1. Introduction
Colorectal cancer (CRC) remains one of the leading causes of cancer-related deaths worldwide. Unfortunately, the incidence and mortality rate of this cancer is expected to rise even further, creating an urgent need to identify specific molecular pathways underlying CRC malignancy that can be targeted for the development of new therapeutic approaches. Recently, protein arginine methyltransferase 5 (PRMT5) has emerged as a key positive modulator of CRC malignancy and its overexpression is highly correlated to poor patient prognosis [
1]. As a member of the superfamily of PRMTs, PRMT5 symmetrically dimethylates the guanidino nitrogen atoms of arginine residues using S-adenosylmethionine (AdoMet) as a methyl donor. Our group and others have previously reported that overexpression of PRMT5 enhanced the proliferative, migratory, and colony-forming abilities of CRC cells, whereas knockdown or pharmacological inhibition of PRMT5 had the opposite effect, making it a highly attractive therapeutic target [
1,
2]. Moreover, accumulating evidence suggests that PRMT5 exerts its pro-tumor functions through methylation of several pleotropic transcription factors (TFs). Through modulation of these TFs, PRMT5 can enact a wider repertoire of transcriptional changes related to many essential cellular processes that go awry during cancer progression [
2,
3,
4,
5]. In this regard, we provided the first evidence of PRMT5 dimethylating the p65 subunit of nuclear kappa B (NF-κB) at arginine 30 (R30) in response to interleukin-1β (IL-1β). This modification potently activated NF-κB signaling, resulting in enhanced DNA-binding ability of p65 and induction of more than 75% of NF-κB target genes involved in inflammation, cell proliferation, survival, and differentiation [
6].
Aberrant activation of the NF-ĸB signaling pathway remains one of the most prominent etiological factors in CRC malignancy [
