Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms


{kind=link}
{kind=link}
{kind=link}
Abstract
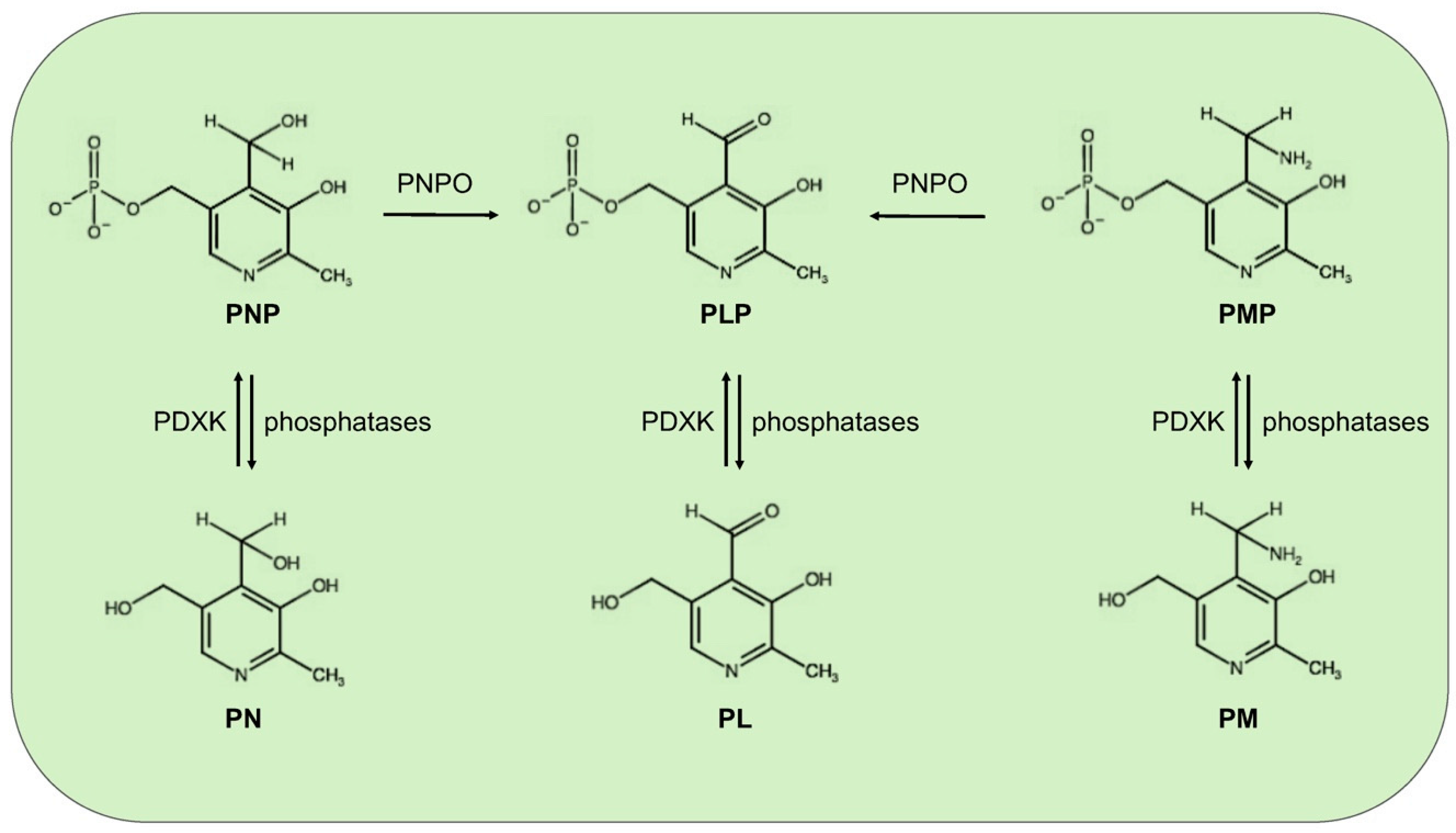
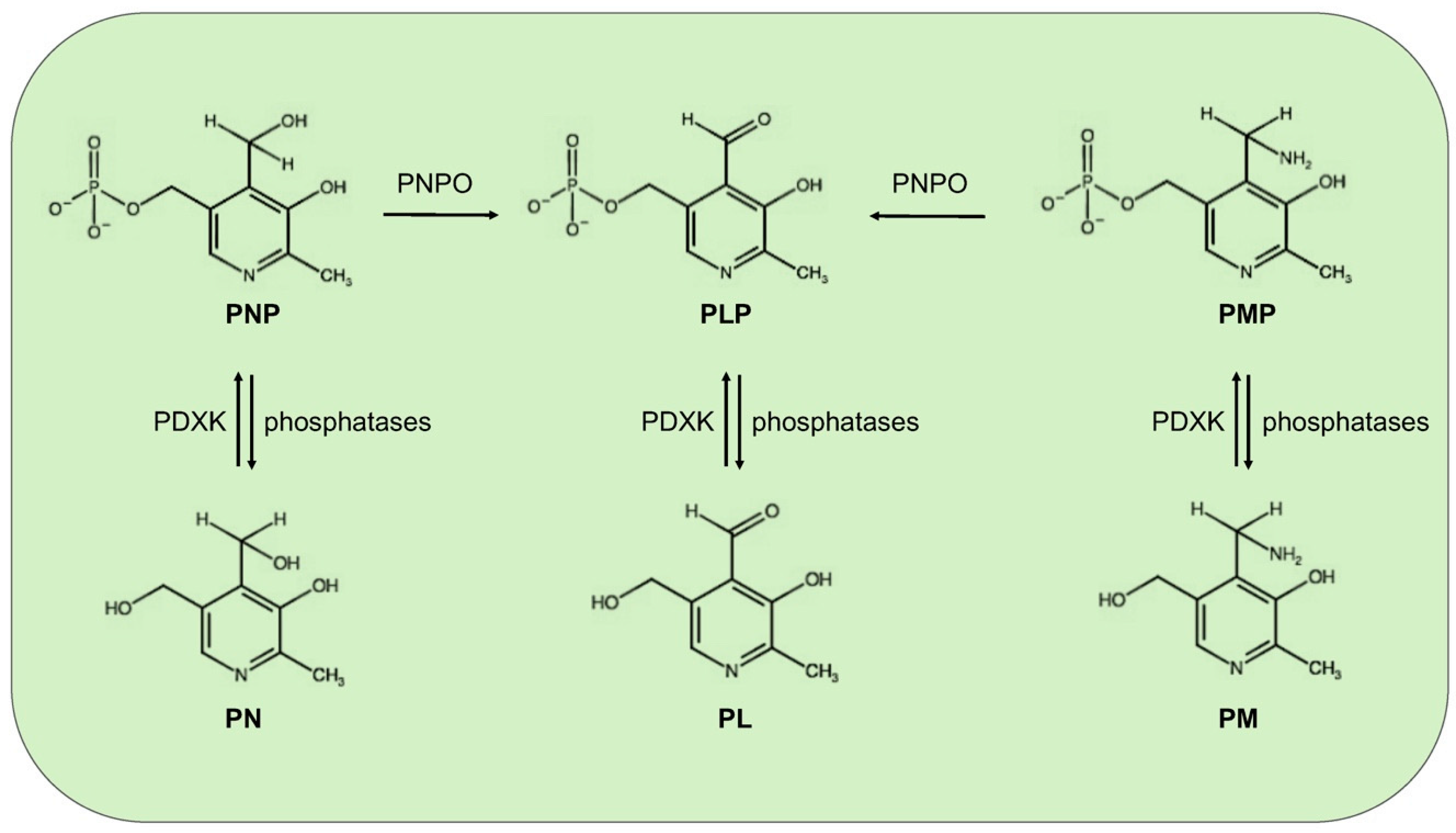
:1. Vitamin B6: Roles and Synthesis
2. Vitamin B6 and Diabetes
3. Is Reduced Vitamin B6 Availability the Cause or the Effect of Diabetes?
3.1. Diabetes Decreases Vitamin B6 Levels
3.2. Reduced Vitamin B6 Levels Trigger Diabetes
