ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
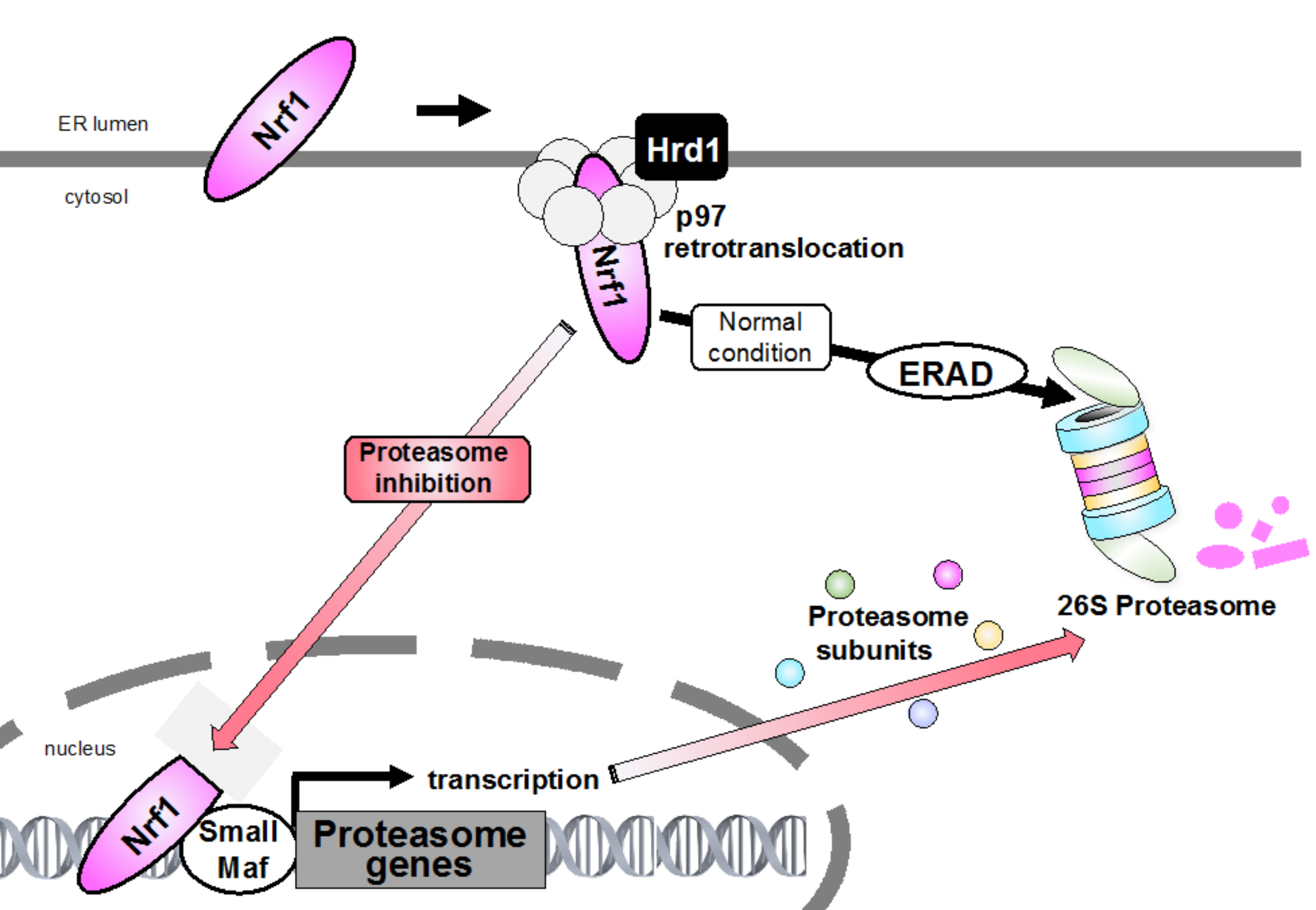
2. ERAD and Proteasome
3. Transcriptional Regulation of Proteasome Genes in Metazoan
3.1. Rpn4 (Yeast)
3.2. SKN-1 (C. elegans)
3.3. CncC (D. melanogaster)
3.4. Nrf Family Members (Mammals)
3.5. Other Transcription Factors that Regulate Proteasome Subunit Gene Expression
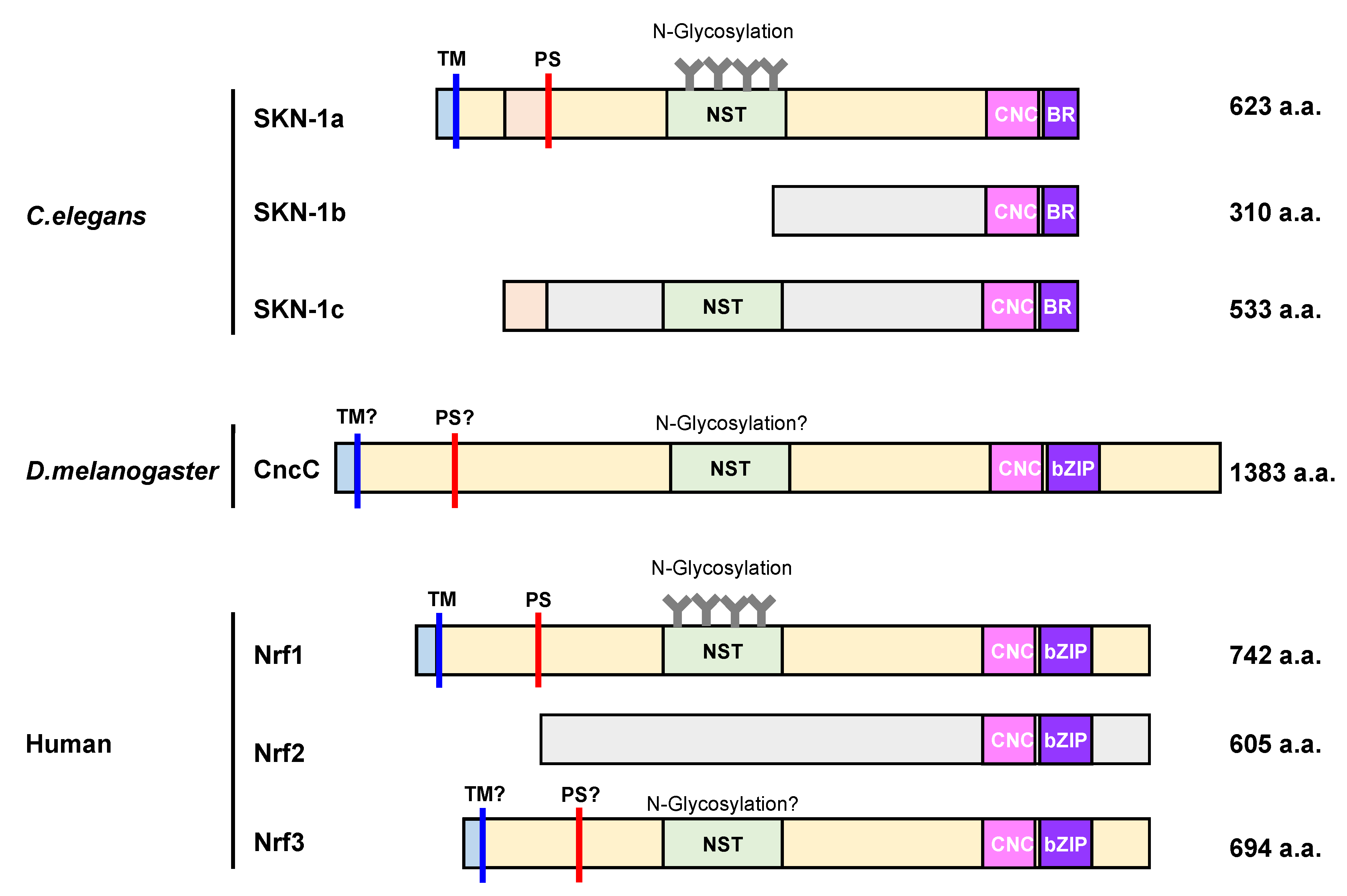
4. Molecular Characteristics of SKN-1, CncC, and Nrf1 Proteins
4.1. Control of Nrf1/SKN-1 Abundance
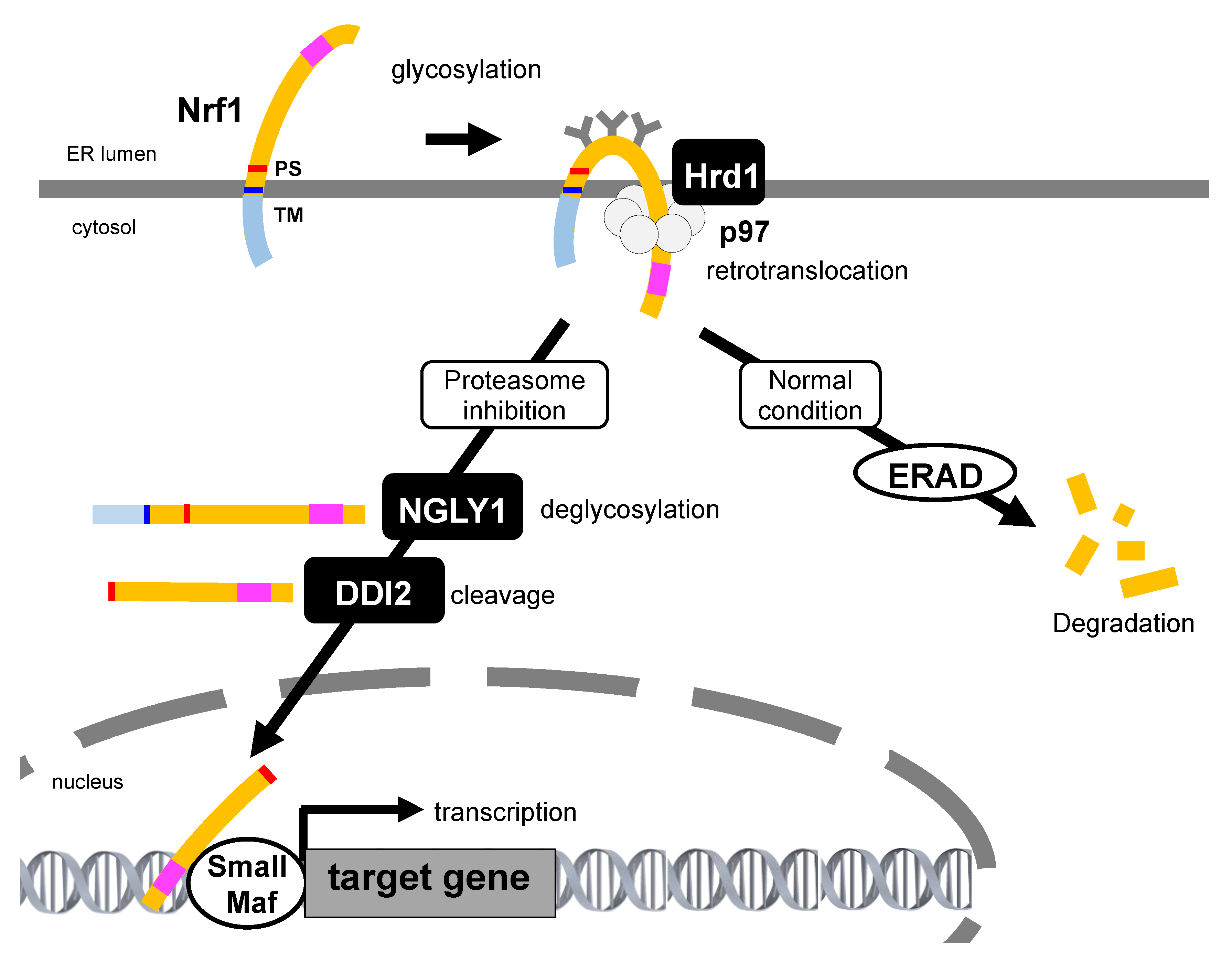
4.2. Retrotranslocation from the ER and Proteolytic Processing Are Required for Nrf1 Activation
4.3. Post-Translational Modification Regulates Nrf1
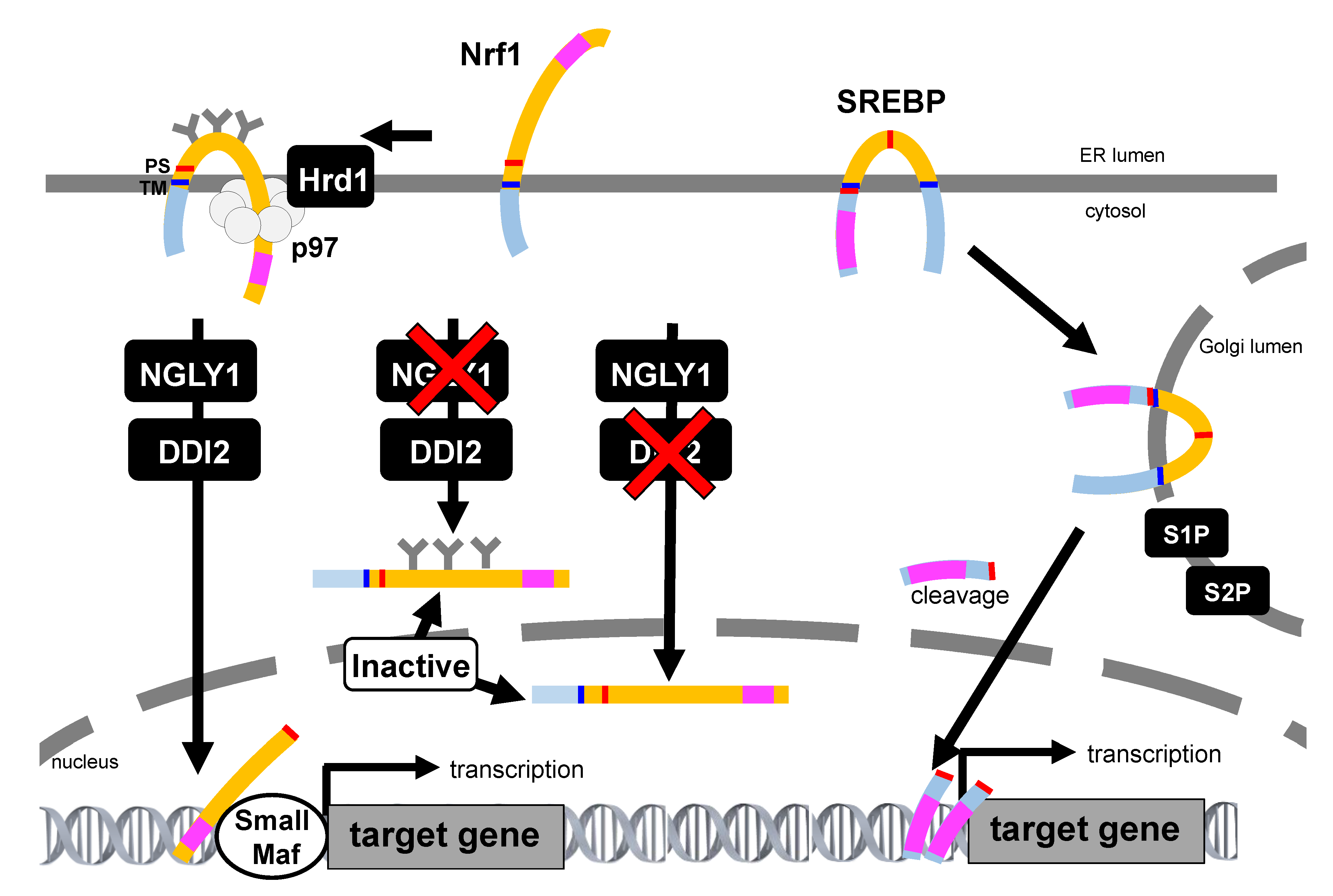
4.3.1. Glycosylation
4.3.2. Phosphorylation
5. Diverse Function and Regulation of Nrf1
6. mTORC1-SREBP Pathway Regulate Nrf1 Activity
7. Relationships of Other Nrf Family Transcription Factors
7.1. Nrf2
7.2. Nrf3
8. Nrf1 Regulation Machinery as a Potential Therapeutic Target
9. Concluding Remarks and Perspectives
Conflicts of Interest
References
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Young, V.R.; Steffee, W.P.; Pencharz, P.B.; Winterer, J.C.; Scrimshaw, N.S. Total human body protein synthesis in relation to protein requirements at various ages. Nature 1975. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E.; Goldberg, A.L. Mechanisms of muscle wasting—The role of the Ubiquitin–Proteasome pathway. N. Engl. J. Med. 1996, 335, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Aaron, C. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Baumeister, W.; Walz, J.; Zühl, F.; Seemüller, E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 1998, 92, 367–380. [Google Scholar] [CrossRef]
- Varshavsky, A. Regulated protein degradation. Trends Biochem. Sci. 2005, 30, 283–286. [Google Scholar] [CrossRef]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim. Biophys. Acta Proteins Proteomics 2012, 1824, 3–13. [Google Scholar] [CrossRef]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef]
- Berner, N.; Reutter, K.-R.; Wolf, D.H. Protein quality control of the endoplasmic reticulum and ubiquitin–proteasome-triggered degradation of aberrant proteins: Yeast pioneers the path. Annu. Rev. Biochem. 2018, 87, 751–782. [Google Scholar] [CrossRef]
- Ariyasu, D.; Yoshida, H.; Hasegawa, Y. Endoplasmic reticulum (Er) stress and endocrine disorders. Int. J. Mol. Sci. 2017, 18, 382. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Kumatori, A.; Tanaka, K.; Inamura, N.; Sone, S.; Ogura, T.; Matsumoto, T.; Tachikawa, T.; Shin, S.; Ichihara, A. Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7071–7075. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Madura, K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005, 65, 5599–5606. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD—Dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Yin, Y.; Gao, G.; Li, S.; Jiang, Y.; Gu, X.; Luo, J. SPD—A web-based secreted protein database. Nucleic Acids Res. 2005, 33, D169–D173. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.; Kim, D.; Jung, K.; Kang, S.; Lee, Y.H. Fungal secretome database: Integrated platform for annotation of fungal secretomes. BMC Genom. 2010, 11, 1–15. [Google Scholar] [CrossRef]
- Mehrtash, A.B.; Hochstrasser, M. Ubiquitin—Dependent protein degradation at the endoplasmic reticulum and nuclear envelope. Semin. Cell Dev. Biol. 2019, 93, 111–124. [Google Scholar] [CrossRef]
- Qi, L.; Tsai, B.; Arvan, P. New insights into the physiological role of endoplasmic reticulum—Associated degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 2571–25718. [Google Scholar]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Somlo, D.; Kim, G.H.; Prescianotto-Baschong, C.; Sun, S.; Beuret, N.; Long, Q.; Rutishauser, J.; Arvan, P.; Spiess, M.; et al. ER–Associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Investig. 2017, 127, 3897–3912. [Google Scholar] [CrossRef]
- Sun, S.; Shi, G.; Han, X.; Francisco, A.B.; Ji, Y.; Mendonça, N.; Liu, X.; Locasale, J.W.; Simpson, K.W.; Duhamel, G.E.; et al. Sel1L is indispensable for mammalian endoplasmic reticulum—Associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 2014, 111, E582–E591. [Google Scholar] [CrossRef]