7,
8]. As a master regulator of innate and inflammatory responses, many key cellular processes that become deregulated in cancer are critically influenced by NF-ĸB-dependent transcription of a diverse array of target genes [
9,
10]. The family of NF-ĸB proteins is divided into two subgroups, comprising of five members. The Rel proteins, which include RelA (p65), RelB and cRel, harboring transactivation domains in their C-termini while the NF-κB1 (p50 and precursor p105) and NF-κB2 (p52 and precursor p100) proteins contain C-terminal ankyrin repeats. These proteins typically exist as dimers (p65:p50 or RelB:p52). In the absence of an activating stimulus, NF-κB is sequestered in the cytoplasm through their complex with the inhibitor of NF-κB (IκB). Activation of the classical NF-κB signaling pathway which involves the prototypical p65:p50 heterodimer, is initiated by a range of extracellular signals including pro-inflammatory cytokines such as IL-1β. These initiating signals then trigger a cascade of events that culminate in phosphorylation of IκBα by the IκB kinase (IKKα/IKKβ) complex, leading to proteasomal ubiquitin-dependent IκBα degradation. Finally, the subsequent liberation of the p65:p50 heterodimer results in its nuclear translocation and activation of NF-κB target gene transcription [
10].
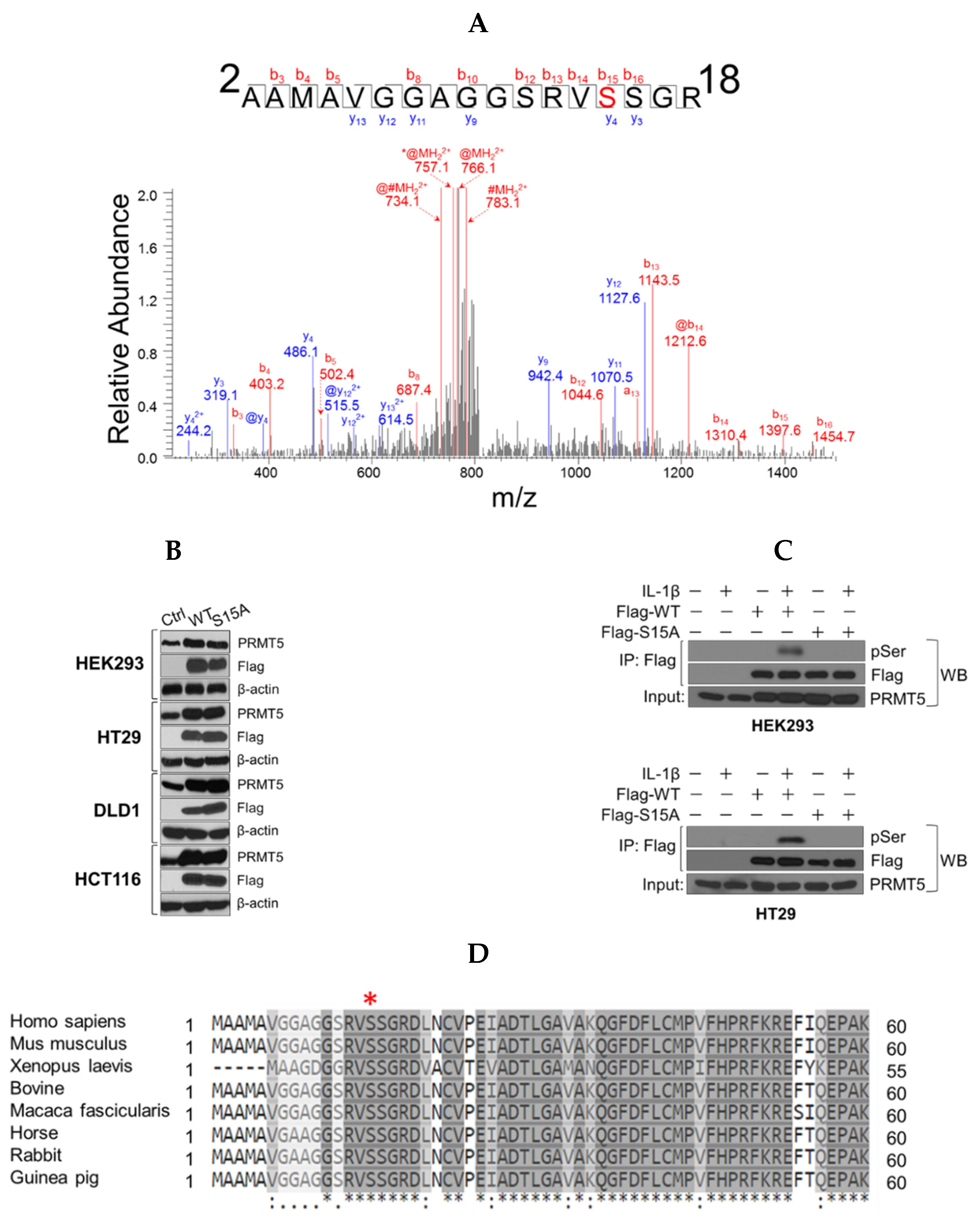
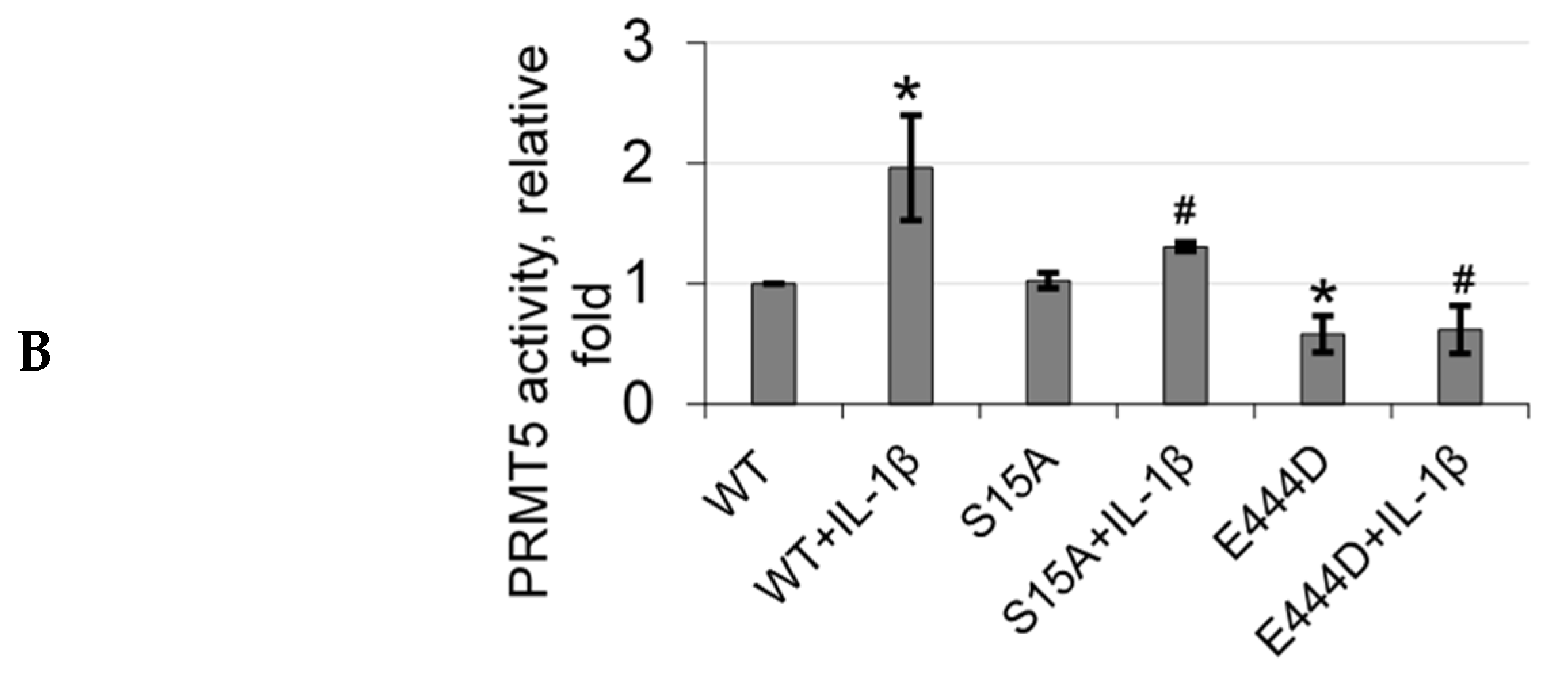
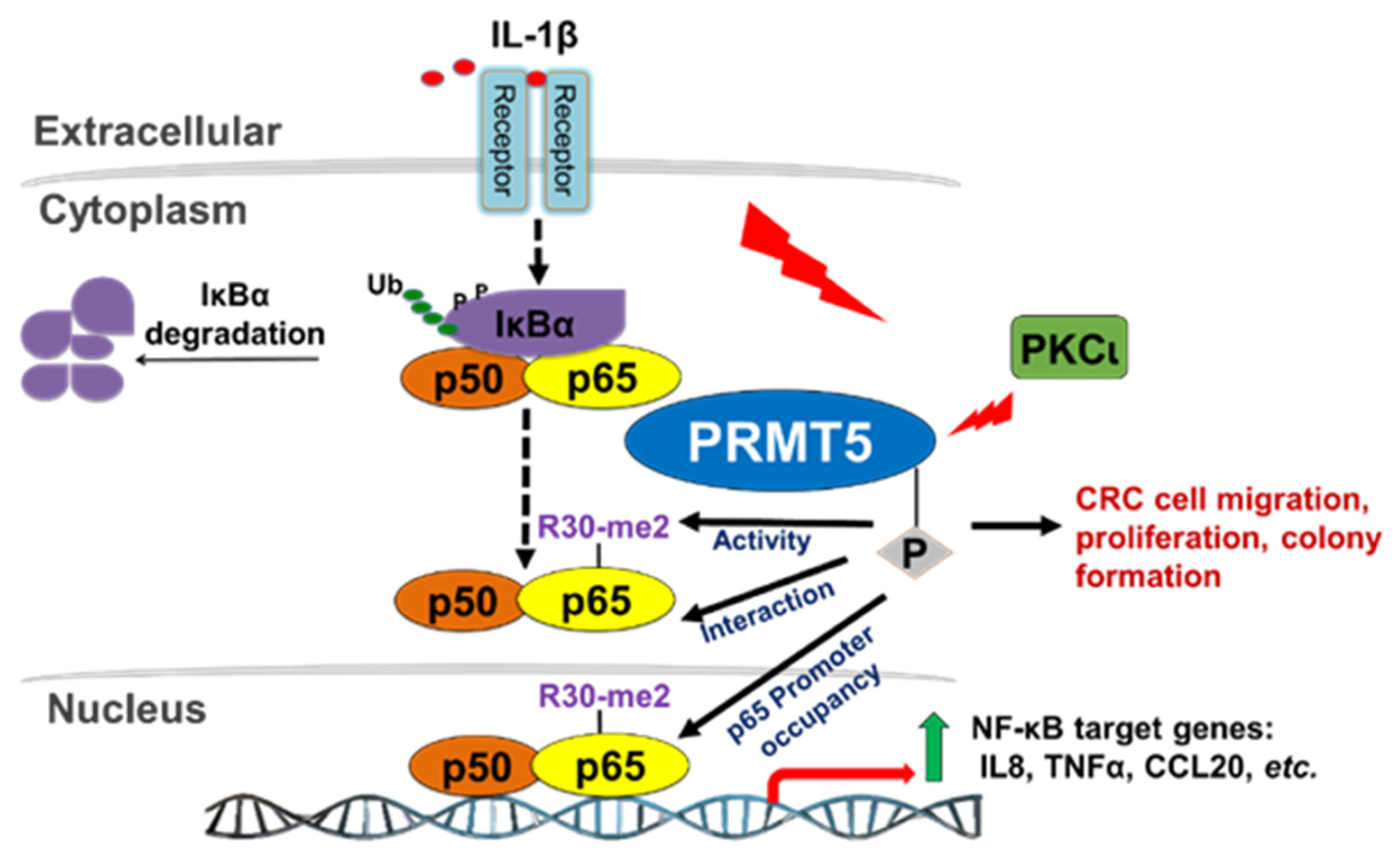
Although we have previously shown that PRMT5 can dynamically regulate p65 via IL-1β-inducible methylation at R30, the precise mechanisms governing this novel PRMT5/p65 signaling axis are still not fully understood. In the present study, we have now uncovered a novel mechanism that PRMT5-mediated activation of NF-κB is dependent on signal-induced phosphorylation of PRMT5 at serine 15 (S15) and propose that blocking this posttranslational modification (PTM) could serve the dual purpose of impairing the tumor-associated functions of PRMT5 via attenuating its activity towards NF-κB as a potential treatment strategy for CRC [
