019, 164, 94–105. [Google Scholar] [CrossRef]
- Li, Y.; Kuscu, C.; Banach, A.; Zhang, Q.; Pulkoski-Gross, A.; Kim, D.; Liu, J.; Roth, E.; Li, E.; Shroyer, K.R.; et al. miR-181a-5p Inhibits Cancer Cell Migration and Angiogenesis via Downregulation of Matrix Metalloproteinase-14. Cancer Res. 2015, 75, 2674–2685. [Google Scholar] [CrossRef]
- Yang, B.; Gao, J.; Rao, Z.; Shen, Q. Clinicopathological and prognostic significance of alpha5beta1-integrin and MMP-14 expressions in colorectal cancer. Neoplasma 2013, 60, 254–261. [Google Scholar] [CrossRef]
- Noel, A.; Jost, M.; Maquoi, E. Matrix metalloproteinases at cancer tumor-host interface. Semin. Cell Dev. Biol. 2008, 19, 52–60. [Google Scholar] [CrossRef]
- Sounni, N.E.; Paye, A.; Host, L.; Noel, A. MT-MMPS as Regulators of Vessel Stability Associated with Angiogenesis. Front. Pharmacol. 2011, 2, 111. [Google Scholar] [CrossRef]
- Cauwe, B.; Van den Steen, P.E.; Opdenakker, G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 113–185. [Google Scholar] [CrossRef]
- Sier, C.F.; Zuidwijk, K.; Zijlmans, H.J.; Hanemaaijer, R.; Mulder-Stapel, A.A.; Prins, F.A.; Dreef, E.J.; Kenter, G.G.; Fleuren, G.J.; Gorter, A. EMMPRIN-induced MMP-2 activation cascade in human cervical squamous cell carcinoma. Int. J. Cancer 2006, 118, 2991–2998. [Google Scholar] [CrossRef]
- Lafleur, M.A.; Handsley, M.M.; Edwards, D.R. Metalloproteinases and their inhibitors in angiogenesis. Expert Rev. Mol. Med. 2003, 5, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.; Ten, D.P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef] [PubMed]
- Duff, S.E.; Li, C.; Garland, J.M.; Kumar, S. CD105 is important for angiogenesis: Evidence and potential applications. FASEB J. 2003, 17, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Bellone, G.; Gramigni, C.; Vizio, B.; Mauri, F.A.; Prati, A.; Solerio, D.; Dughera, L.; Ruffini, E.; Gasparri, G.; Camandona, M. Abnormal expression of Endoglin and its receptor complex (TGF-beta1 and TGF-beta receptor II) as early angiogenic switch indicator in premalignant lesions of the colon mucosa. Int. J. Oncol. 2010, 37, 1153–1165. [Google Scholar] [CrossRef]
- Saad, R.S.; Liu, Y.L.; Nathan, G.; Celebrezze, J.; Medich, D.; Silverman, J.F. Endoglin (CD105) and vascular endothelial growth factor as prognostic markers in colorectal cancer. Mod. Pathol. 2003, 17, 197–203. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.H.; Cheon, D.H.; Kim, T.; Park, Y.E.; Oh, E.S.; Lee, J.E.; Lee, S.T. Processing of syndecan-2 by matrix metalloproteinase-14 and effect of its cleavage on VEGF-induced tube formation of HUVECs. Biochem. J. 2017, 474, 3719–3732. [Google Scholar] [CrossRef]