- Sun, S.; Lourie, R.; Cohen, S.B.; Ji, Y.; Goodrich, J.K.; Poole, A.C.; Ley, R.E.; Denkers, E.Y.; Mcguckin, M.A.; Long, Q.; et al. Epithelial sel1L is required for the maintenance of intestinal homeostasis. Mol. Biol. Cell 2016, 27, 483–490. [Google Scholar] [CrossRef]
- Sha, H.; Sun, S.; Francisco, A.B.; Ehrhardt, N.; Xue, Z.; Liu, L.; Lawrence, P.; Mattijssen, F.; Guber, R.D.; Panhwar, M.S.; et al. The ER-associated degradation adaptor protein sel1l regulates LPL secretion and lipid metabolism. Cell Metab. 2014, 20, 458–470. [Google Scholar] [CrossRef]
- Wangeline, M.A.; Vashistha, N.; Hampton, R.Y. Proteostatic tactics in the strategy of sterol regulation. Annu. Rev. Cell Dev. Biol. 2017, 33, 467–489. [Google Scholar] [CrossRef]
- Wojcikiewicz, R.J.H.; Pearce, M.M.P.; Sliter, D.A.; Wang, Y. When worlds collide: IP3 receptors and the ERAD pathway. Cell Calcium 2009, 46, 147–153. [Google Scholar] [CrossRef]
- Enenkel, C.; Lehmann, A.; Kloetzel, P.M. Subcellular distribution of proteasomes implicates a major location of protein degradation in the nuclear envelope-ER network in yeast. EMBO J. 1998, 7, 6144–6154. [Google Scholar] [CrossRef]
- Palmer, A.; Rivett, A.J.; Thomson, S.; Hendil, K.B.; Butcher, G.W.; Fuertes, G.; Knecht, E. Subpopulations of proteasomes in rat liver nuclei, microsomes and cytosol. Biochem. J. 1996, 316, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Wietrzynski, W.; Lee, C.W.; Schaffer, M.; Beck, F.; Schuller, J.M.; Salomé, P.A.; Plitzko, J.M.; Baumeister, W.; Engel, B.D. Direct visualization of degradation microcompartments at the ER membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Chen, X.; Walters, K.J. Gates, channels, and switches: Elements of the proteasome machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Śledź, P.; Baumeister, W. Structure—Driven developments of 26S proteasome inhibitors. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Yu, H.; Mim, C.; Matouschek, A. regulated protein turnover: Snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 2014, 15, 122–133. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Trempe, J.-F. Reading the ubiquitin postal code. Curr. Opin. Struct. Biol. 2011, 21, 792–801. [Google Scholar] [CrossRef]
- Sahara, K.; Kogleck, L.; Yashiroda, H.; Murata, S. The mechanism for molecular assembly of the proteasome. Adv. Biol. Regul. 2014, 54, 51–58. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, N.; Bustamante, H.; Soza, A.; Burgos, P. Cellular responses to proteasome inhibition: Molecular mechanisms and beyond. Int. J. Mol. Sci. 2019, 20, 3379. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, U.; Robison, B.; Dange, T.; Kahlert, G.; Delaney, J.R.; Kotireddy, S.; Tsuchiya, M.; Tsuchiyama, S.; Murakami, C.J.; Schleit, J.; et al. Elevated proteasome capacity extends replicative lifespan in saccharomyces cerevisiae. PLoS Genet. 2011, 7, e1002253. [Google Scholar] [CrossRef] [PubMed]
- Meiners, S.; Heyken, D.; Weller, A.; Ludwig, A.; Stangl, K.; Kloetzel, P.M.; Krüger, E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J. Biol. Chem. 2003, 278, 21517–21525. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. eLife 2014, 3, e01856. [Google Scholar] [CrossRef]
- Tonoki, A.; Kuranaga, E.; Tomioka, T.; Hamazaki, J.; Murata, S.; Tanaka, K.; Miura, M. Genetic evidence linking age—Dependent attenuation of the 26S proteasome with the aging process. Mol. Cell. Biol. 2009, 29, 1095–1106. [Google Scholar] [CrossRef]
- Vilchez, D.; Morantte, I.; Liu, Z.; Douglas, P.M.; Merkwirth, C.; Rodrigues, A.P.C.; Manning, G.; Dillin, A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 2012, 489, 263–268. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Tzavelas, C.; Pemberton, A.J.; Nezis, I.P.; Rivett, A.J.; Gonos, E.S. Overexpression of proteasome β5 subunit increases the amount of assembled proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem. 2005, 280, 11840–11850. [Google Scholar] [CrossRef]
- Munkácsy, E.; Chocron, E.S.; Quintanilla, L.; Gendron, C.M.; Pletcher, S.D.; Pickering, A.M. Neuronal-specific proteasome augmentation via Prosβ5 overexpression extends lifespan and reduces age-related cognitive decline. Aging Cell 2019, 18, e13005. [Google Scholar] [CrossRef]