11,
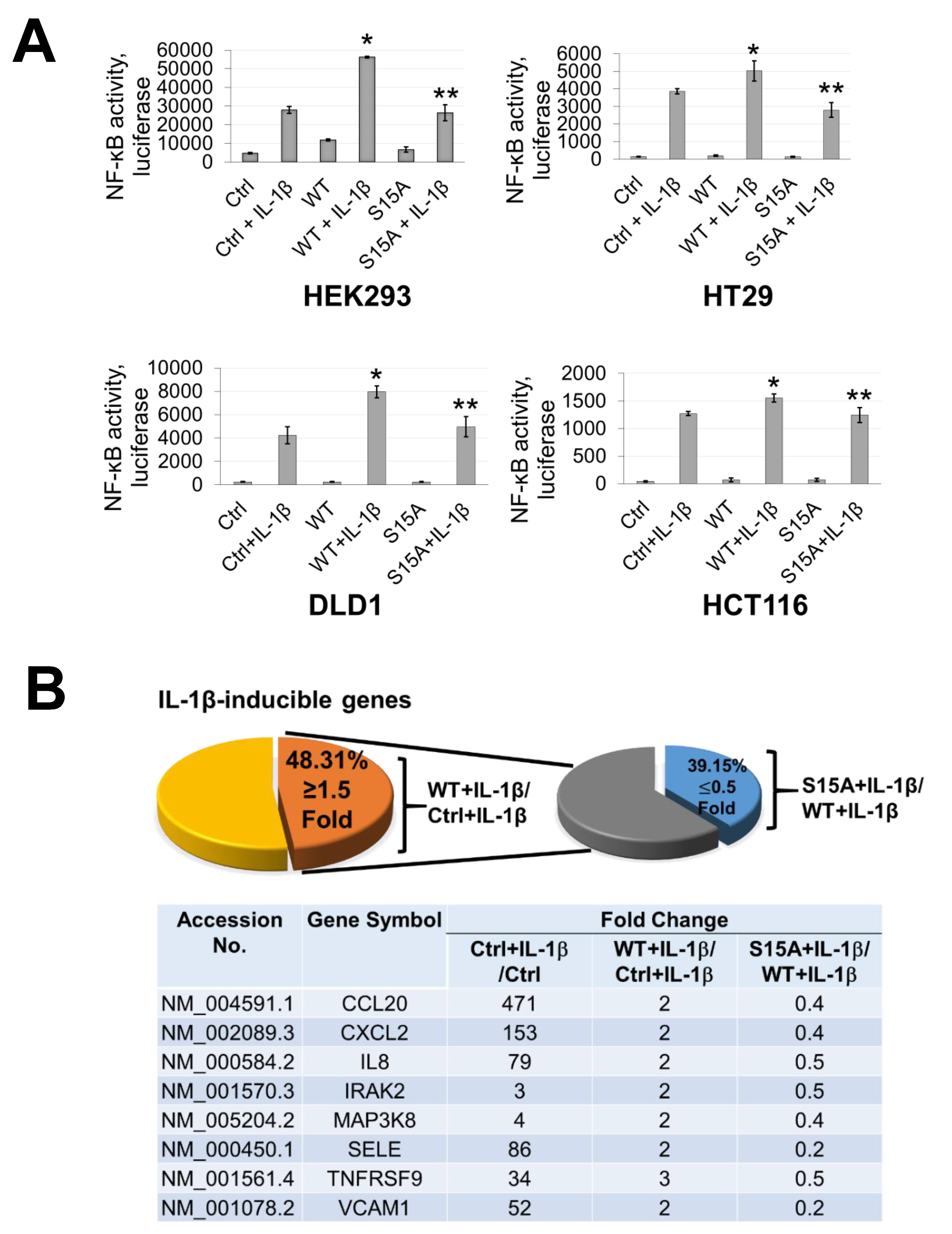
12]. Compared to WT-PRMT5, substitution of serine with alanine at S15 (S15A) dramatically reduced NF-κB transactivation potential which correlated with decreased target gene transcription through attenuated recruitment of p65 to the proximal promoter of its target genes. Moreover, by demonstrating that the S15A mutant of PRMT5 disrupted its interaction with p65, our work provides new insight into the regulation of the PRMT5/NF-κB axis through phospho-dependent protein–protein interactions. These results suggest that crosstalk between kinases and PRMT5 may play a pivotal role in modulating the diverse cellular functions of PRMT5, implicating the potential utility of relevant kinase inhibitors to disrupt PRMT5-NF-κB cooperativity and impede cancer cell growth.
3. Discussion
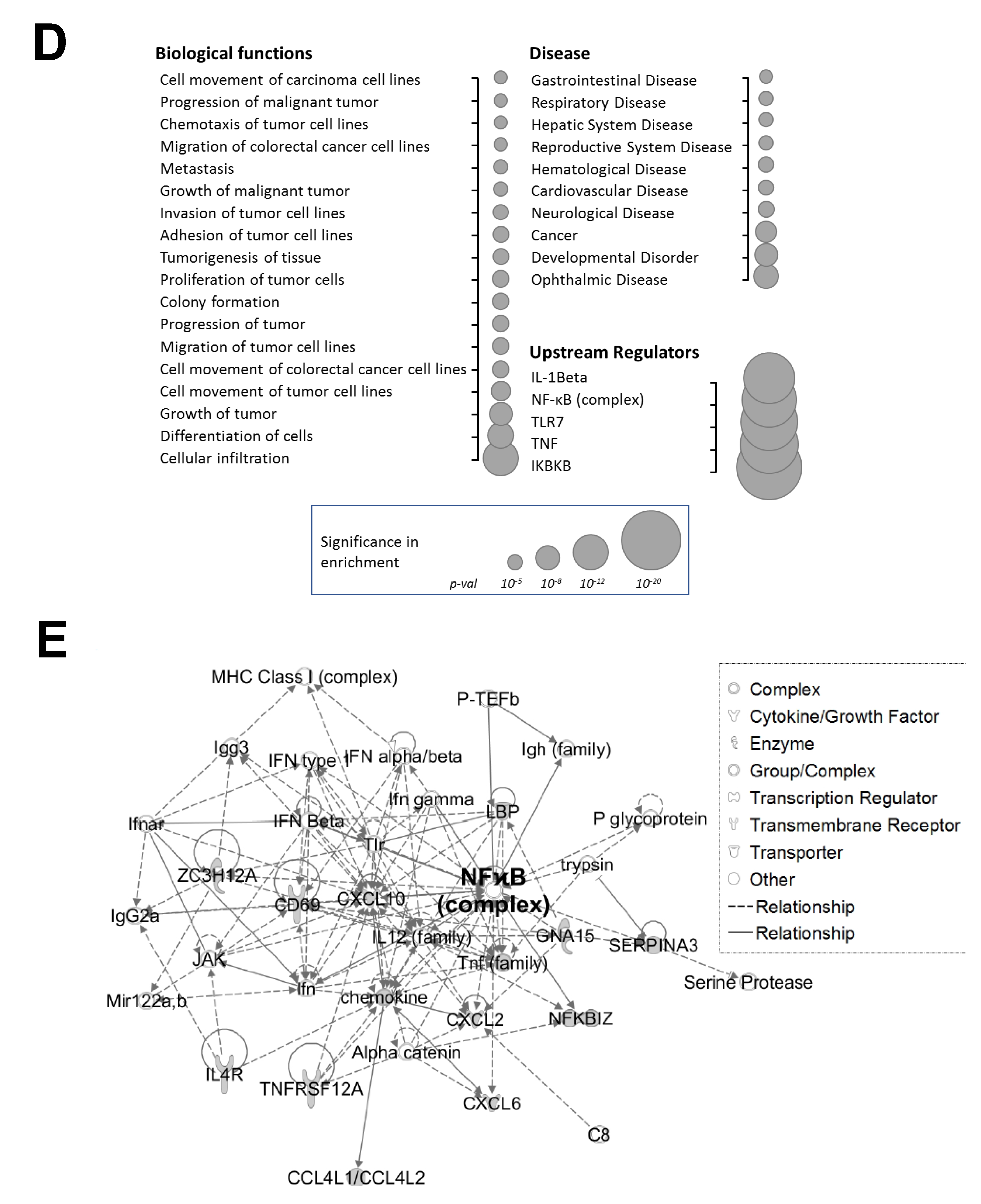
A wealth of reported findings supports the essential biological functions of PRMT5 in cell proliferation, transcriptional activation, signal transduction, and cell differentiation through methylation of its substrate proteins [
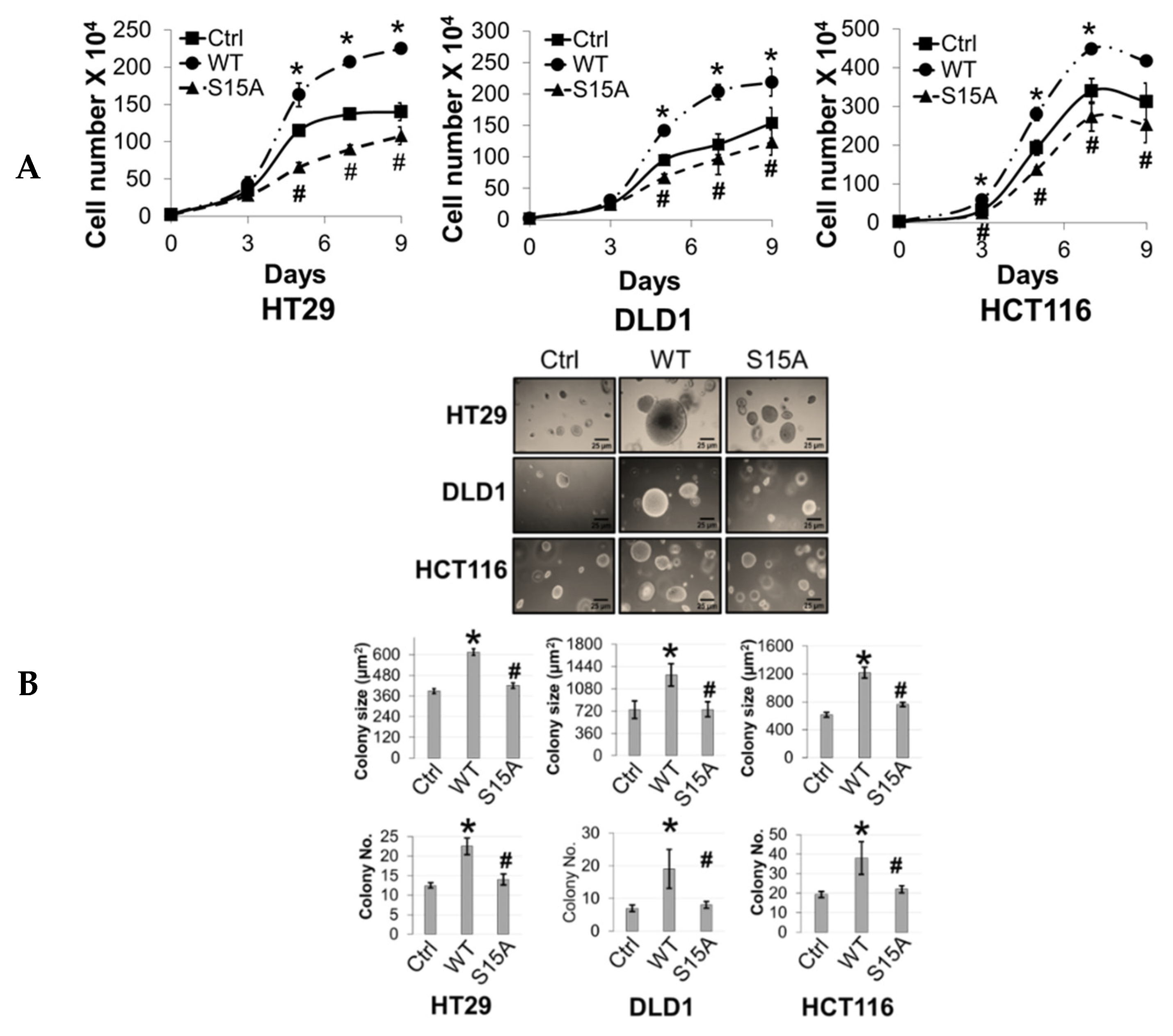
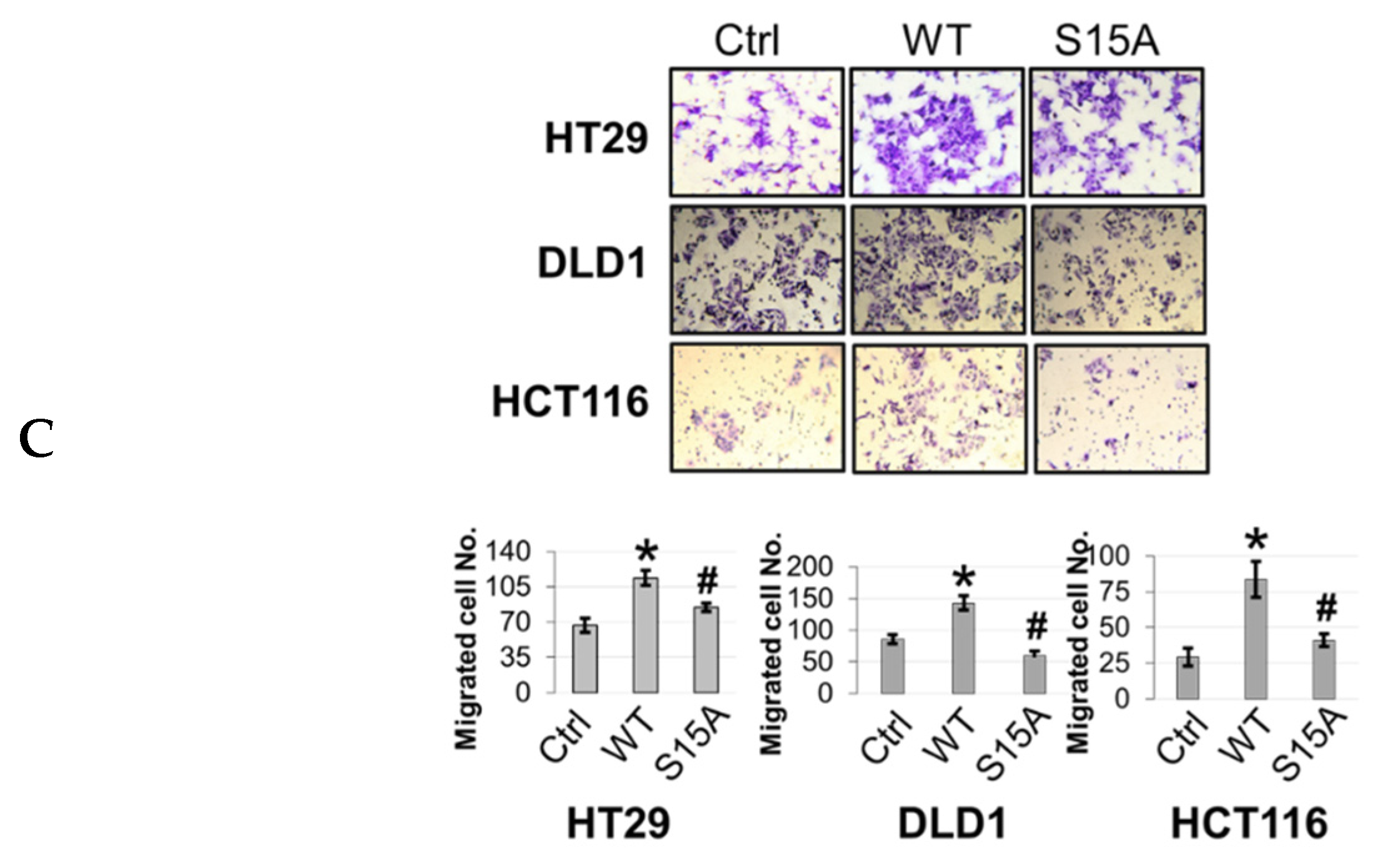
14]. However, far less is known about the factors that underlie its involvement in various signaling axes or the mechanisms that fine-tune its activity towards certain substrates. Nevertheless, this knowledge is critical since these mechanisms form the basis for devising novel therapeutic strategies for targeting PRMT5. In the present study, we report a novel phosphorylation modification on PRMT5 at S15 and mechanistically validated its role in positively regulating the PRMT5/NF-κB signaling