- Corti, F.; Wang, Y.; Rhodes, J.M.; Atri, D.; Archer-Hartmann, S.; Zhang, J.; Zhuang, Z.W.; Chen, D.; Wang, T.; Wang, Z.; et al. Publisher Correction: N-terminal syndecan-2 domain selectively enhances 6-O heparan sulfate chains sulfation and promotes VEGFA165-dependent neovascularization. Nat. Commun. 2019, 10, 2124. [Google Scholar] [CrossRef]
- Dong, Z.; Sun, X.; Xu, J.; Han, X.; Xing, Z.; Wang, D.; Ge, J.; Meng, L.; Xu, X. Serum Membrane Type 1-Matrix Metalloproteinase (MT1-MMP) mRNA Protected by Exosomes as a Potential Biomarker for Gastric Cancer. Med. Sci. Monit. 2019, 25, 7770–7783. [Google Scholar] [CrossRef]
- Kasurinen, A.; Gramolelli, S.; Hagstrom, J.; Laitinen, A.; Kokkola, A.; Miki, Y.; Lehti, K.; Yashiro, M.; Ojala, P.M.; Bockelman, C.; et al. High tissue MMP14 expression predicts worse survival in gastric cancer, particularly with a low PROX1. Cancer Med. 2019, 8, 6995–7005. [Google Scholar] [CrossRef]
- Yoon, W.H.; Jung, Y.J.; Kim, T.D.; Li, G.; Park, B.J.; Kim, J.Y.; Lee, Y.C.; Kim, J.M.; Park, J.I.; Park, H.D.; et al. Gabexate mesilate inhibits colon cancer growth, invasion, and metastasis by reducing matrix metalloproteinases and angiogenesis. Clin. Cancer Res. 2004, 10, 4517–4526. [Google Scholar] [CrossRef]
- Liyanage, C.; Fernando, A.; Batra, J. Differential roles of protease isoforms in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 389–415. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Khalil, R.A. A Disintegrin and Metalloproteinase (ADAM) and ADAM with thrombospondin motifs (ADAMTS) family in vascular biology and disease. Biochem. Pharmacol. 2019, 164, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Hastings, G.; Ortega, M.A.; Lane, T.F.; Oikemus, S.; Lombardo, M.; Iruela-Arispe, M.L. METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J. Biol. Chem. 1999, 274, 23349–23357. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Carpizo, D.R.; Iruela-Arispe, M.L. ADAMTS1/METH1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF165. J. Biol. Chem. 2003, 278, 23656–23665. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Koch, M.; Karl, D.; Torres-Collado, A.X.; Fernando, N.T.; Rothrock, C.; Kuruppu, D.; Ryeom, S.; Iruela-Arispe, M.L.; Yoon, S.S. Variable inhibition of thrombospondin 1 against liver and lung metastases through differential activation of metalloproteinase ADAMTS1. Cancer Res. 2010, 70, 948–956. [Google Scholar] [CrossRef]