- Howell, L.A.; Peterson, A.K.; Tomko, R.J. Proteasome subunit α1 overexpression preferentially drives canonical proteasome biogenesis and enhances stress tolerance in yeast. Sci. Rep. 2019, 9, 12418. [Google Scholar] [CrossRef]
- Nguyen, N.N.; Rana, A.; Goldman, C.; Moore, R.; Tai, J.; Hong, Y.; Shen, J.; Walker, D.W.; Hur, J.H. Proteasome β5 subunit overexpression improves proteostasis during aging and extends lifespan in Drosophila melanogaster. Sci. Rep. 2019, 9, 3170. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Dynamic regulation of the 26S proteasome: From synthesis to degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Shirozu, R.; Yashiroda, H.; Murata, S. Identification of minimum Rpn4-responsive elements in genes related to proteasome functions. FEBS Lett. 2015, 589, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Varshavsky, A. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: A negative feedback circuit. Proc. Natl. Acad. Sci. USA 2001, 98, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, H.; Ju, D.; Xie, Y. Disruption of Rpn4-induced proteasome expression in Saccharomyces cerevisiae reduces cell viability under stressed conditions. Genetics 2008, 180, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liu, Z.L. Comparative transcriptome profiling analyses during the lag phase uncover YAP1, PDR1, PDR3, RPN4, and HSF1 as key regulatory genes in genomic adaptation to the lignocellulose derived inhibitor HMF for Saccharomyces cerevisiae. BMC Genomics 2010, 1, 660. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88, 290–301. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Ruvkun, G. Endoplasmic reticulum-associated SKN-1A/Nrf1 mediates a cytoplasmic unfolded protein response and promotes longevity. eLife 2019, 8, e44425. [Google Scholar] [CrossRef]
- Qu, M.; Li, Y.; Wu, Q.; Xia, Y.; Wang, D. Neuronal ERK signaling in response to graphene oxide in nematode Caenorhabditis elegans. Nanotoxicology 2017, 11, 520–533. [Google Scholar] [CrossRef]
- An, J.H.; Blackwell, T.K. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003, 17, 1882–1893. [Google Scholar] [CrossRef]
- Grimberg, K.B.; Beskow, A.; Lundin, D.; Davis, M.M.; Young, P. Basic Leucine Zipper Protein Cnc-C Is a Substrate and Transcriptional Regulator of the Drosophila 26S Proteasome. Mol. Cell. Biol. 2011, 31, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Pitoniak, A.; Bohmann, D. Mechanisms and functions of Nrf2 signaling in Drosophila. Free Radic. Biol. Med. 2015, 88, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiang, Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem. J. 2016, 473, 961–1000. [Google Scholar] [CrossRef] [PubMed]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 system: An evolutionary journey through stressful space and time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef]
- Waku, T.; Nakamura, N.; Koji, M.; Watanabe, H.; Katoh, H.; Tatsumi, C.; Tamura, N.; Hatanaka, A.; Hirose, S.; Katayama, H.; et al. NRF3-POMP-20S proteasome assembly axis promotes cancer development via ubiquitin-independent proteolysis of p53 and Rb. Mol. Cell. Biol. 2020, 33, 3461–3472. [Google Scholar] [CrossRef]
- Wang, M.; Qiu, L.; Ru, X.; Song, Y.; Zhang, Y. Distinct isoforms of Nrf1 diversely regulate different subsets of its cognate target genes. Sci. Rep. 2019, 9, 2960. [Google Scholar] [CrossRef]
- Xu, H.; Fu, J.; Ha, S.W.; Ju, D.; Zheng, J.; Li, L.; Xie, Y. The CCAAT box-binding transcription factor NF-Y regulates basal expression of human proteasome genes. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 818–825. [Google Scholar] [CrossRef][Green Version]
- Vilchez, D.; Boyer, L.; Morantte, I.; Lutz, M.; Merkwirth, C.; Joyce, D.; Spencer, B.; Page, L.; Masliah, E.; Berggren, W.T.; et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 2012, 489, 304–308. [Google Scholar] [CrossRef]
- Vangala, J.R.; Dudem, S.; Jain, N.; Kalivendi, S.V. Regulation of psmb5 protein and β subunits of mammalian proteasome by constitutively activated signal transducer and activator of transcription 3 (stat3): Potential role in bortezomib-mediated anticancer therapy. J. Biol. Chem. 2014, 289, 12612–12622. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Wang, M.; Xiang, Y.; Qiu, L.; Hu, S.; Zhang, Z.; Mattjus, P.; Zhu, X.; Zhang, Y. Nach is a novel subgroup at an early evolutionary stage of the CNC-bZIP subfamily transcription factors from the marine bacteria to humans. Int. J. Mol. Sci. 2018, 19, 2927. [Google Scholar] [CrossRef]