Mutations in Genes Involved in Vitamin B6 Synthesis Cause Diabetes
4. Mechanisms Underlying the Link between Vitamin B6 Diabetes
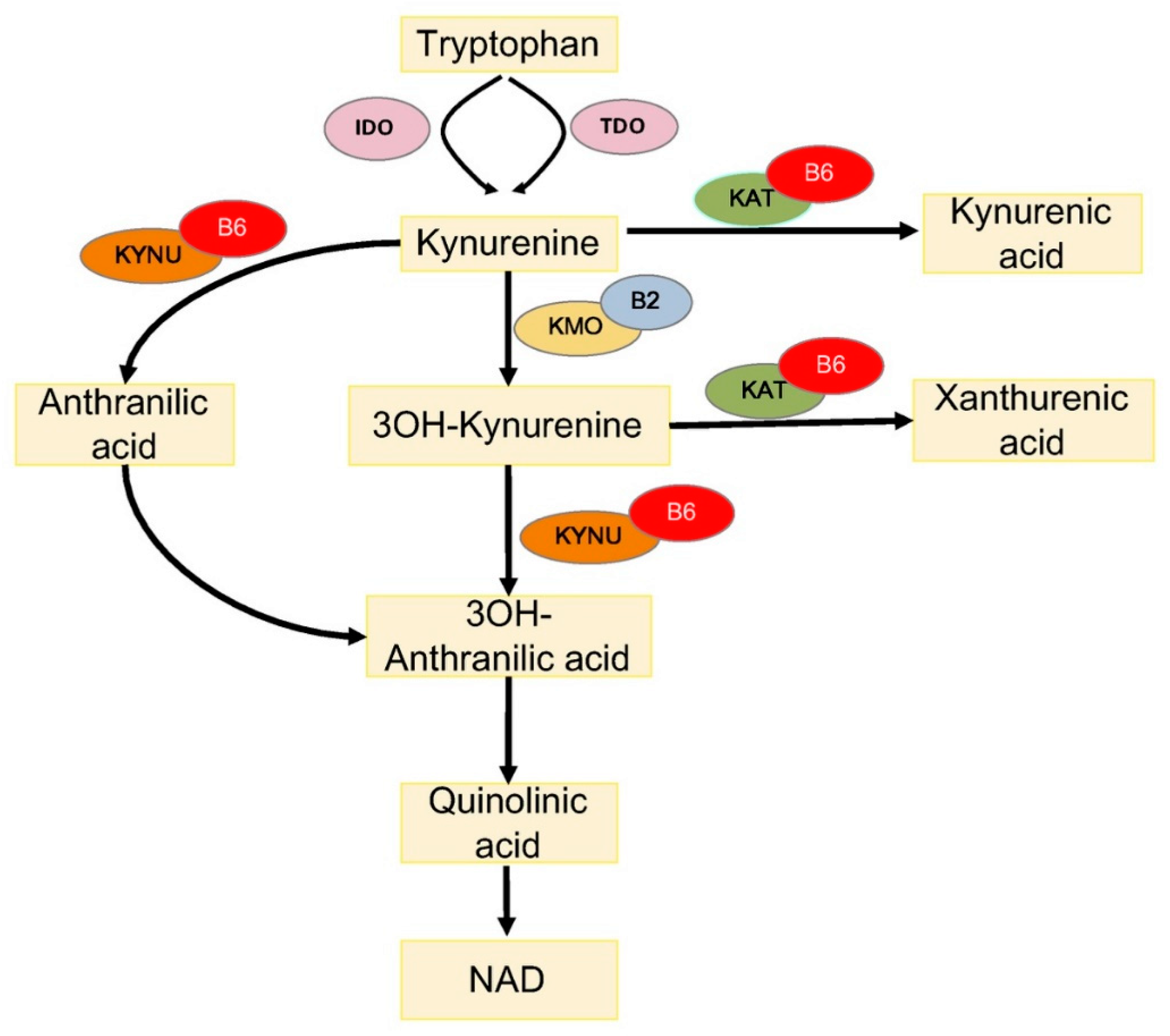
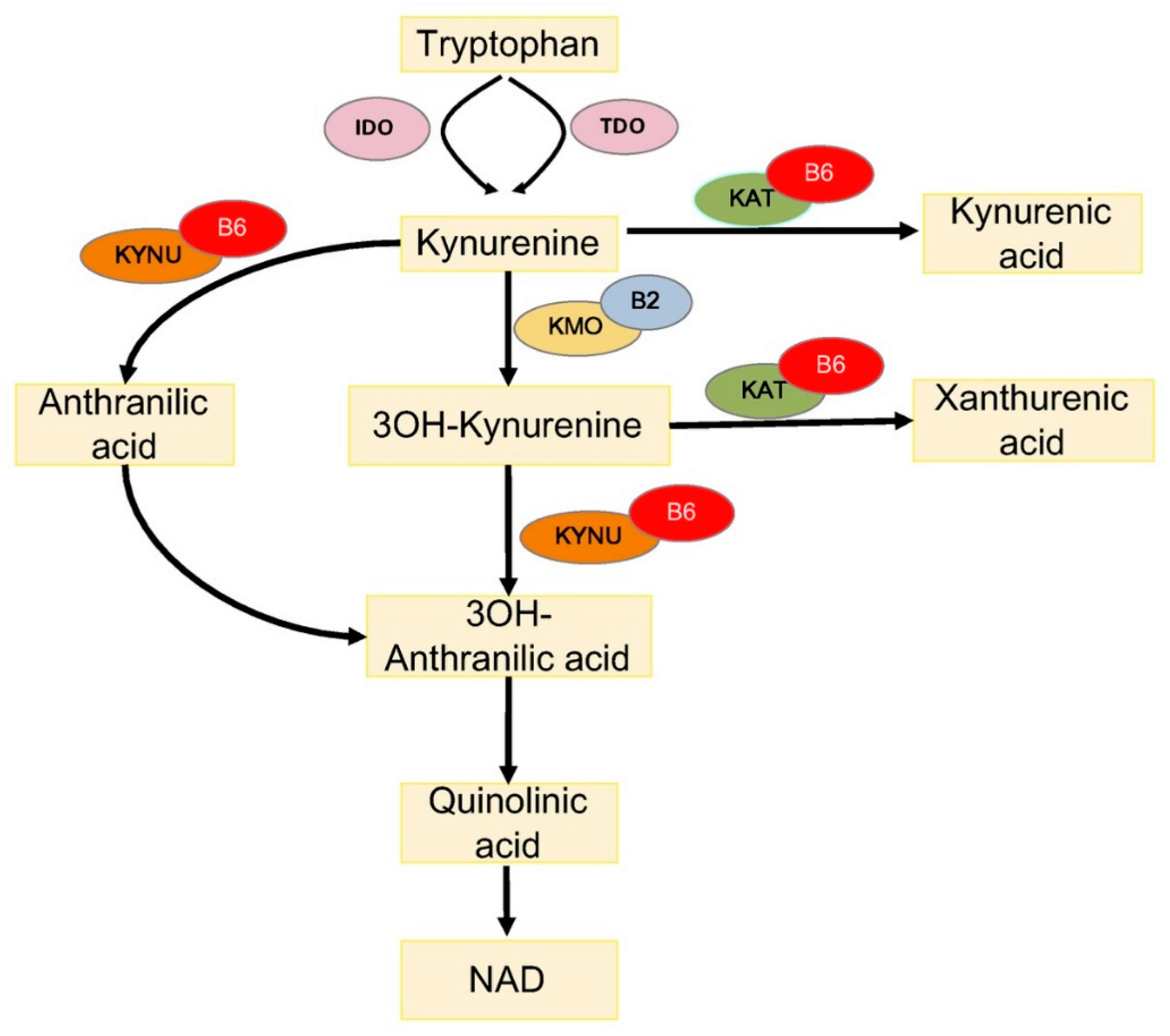
4.1. Vitamin B6 and Tryptophan Metabolism
4.2. Vitamin B6 and Lipid Metabolism
5. Vitamin B6 and Diabetes Complications
6. Vitamin B6 and DNA Damage in Diabetes
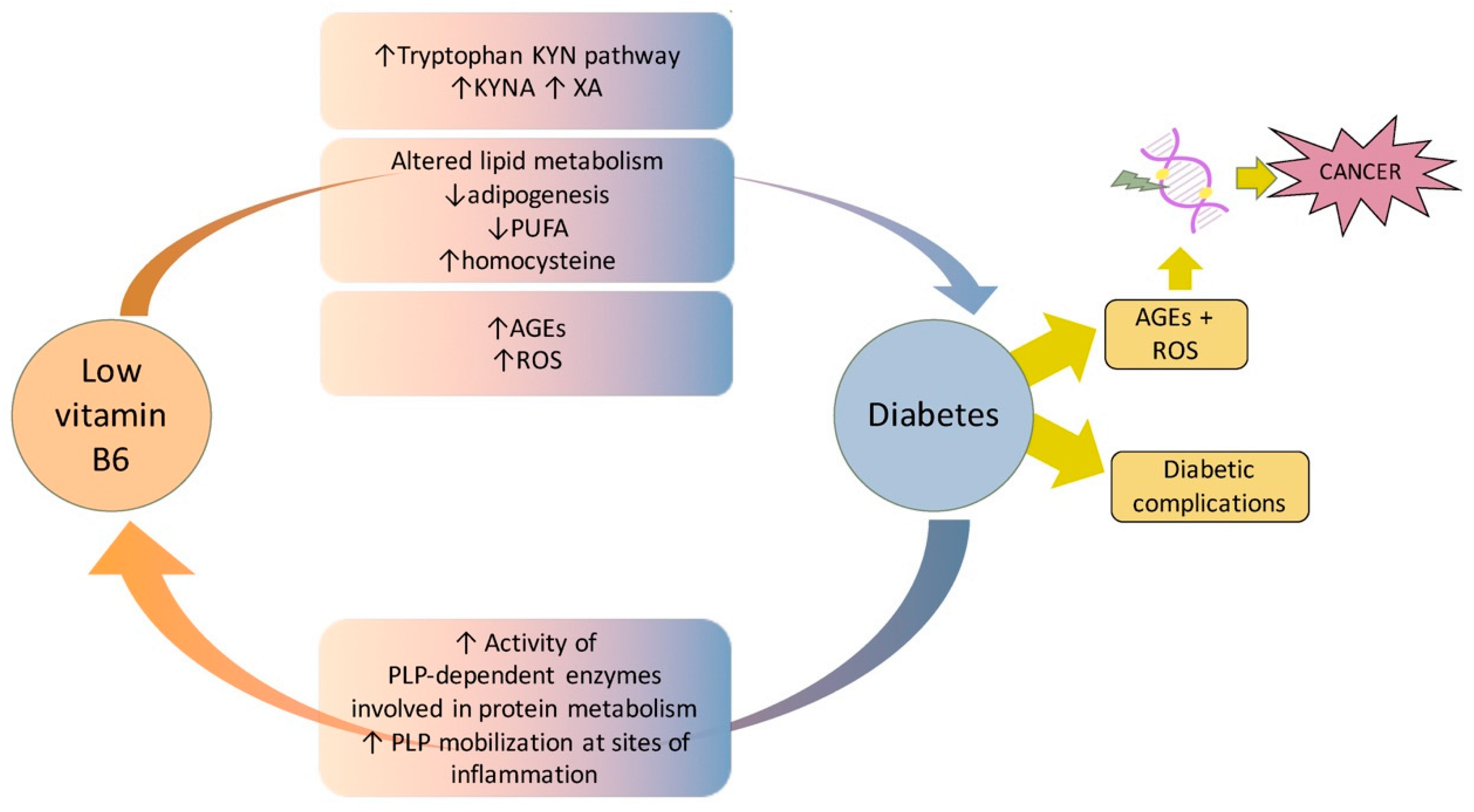