- Filou, S.; Korpetinou, A.; Kyriakopoulou, D.; Bounias, D.; Stavropoulos, M.; Ravazoula, P.; Papachristou, D.J.; Theocharis, A.D.; Vynios, D.H. ADAMTS expression in colorectal cancer. PLoS ONE 2015, 10, e0121209. [Google Scholar] [CrossRef][Green Version]
- Chen, J.; Zhi, Y.; Chang, X.; Zhang, S.; Dai, D. Expression of ADAMTS1 and its correlation with angiogenesis in primary gastric cancer and lymph node metastasis. Dig. Dis. Sci. 2013, 58, 405–413. [Google Scholar] [CrossRef]
- Kumar, S.; Sharghi-Namini, S.; Rao, N.; Ge, R. ADAMTS5 functions as an anti-angiogenic and anti-tumorigenic protein independent of its proteoglycanase activity. Am. J. Pathol. 2012, 181, 1056–1068. [Google Scholar] [CrossRef]
- Sharghi-Namini, S.; Fan, H.; Sulochana, K.N.; Potturi, P.; Xiang, W.; Chong, Y.S.; Wang, Z.; Yang, H.; Ge, R. The first but not the second thrombospondin type 1 repeat of ADAMTS5 functions as an angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 2008, 371, 215–219. [Google Scholar] [CrossRef]
- Huang, J.; Sun, Y.; Chen, H.; Liao, Y.; Li, S.; Chen, C.; Yang, Z. ADAMTS5 acts as a tumor suppressor by inhibiting migration, invasion and angiogenesis in human gastric cancer. Gastric Cancer 2019, 22, 287–301. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, J.; Li, X.; Zhang, C.; Zhang, H.; Jin, J.; Dai, D. Downregulation of ADAMTS8 by DNA Hypermethylation in Gastric Cancer and Its Clinical Significance. BioMed Res. Int. 2016, 2016, 5083841. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tang, J.; Feng, Y.; Li, S.; Xiang, Q.; He, X.; Ren, G.; Peng, W.; Xiang, T. ADAMTS9 is Silenced by Epigenetic Disruption in Colorectal Cancer and Inhibits Cell Growth and Metastasis by Regulating Akt/p53 Signaling. Cell. Physiol. Biochem. 2017, 44, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yuan, S.; Zhao, X.; Luo, T. ADAMTS8 is frequently down-regulated in colorectal cancer and functions as a tumor suppressor. Biochem. Biophys. Res. Commun. 2020, 524, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.C.; Li, J.; Wang, Y.; Li, L.; Zhong, L.; Ma, B.; Su, X.; Ying, J.; Xiang, T.; Rha, S.Y.; et al. The metalloprotease ADAMTS8 displays antitumor properties through antagonizing EGFR-MEK-ERK signaling and is silenced in carcinomas by CpG methylation. Mol. Cancer Res. 2014, 12, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Lin, F.; Zhang, M.; Mu, N.; Ge, X.; Fu, J. Long non-coding RNA AK001058 regulates tumor growth and angiogenesis in colorectal cancer via methylation of ADAMTS12. Am J Transl. Res 2019, 11, 6117–6123. [Google Scholar] [PubMed]