- Bugno, M.; Daniel, M.; Chepelev, N.L.; Willmore, W.G. Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Hamazaki, J.; Murata, S. Transcriptional regulation of the 26S proteasome by Nrf1. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018, 94, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, O.; Skammelsrud, N.; Luna, L.; Nishizawa, M.; Prydz, H.; Kolsto, A.B. Small maf proteins interact with the human transcription factor TCF11/Nrf1/LCR-F1. Nucleic Acids Res. 1996, 24, 4289–4297. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, B.; Eaton, B.A.; Priess, J.R. Skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell 1992, 68, 1061–1075. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Bowerman, B.; Priess, J.R.; Weintraub, H. Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science 1994, 266, 621–628. [Google Scholar] [CrossRef]
- Carroll, A.S.; Gilbert, D.E.; Liu, X.; Cheung, J.W.; Michnowicz, J.E.; Wagner, G.; Ellenberger, T.E.; Blackwell, T.K. SKN-1 domain folding and basic region monomer stabilization upon DNA binding. Genes Dev. 1997, 11, 2227–2238. [Google Scholar] [CrossRef][Green Version]
- Baird, L.; Tsujita, T.; Kobayashi, E.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yamamoto, M. A Homeostatic shift facilitates endoplasmic reticulum proteostasis through transcriptional integration of proteostatic stress response pathways. Mol. Cell. Biol. 2016, 37, e00439-16. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Morita, T.; Kim, M.; Iemura, S.; Natsume, T.; Yamamoto, M.; Kobayashi, A. Dual regulation of the transcriptional activity of Nrf1 by β-TrCP- and Hrd1-dependent degradation mechanisms. Mol. Cell. Biol. 2011, 31, 4500–4512. [Google Scholar] [CrossRef]
- Biswas, M.; Phan, D.; Watanabe, M.; Chan, J.Y. The Fbw7 tumor suppressor regulates nuclear factor E2-related dactor 1 transcription factor turnover through proteasome-mediated proteolysis. J. Biol. Chem. 2011, 286, 39282–39289. [Google Scholar] [CrossRef]
- Fukagai, K.; Waku, T.; Chowdhury, A.M.M.A.; Kubo, K.; Matsumoto, M.; Kato, H.; Natsume, T.; Tsuruta, F.; Chiba, T.; Taniguchi, H.; et al. USP15 stabilizes the transcription factor Nrf1 in the nucleus, promoting the proteasome gene expression. Biochem. Biophys. Res. Commun. 2016, 478, 363–370. [Google Scholar] [CrossRef]
- Choe, K.P.; Przybysz, A.J.; Strange, K. The WD40 Repeat Protein WDR-23 Functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in caenorhabditis elegans. Mol. Cell. Biol. 2009, 29, 2704–2715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lucocq, J.M.; Yamamoto, M.; Hayes, J.D. The NHB1 (N-terminal homology box 1) sequence in transcription factor Nrf1 is required to anchor it to the endoplasmic reticulum and also to enable its asparagine-glycosylation. Biochem. J. 2007, 408, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lucocq, J.M.; Hayes, J.D. The Nrf1 CNC/bZIP protein is a nuclear envelope-bound transcription factor that is activated by t-butyl hydroquinone but not by endoplasmic reticulum stressors. Biochem. J. 2009, 418, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.-C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordoñez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef]
- Northrop, A.; Vangala, J.R.; Feygin, A.; Radhakrishnan, S.K. Disabling the protease DDI2 attenuates the transcriptional activity of NRF1 and potentiates proteasome inhibitor cytotoxicity. Int. J. Mol. Sci. 2020, 21, 327. [Google Scholar] [CrossRef]
- Sha, Z.; Goldberg, A.L. Proteasome-Mediated Processing of Nrf1 Is Essential for Coordinate Induction of All Proteasome Subunits and p97. Curr. Biol. 2014, 24, 1–11. [Google Scholar] [CrossRef]