node through modulation of the PRMT5/p65 interaction, NF-κB target gene expression and proximal promoter occupancy of p65 at its target genes as well as the IL-1β-inducible methyltransferase activity of PRMT5. Moreover, our data indicate that the S15A mutant of PRMT5 could impede the proliferative, migratory, and anchorage-independent growth capabilities of CRC cells. This evidence supports a model in which phosphorylation of PRMT5 may act as a pivotal step in enhancing the NF-κB response observed with PRMT5 overexpression which involves upregulation of the transcription of cancer-associated NF-κB target genes.
Phosphorylation of PRMT5 on other types of amino acids, such as tyrosine and threonine was previously reported. For instance, the Liu group identified tyrosine phosphorylation (Y297, 304, 307) of PRMT5 by mutant Jak2 playing an inhibitory role on PRMT5′s dimethylation of histone substrates. This inhibition also correlated with an enhanced myeloproliferative phenotype [
15]. In a quite different context, threonine phosphorylation of PRMT5 was shown to modulate PRMT5′s function by triggering a PDZ/14-3-3 interaction switch [
16]. In contrast, in our case, we showed for the first time that PRMT5 can be phosphorylated on a serine residue upon IL-1β treatment, leading to the enhancement of its methyltransferase activity and interaction between PRMT5 and its substrate, p65. This sheds important light on how the same but context-dependent modification on different amino acid residues can produce varied outcomes. Intriguingly, our data also implies that cytokines in the tumor microenvironment may affect cancer progression since IL-1β is a pleiotropic pro-inflammatory cytokine abundantly secreted within the tumor microenvironment and critically mediates the induction of a local network of cytokines/chemokines that modulate tumor growth and invasiveness [
17].