- Koo, B.H.; Coe, D.M.; Dixon, L.J.; Somerville, R.P.; Nelson, C.M.; Wang, L.W.; Young, M.E.; Lindner, D.J.; Apte, S.S. ADAMTS9 is a cell-autonomously acting, anti-angiogenic metalloprotease expressed by microvascular endothelial cells. Am. J. Pathol. 2010, 176, 1494–1504. [Google Scholar] [CrossRef]
- Ammendola, M.; Marech, I.; Sammarco, G.; Zuccala, V.; Luposella, M.; Zizzo, N.; Patruno, R.; Crovace, A.; Ruggieri, E.; Zito, A.F.; et al. Infiltrating mast cells correlate with angiogenesis in bone metastases from gastric cancer patients. Int. J. Mol. Sci. 2015, 16, 3237–3250. [Google Scholar] [CrossRef]
- Ammendola, M.; Patruno, R.; Sacco, R.; Marech, I.; Sammarco, G.; Zuccala, V.; Luposella, M.; Zizzo, N.; Gadaleta, C.; Porcelli, M.; et al. Mast cells positive to tryptase and tumour-associated macrophages correlate with angiogenesis in locally advanced colorectal cancer patients undergone to surgery. Expert Opin. Ther. Targets 2016, 20, 533–540. [Google Scholar] [CrossRef]
- Sammarco, G.; Varricchi, G.; Ferraro, V.; Ammendola, M.; De, F.M.; Altomare, D.F.; Luposella, M.; Maltese, L.; Curro, G.; Marone, G.; et al. Mast Cells, Angiogenesis and Lymphangiogenesis in Human Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 2106. [Google Scholar] [CrossRef]
- Sammarco, G.; Gadaleta, C.D.; Zuccala, V.; Albayrak, E.; Patruno, R.; Milella, P.; Sacco, R.; Ammendola, M.; Ranieri, G. Tumor-Associated Macrophages and Mast Cells Positive to Tryptase Are Correlated with Angiogenesis in Surgically-Treated Gastric Cancer Patients. Int. J. Mol. Sci. 2018, 19, 1176. [Google Scholar] [CrossRef]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell. Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ichikawa, Y.; Nakagawa, K.; Kumamoto, T.; Mori, R.; Matsuyama, R.; Takeda, K.; Ota, M.; Tanaka, K.; Tamura, T.; et al. High infiltration of mast cells positive to tryptase predicts worse outcome following resection of colorectal liver metastases. BMC Cancer 2015, 15, 840. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, M.; Bauer, R.; Cubas, C.M.; Udonta, F.; Ben-Batalla, I.; Legler, K.; Hauser, C.; Egberts, J.; Janning, M.; Velthaus, J.; et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat. Commun. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Hsu, I.; Granville, D.J. Granzyme B releases vascular endothelial growth factor from extracellular matrix and induces vascular permeability. Lab. Investig. 