- Nowak, K.; Taubert, R.M.; Haberecht, S.; Venz, S.; Krüger, E. Inhibition of calpain-1 stabilizes TCF11/Nrf1 but does not affect its activation in response to proteasome inhibition. Biosci. Rep. 2018. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Xiang, Y.; Qiu, L.; Zhao, H.; Hayes, J.D. The selective post-translational processing of transcription factor Nrf1 yields distinct isoforms that dictate its ability to differentially regulate gene expression. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Stauffer, W.T.; Arrieta, A.; Blackwood, E.A.; Glembotski, C.C. Sledgehammer to scalpel: Broad challenges to the heart and other tissues yield specific cellular responses via transcriptional regulation of the ER-stress master regulator ATF6α. Int. J. Mol. Sci. 2020, 21, 1134. [Google Scholar] [CrossRef]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 2016, 5, e18357. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 2016, 5, e17721. [Google Scholar] [CrossRef] [PubMed]
- Mótyán, J.A.; Miczi, M.; Tőzsér, J. Dimer interface organization is a main determinant of intermonomeric interactions and correlates with evolutionary relationships of retroviral and retroviral-like ddi1 and ddi2 proteases. Int. J. Mol. Sci. 2020, 21, 1352. [Google Scholar] [CrossRef] [PubMed]
- Serbyn, N.; Noireterre, A.; Bagdiul, I.; Plank, M.; Michel, A.H.; Loewith, R.; Kornmann, B.; Stutz, F. The aspartic protease Ddi1 contributes to DNA-Protein crosslink repair in yeast. Mol. Cell 2020, 77, 1066–1079.e9. [Google Scholar] [CrossRef]
- Svoboda, M.; Konvalinka, J.; Trempe, J.F.; Grantz Saskova, K. The yeast proteases Ddi1 and Wss1 are both involved in the DNA replication stress response. DNA Repair (Amst.) 2019, 80, 45–51. [Google Scholar] [CrossRef]
- Sivá, M.; Svoboda, M.; Veverka, V.; Trempe, J.F.; Hofmann, K.; Kožíšek, M.; Hexnerová, R.; Sedlák, F.; Belza, J.; Brynda, J.; et al. Human DNA-Damage-Inducible 2 Protein Is Structurally and Functionally Distinct from Its Yeast Ortholog. Sci. Rep. 2016, 6, 30443. [Google Scholar] [CrossRef]
- Nowicka, U.; Zhang, D.; Walker, O.; Krutauz, D.; Castañeda, C.A.; Chaturvedi, A.; Chen, T.Y.; Reis, N.; Glickman, M.H.; Fushman, D. DNA-damage-inducible 1 protein (Ddi1) contains an uncharacteristic ubiquitin-like domain that binds ubiquitin. Structure 2015, 23, 542–557. [Google Scholar] [CrossRef]
- Ivantsiv, Y.; Kaplun, L.; Tzirkin-Goldin, R.; Shabek, N.; Raveh, D. Unique role for the UbL-UbA protein Ddi1 in turnover of SCFUfo1 complexes. Mol. Cell. Biol. 2006, 26, 1579–1588. [Google Scholar] [CrossRef]
- Suzuki, T.; Huang, C.; Harada, Y.; Hosomi, A.; Masahara-Negishi, Y.; Seino, J.; Fujihira, H.; Funakoshi, Y.; Suzuki, T.; Dohmae, N. Endo-β-n-acetylglucosaminidase forms N-GlcNAc protein aggregates during ER-associated degradation in NGLY1-defective cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1398–1403. [Google Scholar] [CrossRef]
- Suzuki, T.; Huang, C.; Fujihira, H. The cytoplasmic peptide: N-glycanase (NGLY1)—Structure, expression and cellular functions. Gene 2016, 577, 1–7. [Google Scholar] [CrossRef]
- Owings, K.G.; Lowry, J.B.; Bi, Y.; Might, M.; Chow, C.Y. Transcriptome and functional analysis in a Drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum. Mol. Genet. 2018, 27, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Breen, P.C.; Ruvkun, G. Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 2019, 177, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Valdez, J.L.; Ho, D.V.; Lee, C.S.; Kim, H.M.; Wang, X.; Huang, L.; Chan, J.Y. Nuclear factor-erythroid-2 related transcription factor-1 (Nrf1) is regulated by O-GlcNAc transferase. Free Radic. Biol. Med. 2017, 110, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Sekine, H.; Okazaki, K.; Kato, K.; Alam, M.M.; Shima, H.; Katsuoka, F.; Tsujita, T.; Suzuki, N.; Kobayashi, A.; Igarashi, K.; et al. O -GlcNAcylation signal mediates proteasome inhibitor resistance in cancer cells by stabilizing NRF1. Mol. Cell. Biol. 2018, 38, e00252. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Lü, F.; Liu, X.; Ru, Y.; Ren, Y.; Yao, L.; Zhang, Y. Transcription factor Nrf1 is negatively regulated by its O-GlcNAcylation status. FEBS Lett. 2015, 589, 2347–2358. [Google Scholar] [CrossRef]
- Chepelev, N.L.; Bennitz, J.D.; Huang, T.; McBride, S.; Willmore, W.G. The Nrf1 CNC-bZIP Protein Is Regulated by the Proteasome and Activated by Hypoxia. PLoS ONE 2011, 6, e29167. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Taniguchi, H.; Ito, Y.; Morita, T.; Karim, M.R.; Ohtake, N.; Fukagai, K.; Ito, T.; Okamuro, S.; Iemura, S.-I.; et al. The Casein Kinase 2-Nrf1 Axis Controls the Clearance of Ubiquitinated Proteins by Regulating Proteasome Gene Expression. Mol. Cell. Biol. 2013, 33, 3461–3472. [Google Scholar] [CrossRef]