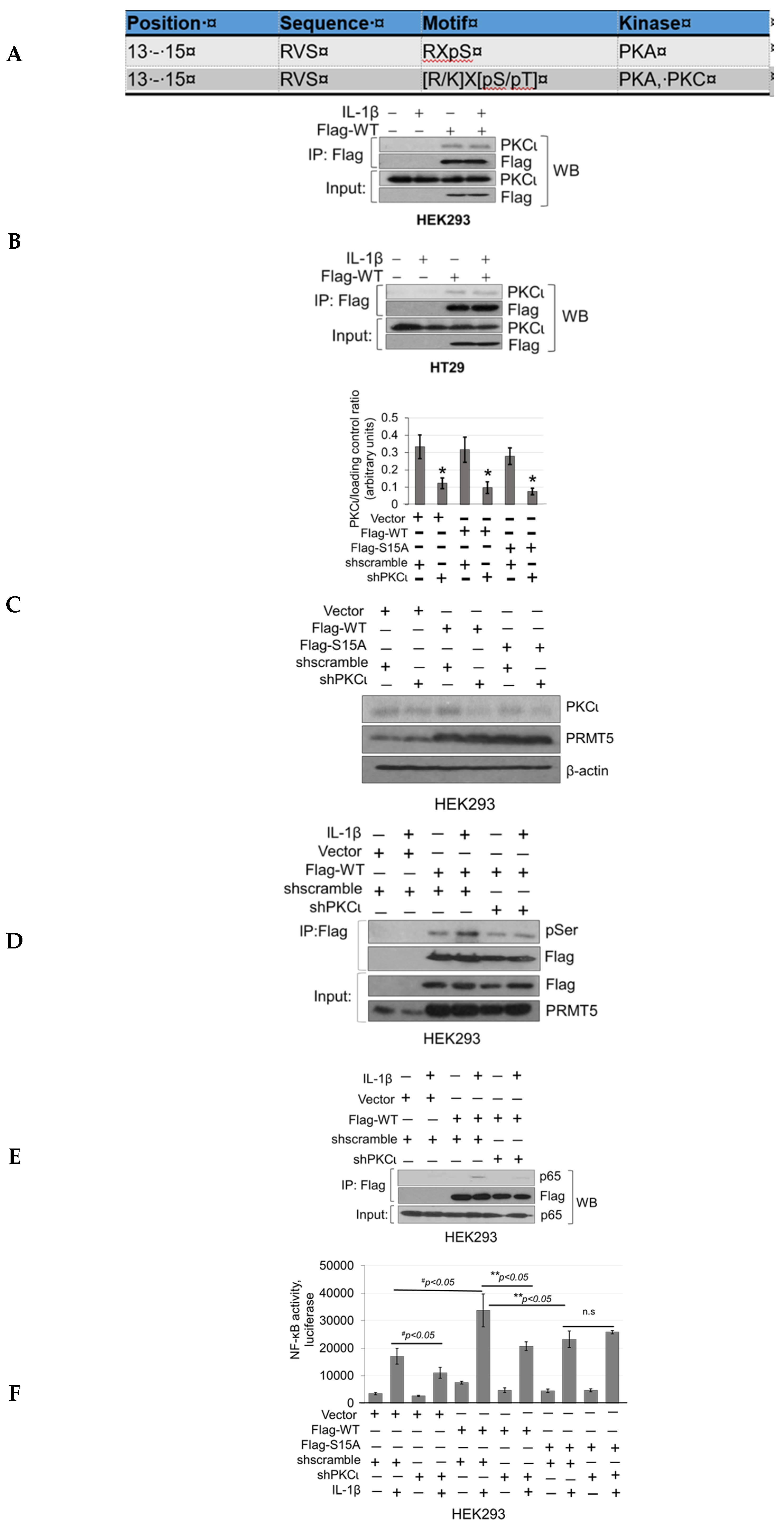
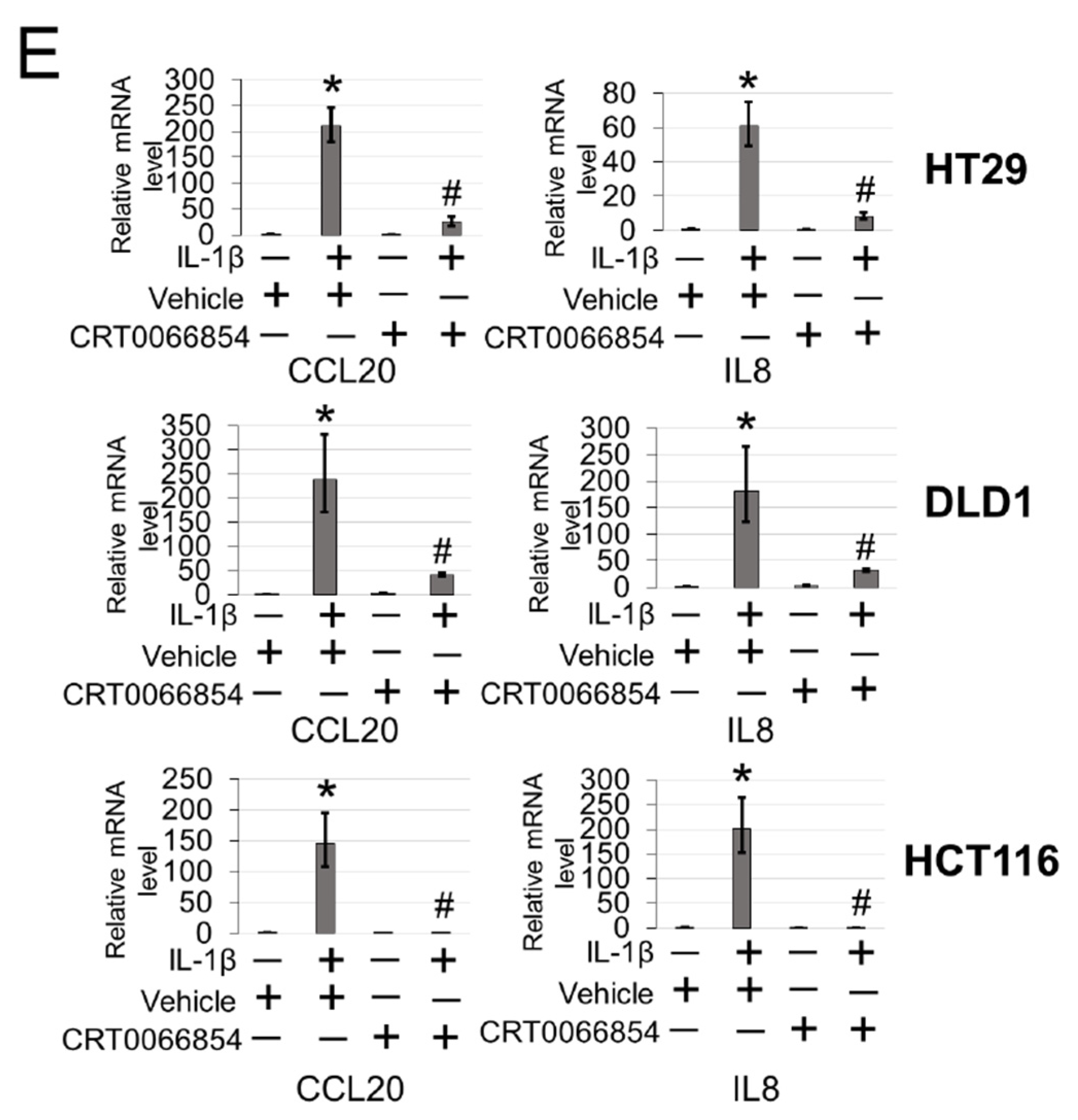
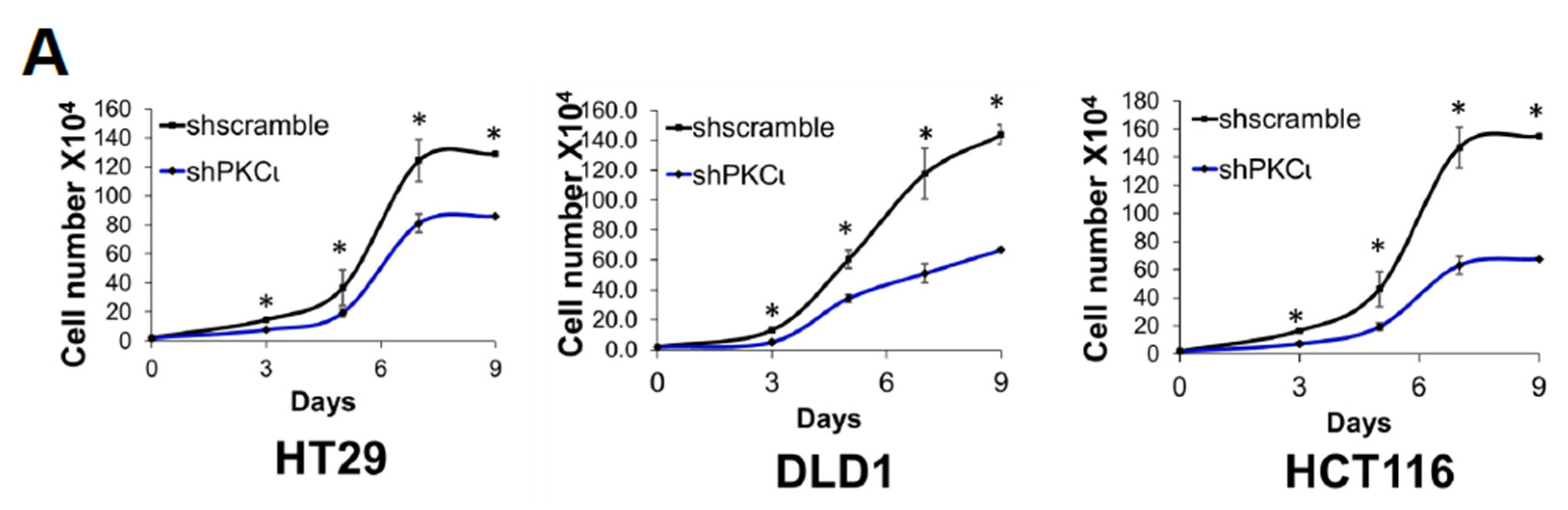
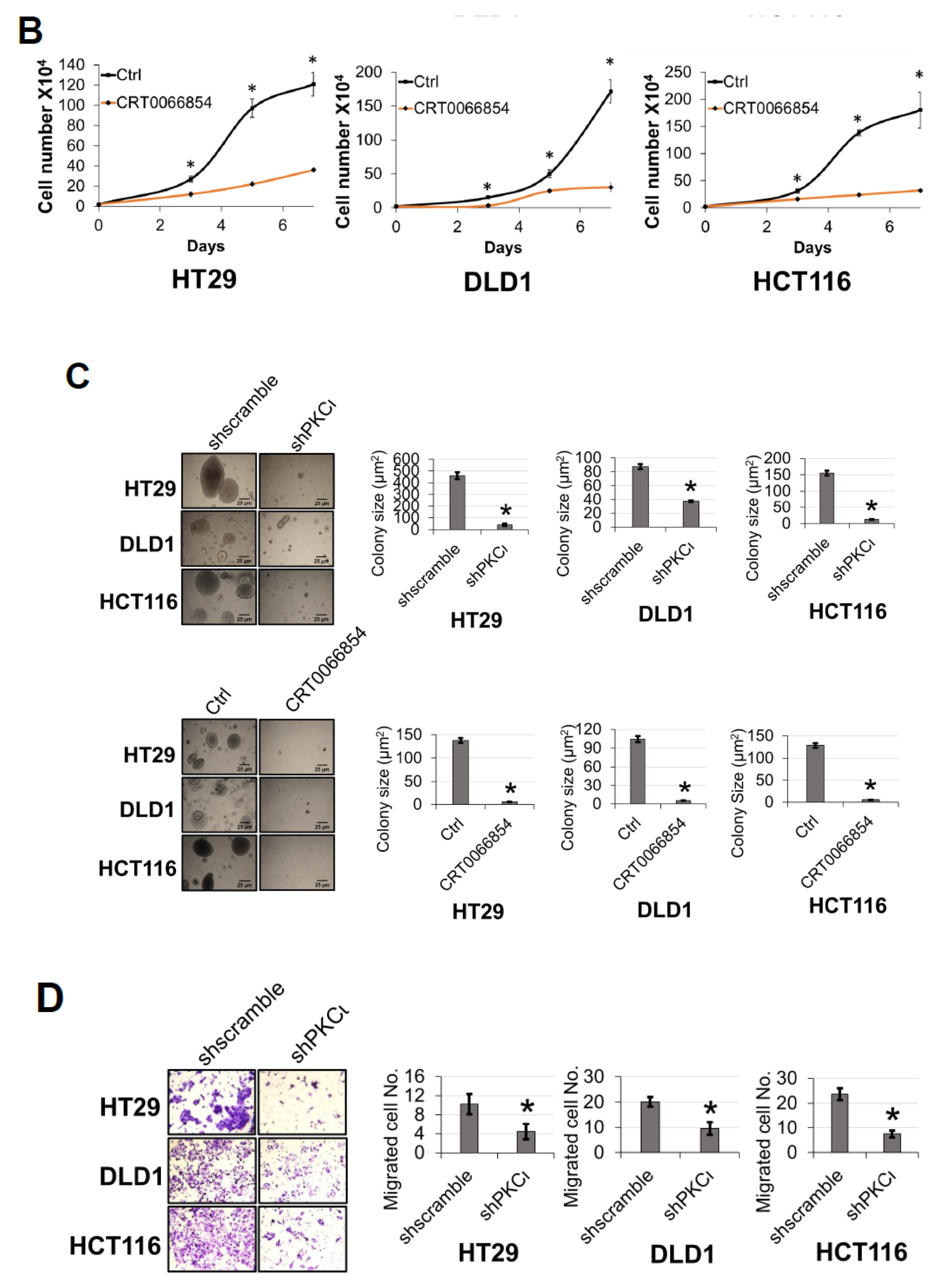
Furthermore, the discovery of PKCι’s link to phosphorylation of PRMT5 on S15 and NF-κB activation is significant. PKCι is an oncogene that has been implicated in the initiation and progression of CRC via regulation of epithelial cell integrity and polarity in a model of colitis-associated colon cancer [
18]. Moreover, mutations and genetic alterations of PKCι are often detected across various cancer types, making it a highly attractive therapeutic target [
19,
20]. Interestingly, we showed that PKCι is also upregulated in CRC patient tissues (
Figure 6H) and its selective inhibition attenuated PRMT5-mediated activation of NF-κB, target gene expression, as well as CRC cell growth and migration. It is worth noting that luciferase and functional assays carried out using the CRT0066854 PKCι inhibitor (
Figure 7 and
Figure 8) sometimes showed a stronger effect compared to shPKCι counterparts. This effect may be due in part to the fact that CRT0066854 inhibits both PKCι and PKCζ isoforms. Moreover, the concentrations (low or high) of CRT0066854 we used may have played a role in the effect that it generated. Additionally, we may appreciate that shRNA knockdown approaches rarely generate a 100% knockdown effect. Taken together, all these potential factors may contribute to the slight differences between using these two different experimental approaches. Although we utilized both pharmacologic and shRNA knockdown approaches, we acknowledge that each approach possesses independent value toward approaching the same biological question but results may not necessarily be perfectly aligned albeit a similar appreciable trend is clear. Overall, in the future, it would be very interesting to determine the anti-tumor efficacy of inhibiting PKCι using relevant CRC mouse models and determine whether these applications could potentially be generalized to other tumor types.