2014, 94, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Ammendola, M.; Sacco, R.; Sammarco, G.; Donato, G.; Zuccala, V.; Romano, R.; Luposella, M.; Patruno, R.; Vallicelli, C.; Verdecchia, G.M.; et al. Mast Cells Positive to Tryptase and c-Kit Receptor Expressing Cells Correlates with Angiogenesis in Gastric Cancer Patients Surgically Treated. Gastroenterol. Res. Pract. 2013, 2013, 703163. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Thomazy, V. Protein crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect. Tissue Res. 2000, 41, 1–27. [Google Scholar] [CrossRef]
- Martinez, J.; Chalupowicz, D.G.; Roush, R.K.; Sheth, A.; Barsigian, C. Transglutaminase-mediated processing of fibronectin by endothelial cell monolayers. Biochemistry 1994, 33, 2538–2545. [Google Scholar] [CrossRef]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell. Sci. 2001, 114, 2755–2773. [Google Scholar]
- Jones, R.A.; Kotsakis, P.; Johnson, T.S.; Chau, D.Y.; Ali, S.; Melino, G.; Griffin, M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006, 13, 1442–1453. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. S3), 4–10. [Google Scholar] [CrossRef] [PubMed]
- Van, C.E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van, C.E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol 2015, 16, 499–508. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Rahbari, N.N.; Kedrin, D.; Incio, J.; Liu, H.; Ho, W.W.; Nia, H.T.; Edrich, C.M.; Jung, K.; Daubriac, J.; Chen, I.; et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci. Transl. Med. 2016, 8, 360ra135. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Suhovskih, A.V.; Aidagulova, S.V.; Kashuba, V.I.; Grigorieva, E.V. Proteoglycans as potential microenvironmental biomarkers for colon cancer. Cell Tissue Res. 2015, 361, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Onyeisi, J.O.S.; Castanho de Almeida Pernambuco Filho, P.; de Araujo, L.S.; Nader, H.B.; Lopes, C.C. Heparan sulfate proteoglycans as trastuzumab targets in anoikis-resistant endothelial cells. J. Cell. Biochem. 2019, 120, 13826–13840. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zang, M.D.; Wang, H.X.; Li, J.F.; Su, L.P.; Yan, M.; Li, C.; Yang, Q.M.; Liu, B.Y.; Zhu, Z.G. Biglycan stimulates VEGF expression in endothelial cells by activating the TLR signaling pathway. Mol. Oncol. 2016, 10, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Coltrini, D.; Ronca, R.; Belleri, M.; Zardi, L.; Indraccolo, S.; Scarlato, V.; Giavazzi, R.; Presta, M. Impact of VEGF-dependent tumour micro-environment on EDB fibronectin expression by subcutaneous human tumour xenografts in nude mice. J. Pathol. 2009, 219, 455–462. [Google Scholar] [CrossRef]