- Biswas, M.; Kwong, E.K.; Park, E.; Nagra, P.; Chan, J.Y. Glycogen synthase kinase 3 regulates expression of nuclear factor-erythroid-2 related transcription factor-1 (Nrf1) and inhibits pro-survival function of Nrf1. Exp. Cell Res. 2013, 319, 1922–1931. [Google Scholar] [CrossRef]
- Ho, D.V.; Chan, J.Y. Induction of Herpud1 expression by ER stress is regulated by Nrf1. FEBS Lett. 2015, 589, 615–620. [Google Scholar] [CrossRef]
- Glover-Cutter, K.M.; Lin, S.; Blackwell, T.K. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 2013, 9, e1003701. [Google Scholar] [CrossRef]
- Paek, J.; Lo, J.Y.; Narasimhan, S.D.; Nguyen, T.N.; Glover-Cutter, K.; Robida-Stubbs, S.; Suzuki, T.; Yamamoto, M.; Blackwell, T.K.; Curran, S.P. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 2012, 16, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.P.; Abate, J.P.; Dilks, K.; Landis, J.; Ashraf, J.; Murphy, C.T.; Blackwell, T.K. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 2009, 8, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Lynn, D.A.; Lo, J.Y.; Paek, J.; Curran, S.P. SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation. Nat. Commun. 2014, 5, 5048. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Ming, M.; Zhao, R.; Pi, J.; Wu, C.; He, Y.Y. Nrf1 CNC-bZIP protein promotes cell survival and nucleotide excision repair through maintaining glutathione homeostasis. J. Biol. Chem. 2012, 287, 18788–18795. [Google Scholar] [CrossRef]
- Taniguchi, H.; Okamuro, S.; Koji, M.; Waku, T.; Kubo, K.; Hatanaka, A.; Sun, Y.; Chowdhury, A.M.M.A.; Fukamizu, A.; Kobayashi, A. Possible roles of the transcription factor Nrf1 (NFE2L1) in neural homeostasis by regulating the gene expression of deubiquitinating enzymes. Biochem. Biophys. Res. Commun. 2017, 484, 176–183. [Google Scholar] [CrossRef]
- Lacher, S.E.; Alazizi, A.; Wang, X.; Bell, D.A.; Pique-Regi, R.; Luca, F.; Slattery, M. A hypermorphic antioxidant response element is associated with increased MS4A6A expression and Alzheimer’s disease. Redox Biol. 2018, 14, 686–693. [Google Scholar] [CrossRef]
- Yang, K.; Huang, R.; Fujihira, H.; Suzuki, T.; Yan, N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J. Exp. Med. 2018, 215, 2600–2616. [Google Scholar] [CrossRef]
- Kim, H.M.; Han, J.W.; Chan, J.Y. Nuclear factor Erythroid-2 like 1 (NFE2L1): Structure, function and regulation. Gene 2016, 584, 17–25. [Google Scholar] [CrossRef]
- Bott, L.C.; Badders, N.M.; Chen, K.L.; Harmison, G.G.; Bautista, E.; Shih, C.C.Y.; Katsuno, M.; Sobue, G.; Taylor, J.P.; Dantuma, N.P.; et al. A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2016, 25, 1979–1989. [Google Scholar] [CrossRef]
- Vangala, J.R.; Radhakrishnan, S.K. Nrf1-mediated transcriptional regulation of the proteasome requires a functional TIP60 complex. J. Biol. Chem. 2019, 294, 2036–2045. [Google Scholar] [CrossRef]
- Ebstein, F.; Poli Harlowe, M.C.; Studencka-Turski, M.; Krüger, E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front. Immunol. 2019, 10, 2756. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Goldberg, A.L. Reply to Vangala et al.: Complete inhibition of the proteasome reduces new proteasome production by causing Nrf1 aggregation. Curr. Biol. 2016, 26, R836–R837. [Google Scholar] [CrossRef] [PubMed]
- Kitiphongspattana, K.; Mathews, C.E.; Leiter, E.H.; Gaskins, H.R. Proteasome inhibition alters glucose-stimulated (pro)insulin secretion and turnover in pancreatic β-cells. J. Biol. Chem. 2005, 280, 15727–15734. [Google Scholar] [CrossRef] [PubMed]
- Sotzny, F.; Schormann, E.; Kühlewindt, I.; Koch, A.; Brehm, A.; Goldbach-Mansky, R.; Gilling, K.E.; Krüger, E. TCF11/Nrf1-Mediated induction of proteasome expression prevents cytotoxicity by rotenone. Antioxid. Redox Signal. 2016, 25, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109. [Google Scholar] [CrossRef] [PubMed]