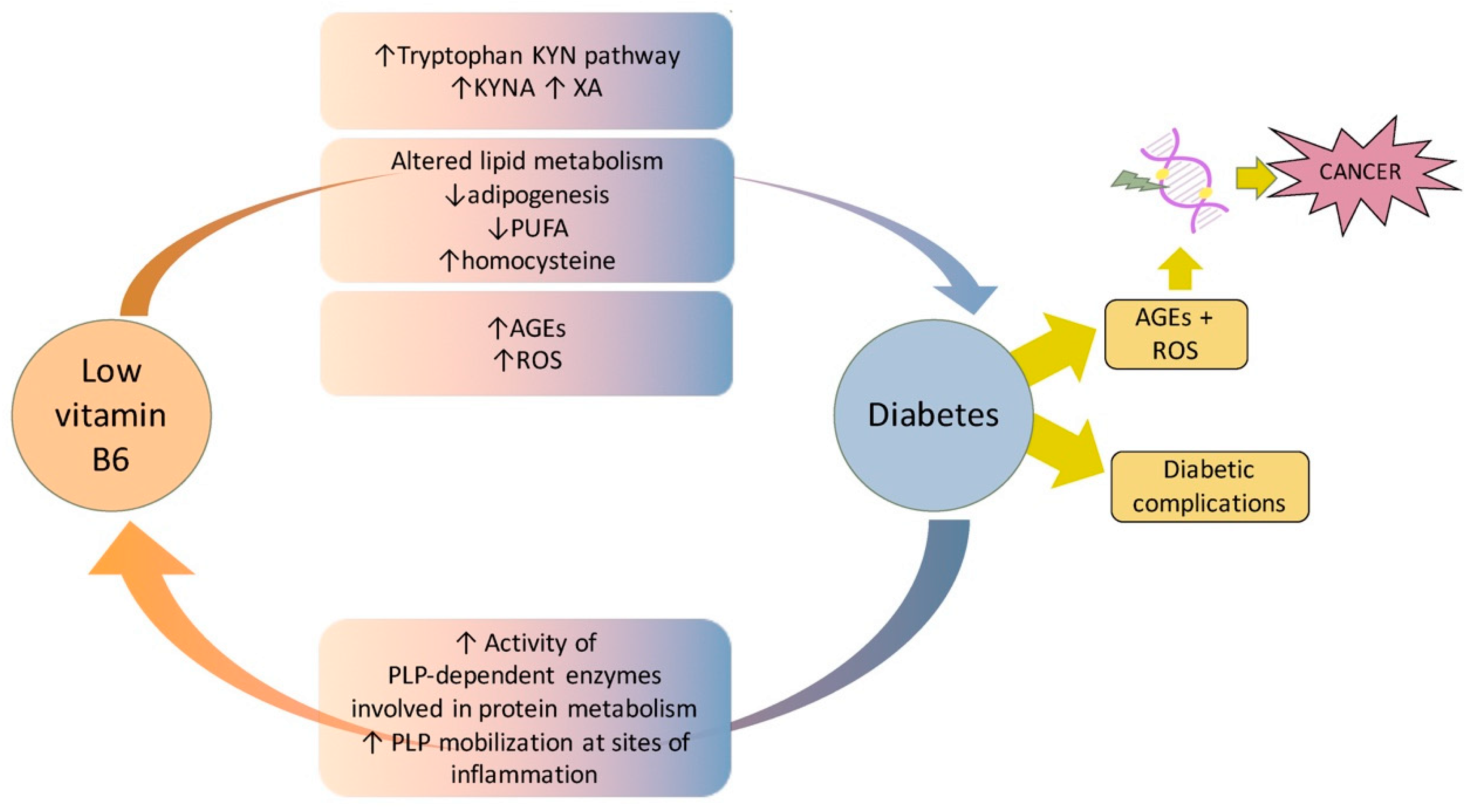
7. Vitamin B6 Diabetes and Cancer
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PLP | pyridoxal 5′-phosphate |
| AGEs | Advanced Glycation End products |
| CABs | Chromosome aberrations |
References
- Hellmann, H.; Mooney, S. Vitamin B6: A molecule for human health? Molecules 2010, 15, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Percudani, R.; Peracchi, A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef]