- Compagni, A.; Wilgenbus, P.; Impagnatiello, M.A.; Cotten, M.; Christofori, G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000, 60, 7163–7169. [Google Scholar]
- Murakami, M.; Simons, M. Fibroblast growth factor regulation of neovascularization. Curr. Opin. Hematol. 2008, 15, 215–220. [Google Scholar] [CrossRef]
- Rusnati, M.; Borsotti, P.; Moroni, E.; Foglieni, C.; Chiodelli, P.; Carminati, L.; Pinessi, D.; Annis, D.S.; Paiardi, G.; Bugatti, A.; et al. The calcium-binding type III repeats domain of thrombospondin-2 binds to fibroblast growth factor 2 (FGF2). Angiogenesis 2019, 22, 133–144. [Google Scholar] [CrossRef]
- Margosio, B.; Rusnati, M.; Bonezzi, K.; Cordes, B.L.; Annis, D.S.; Urbinati, C.; Giavazzi, R.; Presta, M.; Ribatti, D.; Mosher, D.F.; et al. Fibroblast growth factor-2 binding to the thrombospondin-1 type III repeats, a novel antiangiogenic domain. Int. J. Biochem. Cell Biol. 2008, 40, 700–709. [Google Scholar] [CrossRef]
- Giacomini, A.; Chiodelli, P.; Matarazzo, S.; Rusnati, M.; Presta, M.; Ronca, R. Blocking the FGF/FGFR system as a “two-compartment” antiangiogenic/antitumor approach in cancer therapy. Pharmacol. Res. 2016, 107, 172–185. [Google Scholar] [CrossRef]
- Fuster, M.M.; Wang, L.; Castagnola, J.; Sikora, L.; Reddi, K.; Lee, P.H.; Radek, K.A.; Schuksz, M.; Bishop, J.R.; Gallo, R.L.; et al. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J. Cell Biol. 2007, 177, 539–549. [Google Scholar] [CrossRef]
- Lieu, C.; Heymach, J.; Overman, M.; Tran, H.; Kopetz, S. Beyond VEGF: Inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin. Cancer Res. 2011, 17, 6130–6139. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Watanabe, M.S.; Minoshima, Y.; Matsui, J.; Funahashi, Y. Activated FGF2 signaling pathway in tumor vasculature is essential for acquired resistance to anti-VEGF therapy. Sci. Rep. 2020, 10, 2939. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yu, Z.; Li, Z.; Tang, J.; Lai, X.; Liu, L. Expression of angiogenic growth factors VEGF, bFGF and ANG1 in colon cancer after bevacizumab treatment in vitro: A potential self-regulating mechanism. Oncol. Rep. 2017, 37, 601–607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von, M.M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Grothey, A.; Van, C.E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Hellberg, C.; Ostman, A.; Heldin, C.H. PDGF and vessel maturation. Recent Results Cancer Res. 2010, 180, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Appiah-Kubi, K.; Wang, Y.; Wu, M.; Tao, Y.; Wu, Y.; Chen, Y. The clinical significance of platelet-derived growth factors (PDGFs) and their receptors (PDGFRs) in gastric cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2018, 127, 15–28. [Google Scholar] [CrossRef]