- Steinbaugh, M.J.; Narasimhan, S.D.; Robida-Stubbs, S.; Moronetti Mazzeo, L.E.; Dreyfuss, J.M.; Hourihan, J.M.; Raghavan, P.; Operaña, T.N.; Esmaillie, R.; Blackwell, T.K. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. eLife 2015, 4, e07836. [Google Scholar] [CrossRef] [PubMed]
- Tullet, J.M.A.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Keap1/Nrf2 Signaling regulates oxidative stress tolerance and lifespan in drosophila. Dev. Cell 2008, 14, 76–85. [Google Scholar] [CrossRef]
- Lee, C.S.; Lee, C.; Hu, T.; Nguyen, J.M.; Zhang, J.; Martin, M.V.; Vawter, M.P.; Huang, E.J.; Chan, J.Y. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 8408–8413. [Google Scholar] [CrossRef]
- Lee, C.S.; Ho, D.V.; Chan, J.Y. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J. 2013, 280, 3609–3620. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.B.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, S.; Zhang, Y. Nrf1 is paved as a new strategic avenue to prevent and treat cancer, neurodegenerative and other diseases. Toxicol. Appl. Pharmacol. 2018, 360, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zheng, H.; Cui, Q.; Chen, C.; Bao, S.; Sun, J.; Li, L.; Yang, B.; Wang, H.; Hou, Y.; et al. Nfe2l1-silenced insulinoma cells acquire aggressiveness and chemoresistance. Endocr. Relat. Cancer 2018, 25, 185–200. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Higashi, C.; Fukutomi, T.; Katsuoka, F.; Tsujita, T.; Yagishita, Y.; Matsuyama, Y.; Motohashi, H.; Uruno, A.; Yamamoto, M. Transcription factor NF-E2-related factor 1 impairs glucose metabolism in mice. Genes Cells 2014, 19, 650–665. [Google Scholar] [CrossRef]
- Zhang, Y.; Manning, B.D. mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle 2015, 14, 2011–2017. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.H.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef]
- Fok, W.C.; Chen, Y.; Bokov, A.; Zhang, Y.; Salmon, A.B.; Diaz, V.; Javors, M.; Wood, W.H.; Zhang, Y.; Becker, K.G.; et al. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS ONE 2014, 9, e83988. [Google Scholar] [CrossRef]
- Zhang, Y.; Bokov, A.; Gelfond, J.; Soto, V.; Ikeno, Y.; Hubbard, G.; Diaz, V.; Sloane, L.; Maslin, K.; Treaster, S.; et al. Rapamycin extends life and health in C57BL/6 mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 119–130. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Hataya, N.; Katsuoka, F.; Yamamoto, M. NF-E2-Related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the Lipin1 and PGC-1 genes. Mol. Cell. Biol. 2012, 32, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgül, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Lü, F.; Stewart, D.; Zhang, Y. Mechanisms underlying chemopreventive effects of flavonoids via multiple signaling nodes within Nrf2-ARE and AhR-XRE gene regulatory networks. Curr. Chem. Biol. 2013, 7, 151–176. [Google Scholar] [CrossRef]
- Liu, P.; Kerins, M.J.; Tian, W.; Neupane, D.; Zhang, D.D.; Ooi, A. Differential and overlapping targets of the transcriptional regulators NRF1, NRF2, and NRF3 in human cells. J. Biol. Chem. 2019, 294, 18131–18149. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/ -TrCP Promotes Glycogen Synthase Kinase 3-Dependent degradation of the Nrf2 transcription factor in a Keap1-Independent manner. Mol. Cell. Biol. 2011, 20, 144–151. [Google Scholar] [CrossRef]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Wang, W.; Chan, J.Y. Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain: Inhibition of nuclear translocation and transacting function. J. Biol. Chem. 2006, 281, 19676–19687. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Zhu, Y.P.; Zheng, Z.; Hu, S.; Ru, X.; Fan, Z.; Qiu, L.; Zhang, Y. Unification of opposites between two antioxidant transcription factors nrf1 and nrf2 in mediating distinct cellular responses to the endoplasmic reticulum stressor tunicamycin. Antioxidants 2020, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK—Dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum Stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Kwong, M.; Lu, R.; Chang, J.; Wang, B.; Yen, T.S.B.; Kan, Y.W. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998, 17, 1779–1787. [Google Scholar] [CrossRef]
- Leung, L.; Kwong, M.; Hou, S.; Lee, C.; Chan, J.Y. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 2003, 278, 48021–48029. [Google Scholar] [CrossRef]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef]