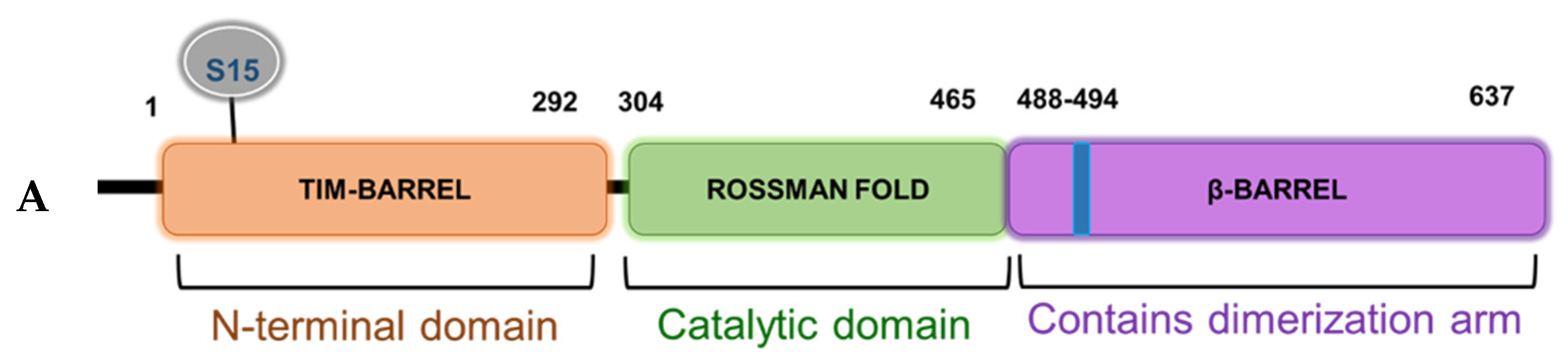
From a structural perspective, PRMT5 is commonly found complexed with other proteins and these interactions have been shown to dictate both its catalytic activity and substrate specificity [
21,
22]. Interestingly, the N-terminal domain (residues 13–292) of PRMT5 adopts a TIM barrel structure, which has been implicated as essential for co-factor binding (e.g., MEP50) as well as acting as a scaffold for the binding of adaptor proteins (e.g., pICLn, Riok1, etc.) [
21,
22,
23]. However, the exact mechanisms underlying structural changes in the PRMT5 protein that lead to altered activity, cofactor binding, and/or substrate selectivity are still largely unknown. In keeping with these observations, the location of the highly conserved S15 residue within the TIM Barrel region as well as its role in mediating a phospho-dependent interaction axis between PRMT5 and its substrate p65 support a possible regulatory role for this segment. Future molecular dynamics simulation as well as co-crystallization studies may provide critical structural insights into S15 phosphorylation-induced conformational changes of PRMT5. These may serve as a basis for understanding mechanisms that dictate PRMT5′s activity and discrete binding to p65 and other binding partners.
In conclusion, both NF-κB and PRMT5 are individually known to play critical roles in cancer progression, and the mechanisms underlying the cooperativity between the two are inherently critical and complicated. Based on our results, we presented a hypothetical model in which IL-1β stimulation activates the NF-κB pathway and induces PKCι-mediated phosphorylation of PRMT5. Insights from this study are, therefore, of particular importance in terms of enhancing our understanding of the role of phosphorylation sites such as S15 on PRMT5 in modulating the interaction between PRMT5 and NF-κB. In the future, these findings may also open up novel therapeutic avenues for CRC patients based on disrupting the PKCι/PRMT5/NF-κB signaling axis.
4. Experimental Procedures
4.1. Liquid Chromatography-Tandem Mass Spectrometric Analysis
Coomassie-stained SDS-PAGE gel band containing Flag-PRMT5 protein was subjected to in-gel tryptic digestion. Flag-PRMT5 gel pieces were subjected to destaining and reduction of cysteine residues using 50% acetonitrile in 100 mM ammonium bicarbonate and 100% acetonitrile followed by treatment, followed by 20 mM DTT at room temperature for 60 min. Alkylation with 55 mM iodoacetamide for 30 min was performed in the dark. The solution was removed, and the gel pieces were washed with 100 mM ammonium bicarbonate and dehydrated in acetonitrile. Gel pieces were then dried in a SpeedVac centrifuge, and proteolytically digested by rehydration overnight at 37 °C in 50 mM ammonium bicarbonate containing sequencing grade modified trypsin (Promega, Madison, WI, USA). Extracted peptides were treated with 50% acetonitrile in 5% formic acid, dried, and reconstituted in 0.1% formic acid for mass spectrometry analysis. Analysis of proteolytic digests was performed by using an LTQ Orbitrap XL linear ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), coupled with an Ultimate 3000 HPLC system (Dionex, Sunnyvale, CA, USA). The digests were injected onto a reverse-phase C18 column (0.075 × 150 mm, Dionex) equilibrated with 0.1% formic acid/4% acetonitrile (v/v). A linear gradient of acetonitrile from 4% to 40% in water in the presence of 0.1% formic acid over a period of 45 min was used at a flow rate of 300 nL/min. The spectra were acquired by data dependent methods, consisting of a full scan (m/z 400–2000) and then tandems on the five most abundant precursor ions. The previously selected precursor ions were scanned once during 30 s and then were excluded for 30 s. The obtained data were analyzed by Mascot software (Matrix Science, Chicago, IL, USA) against customized PRMT5 protein database with the setting of 10 ppm for precursor ions and 0.8 Da for product ions. Carbamidomethylation of cysteine was set as fixed modification, while oxidation of methionine, phosphorylation of serine, threonine, and tyrosine were set as variable modifications. The tandem mass spectra of candidate-modified peptides were further interpreted manually.
4.2. Cell Lines and Transfections
The HEK293C6 (HEK293) cell line was previously described [
24]. CRC cell lines HT29, DLD1, and HCT116 were procured from ATCC (Manassas, VA, USA) and were cultivated in RPMI (Roswell Park Memorial Institute, Buffalo, NY, USA) 1640 medium supplemented with 100 units/mL penicillin, 100 g/mL streptomycin, and 5% fetal bovine serum (FBS). The FLAG-S15A and FLAG-E444D mutants of PRMT5 were generated using the QuikChange II XL Site-Directed Mutagenesis Kit from Agilent Technologies. Primers were designed using the Agilent Technologies QuikChange Primer Design online software. To generate stable cell lines using lentiviruses, various PRMT5 constructs including FLAG-WT-PRMT5 were cloned into the lentiviral backbone pLVX-IRES-puro vector and transfected into HEK293T packaging cell line along with relevant lentivirus packaging and helper plasmids using Lipofectamine and PLUS reagents (Life Technologies/Invitrogen, Carlsbad, CA, USA). Similarly, the pool of five shRNA constructs targeting PKCι (Sigma-Aldrich, St. Louis, MO, USA) was used to transfect HEK293T cells for lentivirus generation. HEK293C6 and CRC cell lines were subsequently infected with relevant lentiviruses as previously described [
2,
6]. All stable cell lines were selected with 1 μg/mL of puromycin.
4.3. Western Blotting and Antibodies
Cells were cultured to about 90–95% confluence before treatment with 10 ng/mL of IL-1β. Whole cell samples were collected and lysed using Radio Immunoprecipitation Assay buffer (RIPA buffer: 150 mM NaCl, 0.1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl pH 8.0, and protease inhibitors). Whole cell lysates were then separated by SDS/PAGE gels, and further assessed by Western blotting. Different antibodies were used to detect the target proteins of interest, obtained from the following commercial sources: anti-PRMT5 (Abcam, Cambridge, UK; ab109451), anti-FLAG (Sigma-Aldrich, St. Louis, MO, USA; F1804), and anti-p65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; sc-109); anti-PKCι (Proteintech, Chicago, IL, USA; 66493-1-Ig). For cell fractionation experiments, cytoplasmic and nuclear fractions were subject to SDS-PAGE and probed with anti-LaminB1 (Proteintech, Chicago, IL, USA; 12987-1-AP); anti-α-tubulin (Cell Signal, Danvers, MA, USA; 2144S).
4.4. Luciferase Assays
NF-κB luciferase assays were conducted by transiently transfecting the κB-luciferase construct p5XIP10 κB into parental control or FLAG-PRMT5 stable cell lines after which luciferase activity was quantified 48 h later using the Luciferase Assay System with Reporter Lysis Buffer kit (Promega, Fitchburg, WI, USA; E4030). The κB-luciferase plasmid p5XIP10 κB contains five tandem copies of the NF-κB DNA binding site derived from the IP10 gene (an established target gene of NF-κB) upstream of a luciferase reporter gene. Luciferase activity was measured using a Synergy H1 Multi-Mode Reader (BioTek Instruments Inc., Winooski, VT, USA).
4.5. Co-Immunoprecipitations
Cells stably expressing FLAG-PRMT5 proteins were cultured to ≈95% confluency then lysed in co-immunoprecipitation buffer (1% Triton X-100 (
v/
v), 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM sodium orthovanadate, 20 µM aprotinin, 1 mM phenylmethanesulfonyl fluoride, and 1 mM pepstatin A). FLAG-PRMT5 proteins were then immunoprecipitated with anti-FLAG-M2 beads (Sigma-Aldrich, St. Louis, MO), using immunoprecipitation methods previously described [
2,
6]. Briefly, cell lysates with equivalent amounts of protein were incubated with anti-FLAG-M2 beads at 4 °C overnight. Beads were then washed, and FLAG-tagged proteins were eluted and separated by SDS/PAGE [
2,
6].