- Üçüncü, M.; Serilmez, M.; Sari, M.; Bademler, S.; Karabulut, S. The Diagnostic Significance of PDGF, EphA7, CCR5, and CCL5 Levels in Colorectal Cancer. Biomolecules 2019, 9, 464. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Nakamura, M.; Andersson, P.; Rouhi, P.; Yang, X.; Jensen, L.; Lim, S.; Feng, N.; et al. Tumour PDGF-BB expression levels determine dual effects of anti-PDGF drugs on vascular remodelling and metastasis. Nat. Commun. 2013, 4, 2129. [Google Scholar] [CrossRef]
- Schiffmann, L.M.; Brunold, M.; Liwschitz, M.; Goede, V.; Loges, S.; Wroblewski, M.; Quaas, A.; Alakus, H.; Stippel, D.; Bruns, C.J.; et al. A combination of low-dose bevacizumab and imatinib enhances vascular normalisation without inducing extracellular matrix deposition. Br. J. Cancer 2017, 116, 600–608. [Google Scholar] [CrossRef]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yu, Q. Angiopoietin-1, unlike angiopoietin-2, is incorporated into the extracellular matrix via its linker peptide region. J. Biol. Chem. 2001, 276, 34990–34998. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Tanaka, K.; Kitajima, T.; Shimura, T.; Kawamura, M.; Kawamoto, A.; Okugawa, Y.; Saigusa, S.; Hiro, J.; Inoue, Y.; et al. Elevated serum angiopoietin-like protein 2 correlates with the metastatic properties of colorectal cancer: A serum biomarker for early diagnosis and recurrence. Clin. Cancer Res. 2014, 20, 6175–6186. [Google Scholar] [CrossRef] [PubMed]
- Blank, S.; Deck, C.; Dreikhausen, L.; Weichert, W.; Giese, N.; Falk, C.; Schmidt, T.; Ott, K. Angiogenic and growth factors in gastric cancer. J. Surg. Res. 2015, 194, 420–429. [Google Scholar] [CrossRef]
- Falcon, B.L.; Hashizume, H.; Koumoutsakos, P.; Chou, J.; Bready, J.V.; Coxon, A.; Oliner, J.D.; McDonald, D.M. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am. J. Pathol. 2009, 175, 2159–2170. [Google Scholar] [CrossRef]
- Coutelle, O.; Schiffmann, L.M.; Liwschitz, M.; Brunold, M.; Goede, V.; Hallek, M.; Kashkar, H.; Hacker, U.T. Dual targeting of Angiopoetin-2 and VEGF potentiates effective vascular normalisation without inducing empty basement membrane sleeves in xenograft tumours. Br. J. Cancer 2015, 112, 495–503. [Google Scholar] [CrossRef]