- Zhang, Y.; Kobayashi, A.; Yamamoto, M.; Hayes, J.D. The Nrf3 transcription factor is a membrane-bound glycoprotein targeted to the endoplasmic reticulum through its N-terminal homology box 1 sequence. J. Biol. Chem. 2009, 284, 3195–3210. [Google Scholar] [CrossRef]
- Nouhi, Z.; Chevillard, G.; Derjuga, A.; Blank, V. Endoplasmic reticulum association and N-linked glycosylation of the human Nrf3 transcription factor. FEBS Lett. 2007, 581, 5401–5406. [Google Scholar] [CrossRef]
- Chowdhury, A.M.M.A.; Katoh, H.; Hatanaka, A.; Iwanari, H.; Nakamura, N.; Hamakubo, T.; Natsume, T.; Waku, T.; Kobayashi, A. Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Sci. Rep. 2017, 7, 12494. [Google Scholar] [CrossRef]
- Kannan, M.B.; Dodard-Friedman, I.; Blank, V. Stringent control of NFE2L3 (Nuclear Factor, Erythroid 2-Like 3; NRF3) protein degradation by FBW7 (F-box/WD Repeatcontaining–Protein 7) and glycogen synthase kinase 3 (GSK3). J. Biol. Chem. 2015, 290, 26292–26302. [Google Scholar] [CrossRef]
- Kobayashi, A.; Waku, T. New addiction to the NRF2-related factor NRF3 in cancer cells: Ubiquitin-independent proteolysis through the 20S proteasome. Cancer Sci. 2020, 111, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.J.; Li, J. Proteasome inhibitors: Harnessing proteostasis to combat disease. Molecules 2020, 25, 671. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell 2017, 168, 890903.e15. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.; Müller-Ide, H.; Rückrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk. Res. 2014, 38, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Fassmannová, D.; Sedlák, F.; Sedláček, J.; Špička, I.; Grantz Šašková, K. Nelfinavir Inhibits the TCF11/Nrf1-Mediated proteasome recovery pathway in multiple myeloma. Cancers 2020, 12, 1065. [Google Scholar] [CrossRef]
- Tsujita, T.; Baird, L.; Furusawa, Y.; Katsuoka, F.; Hou, Y.; Gotoh, S.; Kawaguchi, S.I.; Yamamoto, M. Discovery of an NRF1-specific inducer from a large-scale chemical library using a direct NRF1-protein monitoring system. Genes Cells 2015, 20, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Iaconelli, J.; Ibrahim, L.; Chen, E.; Hull, M.; Schultz, P.G.; Bollong, M.J. Small-Molecule stimulators of NRF1 transcriptional activity. ChemBioChem 2019, 116, 6435–6440. [Google Scholar] [CrossRef]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-β generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef]
- Li, F.; Gao, B.; Dong, H.; Shi, J.; Fang, D. Icariin induces Synoviolin expression through NFE2L1 to protect neurons from ER stress-induced apoptosis. PLoS ONE 2015, 10, e0119955. [Google Scholar] [CrossRef]
- Zhao, R.; Hou, Y.; Xue, P.; Woods, C.G.; Fu, J.; Feng, B.; Guan, D.; Sun, G.; Chan, J.Y.; Waalkes, M.P.; et al. Long isoforms of NRF1 contribute to arsenic-induced antioxidant response in human keratinocytes. Environ. Health Perspect. 2011, 119, 56–62. [Google Scholar] [CrossRef]
- Yip, M.C.J.; Bodnar, N.O.; Rapoport, T.A. Ddi1 is a ubiquitin-dependent protease. Proc. Natl. Acad. Sci. USA 2020, 117, 7776–7781. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.L. The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 2009, 118, 329–347. [Google Scholar] [CrossRef] [PubMed]
- Galves, M.; Rathi, R.; Prag, G.; Ashkenazi, A. Ubiquitin signaling and degradation of aggregate-prone proteins. Trends Biochem. Sci. 2019, 44, 872–884. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamazaki, J.; Murata, S. ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond. Int. J. Mol. Sci. 2020, 21, 3683. https://doi.org/10.3390/ijms21103683
Hamazaki J, Murata S. ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond. International Journal of Molecular Sciences. 2020; 21(10):3683. https://doi.org/10.3390/ijms21103683
Chicago/Turabian StyleHamazaki, Jun, and Shigeo Murata. 2020. "ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond" International Journal of Molecular Sciences 21, no. 10: 3683. https://doi.org/10.3390/ijms21103683
APA StyleHamazaki, J., & Murata, S. (2020). ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond. International Journal of Molecular Sciences, 21(10), 3683. https://doi.org/10.3390/ijms21103683