4.6. Cell Proliferation and Anchorage-Independent Growth Assays
For cell proliferation assays, CRC cells overexpressing FLAG-PRMT5 constructs were seeded in triplicate at 2 × 10
4 cells/well in a 6-well plate. Cells were counted at days 3, 5, 7, and 9 post-seeding using a cell counting chamber. For anchorage-independent growth assays, type VII agarose (Sigma-Aldrich, St. Louis, MO) was used to prepare 2.4% and 1.2% bottom and top agar layers, respectively. Then, 2 × 10
5 cells were resuspended in the top layer and plated onto the bottom layer. Cells were then cultured for 12–14 days at 37 °C and 5% CO
2. Images of colonies were captured using a Canon EOS Rebel T3i Digital SLR camera and colony size and number were quantified using the ImageJ v.1.52a software (Madison, WI, USA;
http://imagej.nih.gov/ij/).
4.7. Migration Assay
Migration assays were conducted using Boyden chambers. Briefly, a Boyden chamber consists of 8 μm pore size cell culture inserts in a 24-well plate. Each insert was coated with gelatin on the side facing the lower chamber. A total of 2 × 105 cells were seeded in the top of the insert (upper chamber) in serum-free media while serum-rich media (10% serum) was supplied in the well below as a chemoattractant. After 48 h, migrated cells were fixed with 4% formaldehyde and stained with crystal violet. Stained cells were visualized with a light microscope and quantified. Images were captured using a Canon EOS Rebel T3i Digital SLR camera.
4.8. Illumina Microarrays and Quantitative PCR (qPCR)
Microarray and qPCR experiments were carried out as previously described [
6]. Briefly, parental or 293 cells with WT-PRMT5 or S15A overexpression were cultured to ≈90% confluence and total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Total isolated RNA was used to prepare cDNA using the SuperScript III First-Strand Synthesis PCR System (Invitrogen, Carlsbad, CA). cDNA was labeled with biotin-UTP using the Illumina Total Prep RNA amplification kit (Ambion/Applied Biosystems, Foster City, CA, USA), hybridized to Illumina Human Ref-v3 v1 Expression Bead Chips and then scanned in a Bead Array reader using standard Illumina protocols (Illumina, San Diego, CA, USA). Illumina’s Bead Studio software was used for data analysis. qPCR was carried out using FastStart Universal SYBR Green Master ROX (Roche, Basel, Switzerland). Primers were designed by the Primer Express 3.0 software. Primer information is listed below: TNF-Forward: 5′-TGGCCCAGGCAGTCAGA-3′; TNF-Reverse: 5′-GGTTTGCTACAACATGGGCTACA-3′; IL8-Forward: 5′-TCCTGATTTCTGCAGCTCTGT-3′; IL8-Reverse: 5′-AAATTTGGGGTGGAAAGGTT-3′; CCL20-Forward: 5′-GTGCTGCTACTCCACCTCTG-3′; CCL20-Reverse: 5′-CGTGTGAAGCCCACAATAAA-3′.
4.9. Chromatin Immunoprecipitation (ChIP)
Cells were left untreated or stimulated with 10 ng/mL of IL-1β for 1 and 4 h, and cross-linked with 1% formaldehyde for 10 min at room temperature (R.T.). The cross-linking was stopped by adding glycine and cells were then washed with cold× phosphate-buffered saline (1× PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4), scraped, and pelleted by centrifugation at 2000 rpm. Cells were lysed in Farnham Lysis buffer (mM PIPES pH 8.0/85 mM KCl/0.5% Tween 20) supplemented with protease inhibitors followed by chromatin shearing to yield fragments of 200–1000 bp using a sonifier (Fisher Scientific, Hampton, NH, USA) equipped with a microtip (40 s on/50 s off, 4 min at 40% power output). Sonicated lysates were centrifuged, and the resulting supernatant was diluted 5-fold with ChIP dilution buffer containing 16.7 mM Tris HCl pH 8.1, 167 mM NaCl, 0.01% SDS, 1.1% (v/v) Triton X-100, and 1.2 mM EDTA. Diluted lysates were precleared for 1 h with protein A/G agarose. Immunoprecipitations were performed using ChIP-grade anti-RelA (Abcam, ab7970, Cambridge, United Kingdom) antibody at 4 °C overnight. Immune complexes were collected with protein A/G agarose, washed with low-salt wash buffer [20 mM Tris HCl pH 8.1, 150 mM NaCl, 0.1% SDS, 1% (v/v) Triton X-100, and 2 mM EDTA], high-salt wash buffer [20 mM Tris HCl pH 8.1, 500 mM NaCl, 0.1% SDS, 1% (v/v) Triton X-100, and 2 mM EDTA], LiCl wash buffer [10 mM Tris HCl pH 8.1, 250 mM LiCl, 1% (w/v) sodium deoxycholate, 1% (v/v) IGEPAL-CA630, and 1 mM EDTA], and TE buffer (10 mM Tris HCl pH 8.0 and 1 mM EDTA). Protein-DNA complexes were eluted from antibodies with elution buffer containing 1% SDS and 0.1 M NaHCO3, incubated in the presence of 192 mM NaCl for 4 h at 65 °C, and digested with proteinase K for 1 h at 45 °C. DNA was recovered using the Qiagen quick DNA purification kit and IL8 gene-specific ChIP primers (Qiagen, Hilden, Germany) were used in the PCR analyses.
4.10. AlphaLISA Assay
FLAG-WT-PRMT5 or S15A mutant enzyme was purified from 293 cells using anti-FLAG-M2 beads (Sigma-Aldrich, St. Louis, MO) as described in co-immunoprecipitation methods above. The enzyme prep was diluted in assay buffer (30 mM Tris, pH 8.0, 1mM DTT, 0.01% BSA, 0.01% Tween-20) prior to use. SAM (New England Biolabs, Ipswich, MA, USA) and unmethylated histone H4R3 (Anaspec, Fremont, CA, USA) were used as the methyl group donor and PRMT5 enzyme substrate, respectively. Acceptor beads diluted in 1× Epigenetics buffer (PerkinElmer, Waltham, MA, USA) were added at a final concentration of 20 μg/mL to the reaction mixture and the plate was incubated at R.T. for 1 h. Donor beads diluted in 1× Epigenetics buffer (PerkinElmer, Waltham, MA) were then added at a final concentration of 20 μg/mL and the plate was further incubated at R.T. for 30 min. All reactions were performed in triplicate and repeated three independent times. The plates were read using an EnVision
® Reader [
2].
4.11. Ingenuity Pathway Analysis (IPA)
Groups of genes regulated by S15A were analyzed by the IPA software 8.7 (Qiagen, Hilden, Germany). The setting and filter were as follows: reference set: Ingenuity Knowledge Base (Genes _ Endogenous Chemicals); Relationship to include: Direct and Indirect; Includes Endogenous Chemicals; Filter Summary: Consider only molecules where species _ Human OR Rat OR Mouse. The p values for the enrichment test were calculated using Fisher’s exact test, right-tailed. Log10 (p) was visualized to the left of the p value. p < 0.05 was considered to be statistically significant.
4.12. Cell Fractionation Assay
Fractionation experiments were conducted according to the manufacturer’s instructions for the nuclear extract kit (Active Motif, Carlsbad, CA, USA). Briefly, cells were grown to about 80% confluence, washed with ice-cold 1× PBS/Phosphatase inhibitors, collected and pelleted for 5 min at 500 rpm. Cell pellets were gently resuspended in 1× hypotonic buffer and incubated for 15 min on ice. The cytoplasmic fraction was collected as the lysate following centrifuging for 30 s at 14,000× g. The remaining cell pellet was further lysed in Complete Lysis Buffer, incubated on ice for 30 min and centrifuged for 10 min at 14,000× g to collect the nuclear fraction. The cytoplasmic and nuclear proteins were then subjected to Western blot analyses.
4.13. Statistical Analysis
Statistical analyses were performed using Prism 6 software (GraphPad, San Diego, CA, USA). Data represent the mean ±S.D. or ±SEM as indicated. A two-tailed Student’s t-test was used when comparing two means between groups as specified. All statistics were carried out for triplicate experiments and a p < 0.05 was considered statistically significant.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