- Hidalgo, M.; Martinez-Garcia, M.; Le, T.C.; Massard, C.; Garralda, E.; Boni, V.; Taus, A.; Albanell, J.; Sablin, M.P.; Alt, M.; et al. First-in-Human Phase I Study of Single-agent Vanucizumab, A First-in-Class Bispecific Anti-Angiopoietin-2/Anti-VEGF-A Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1536–1545. [Google Scholar] [CrossRef]




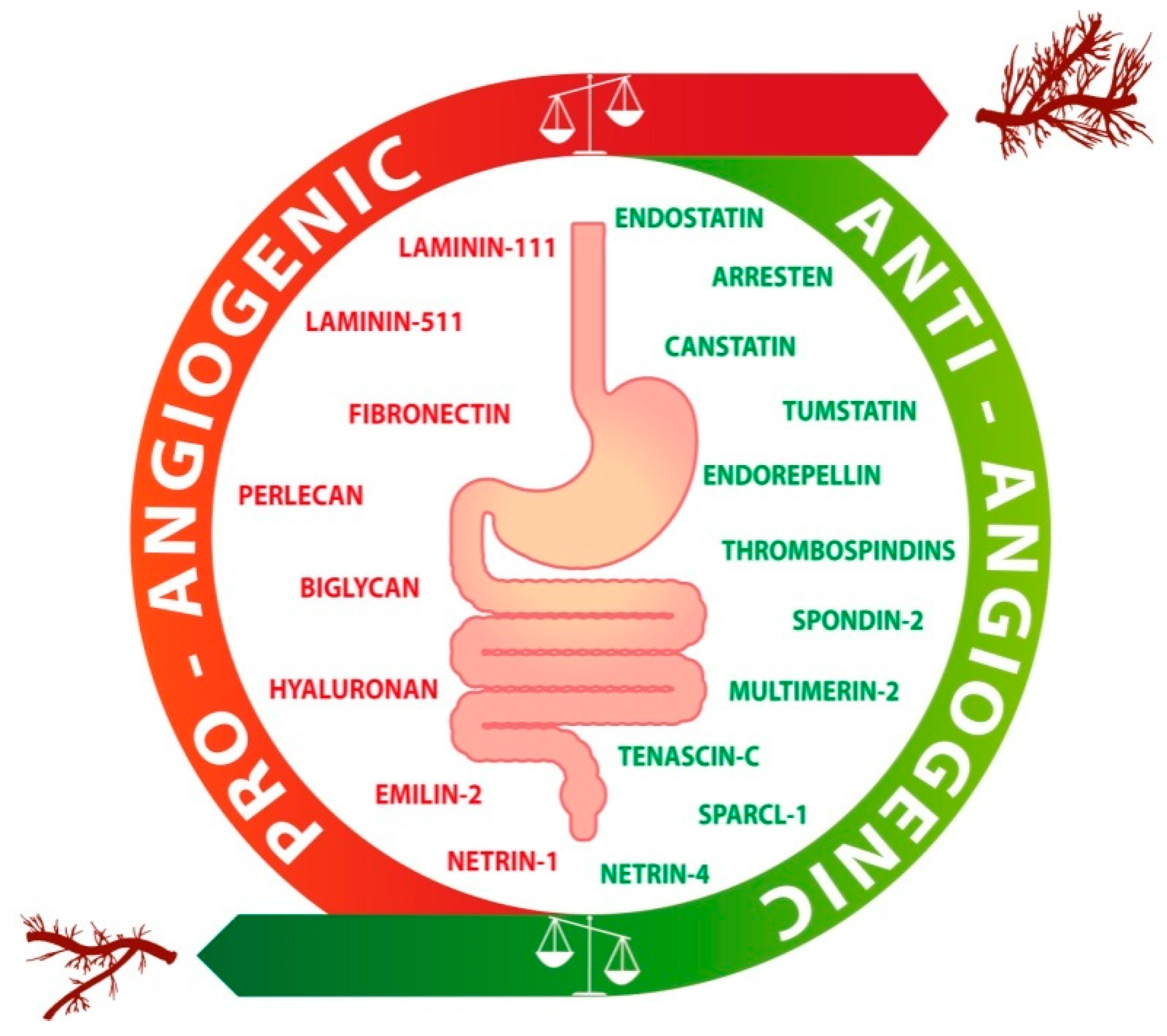{kind=link}
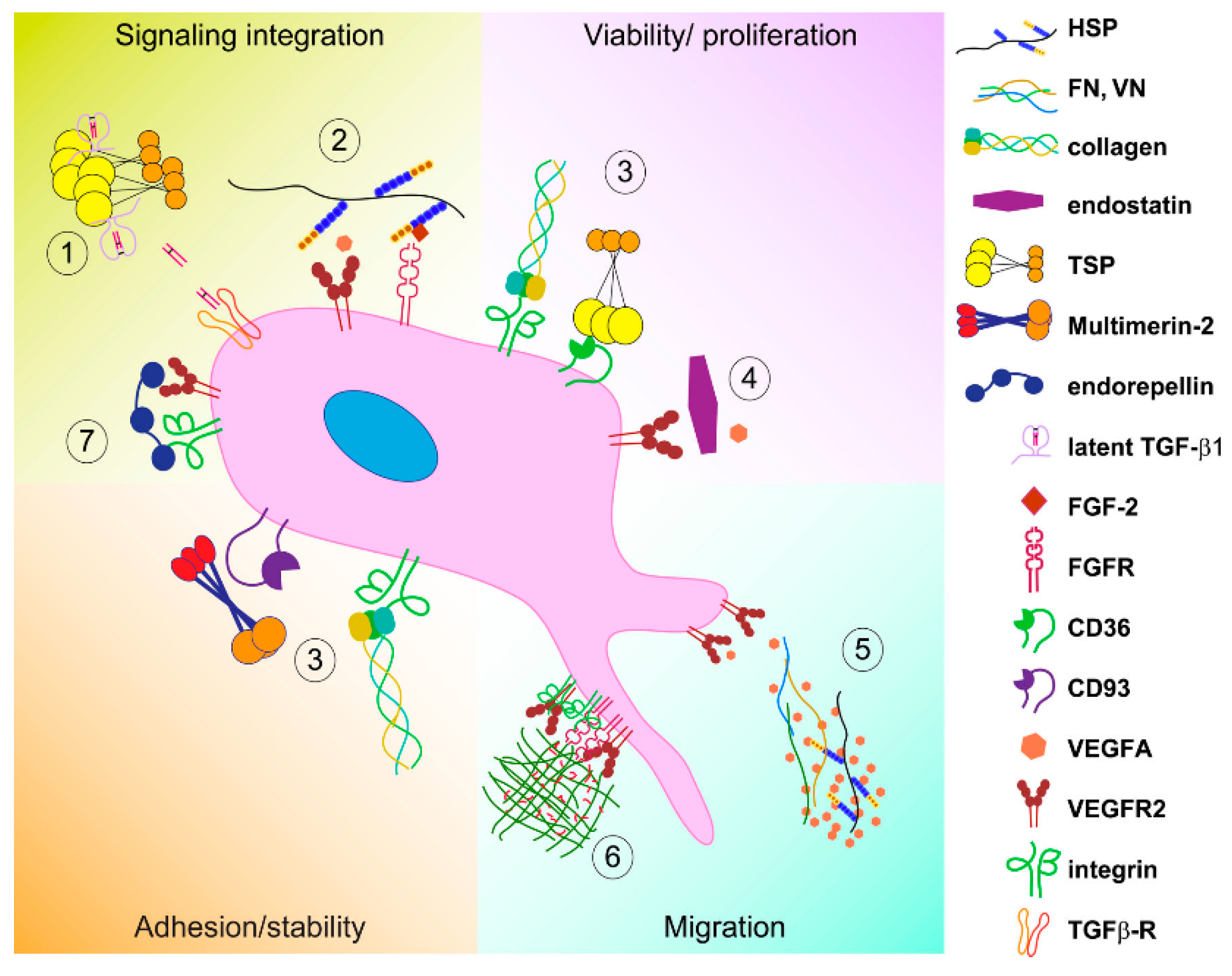{kind=link}
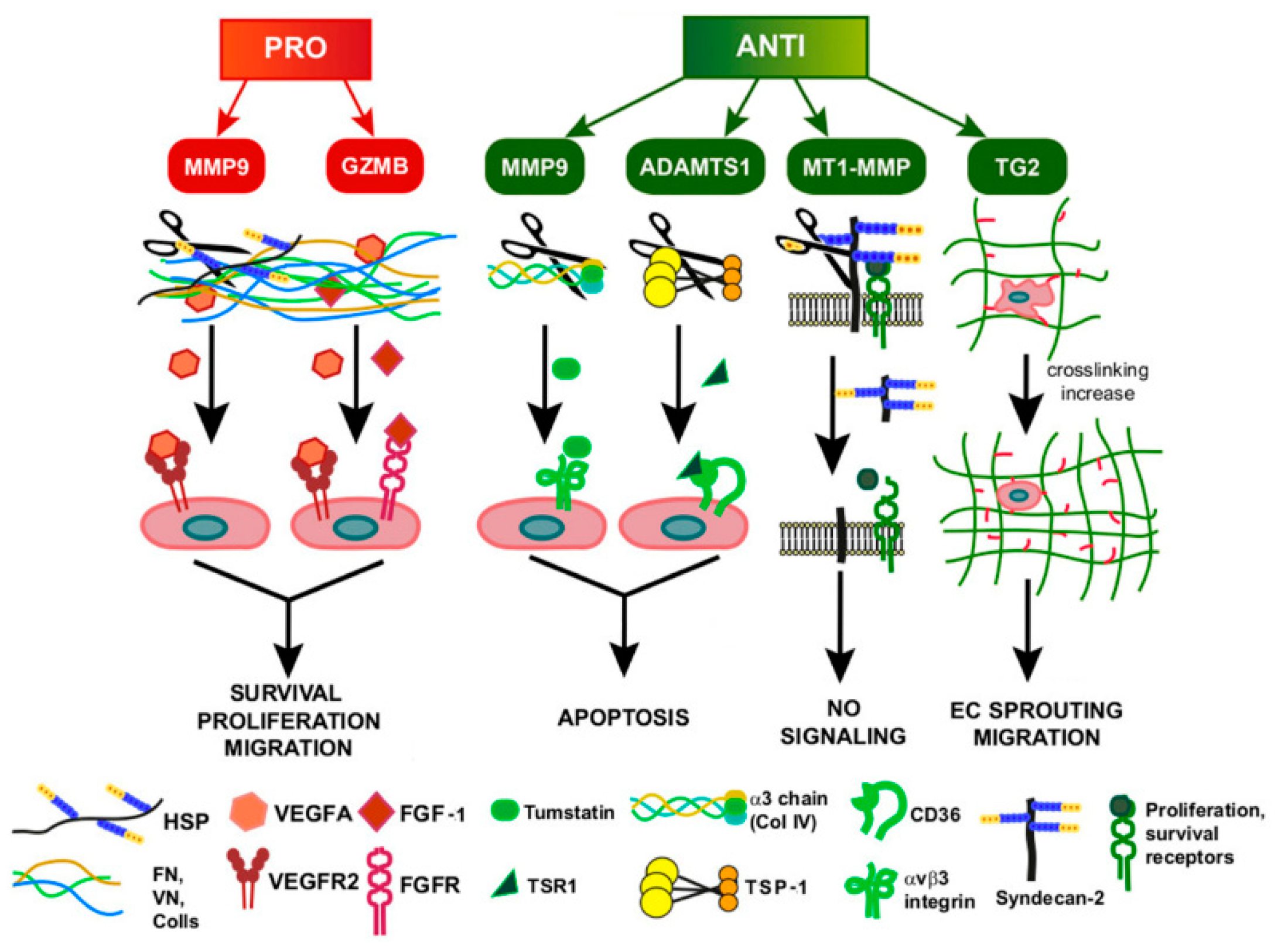{kind=link}
| Antiangiogenic Drug | Target | Type of Molecule | Cancer Type | Clinical Use | Ref |
|---|---|---|---|---|---|
| Bevacizumab | VEGFA | Blocking humanized mAb | mCRC | 1st line alone or in combination with chemotherapy | [34] |
| Aflibercept | VEGFA | Trapping Recombinant fusion protein (VEGFR1-VEGFR2) | mCRC | 2nd line | [293] |
| Sunitinib | VEGFR2 | Small RTK inhibitor | GC | 2nd line | [294] |
| Ramucirumb | VEGFR2 | Blocking humanized mAb | mCRC/advanced GC | 2nd line alone or in combination with chemotherapy | [295,296] |
| Regorafenib | FGFR2/VEGFR2 | Small RTK inhibitor | mCRC/GC | After the failure of other lines of therapies | [314,315] |
| Imatinib | PDGFR | Small RTK inhibitor | mCRC/advanced GC | Adjuvant chemotherapy in patients with wt PDGFR | [320] |
| Vanucizumab | ANG2/VEGFA | Bi-specific mAb | CRC | Not approved | [327] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreuzzi, E.; Capuano, A.; Poletto, E.; Pivetta, E.; Fejza, A.; Favero, A.; Doliana, R.; Cannizzaro, R.; Spessotto, P.; Mongiat, M. Role of Extracellular Matrix in Gastrointestinal Cancer-Associated Angiogenesis. Int. J. Mol. Sci. 2020, 21, 3686. https://doi.org/10.3390/ijms21103686
Andreuzzi E, Capuano A, Poletto E, Pivetta E, Fejza A, Favero A, Doliana R, Cannizzaro R, Spessotto P, Mongiat M. Role of Extracellular Matrix in Gastrointestinal Cancer-Associated Angiogenesis. International Journal of Molecular Sciences. 2020; 21(10):3686. https://doi.org/10.3390/ijms21103686
Chicago/Turabian StyleAndreuzzi, Eva, Alessandra Capuano, Evelina Poletto, Eliana Pivetta, Albina Fejza, Andrea Favero, Roberto Doliana, Renato Cannizzaro, Paola Spessotto, and Maurizio Mongiat. 2020. "Role of Extracellular Matrix in Gastrointestinal Cancer-Associated Angiogenesis" International Journal of Molecular Sciences 21, no. 10: 3686. https://doi.org/10.3390/ijms21103686
APA StyleAndreuzzi, E., Capuano, A., Poletto, E., Pivetta, E., Fejza, A., Favero, A., Doliana, R., Cannizzaro, R., Spessotto, P., & Mongiat, M. (2020). Role of Extracellular Matrix in Gastrointestinal Cancer-Associated Angiogenesis. International Journal of Molecular Sciences, 21(10), 3686. https://doi.org/10.3390/ijms21103686