- Di Salvo, M.L.; Contestabile, R.; Safo, M.K. Vitamin B(6) salvage enzymes: Mechanism, structure and regulation. Biochim. Biophys. Acta 2011, 1814, 1597–1608. [Google Scholar] [CrossRef]
- Bilski, P.; Li, M.Y.; Ehrenshaft, M.; Daub, M.E.; Chignell, C.F. Vitamin B6 (pyridoxine) and its derivatives are efficient singlet oxygen quenchers and potential fungal antioxidants. Photochem. Photobiol. 2000, 71, 129–134. [Google Scholar] [CrossRef]
- Booth, A.A.; Khalifah, R.G.; Todd, P.; Hudson, B.G. In vitro kinetic studies of formation of antigenic advanced glycation end products (AGEs). Novel inhibition of post-Amadori glycation pathways. J. Biol. Chem. 1997, 272, 5430–5437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrecht, G.; Braun, K.; Damer, M.; Ganso, M.; Hildebrandt, C.; Ullmann, H.; Kassack, M.U.; Nickel, P. Structure-activity relationships of suramin and pyridoxal-5′-phosphate derivatives as P2 receptor antagonists. Curr. Pharm. Des. 2002, 8, 2371–2399. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.B. Two interconnected B vitamins: Riboflavin and pyridoxine. Physiol. Rev. 1989, 69, 1170–1198. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M. Recent advances in carrier-mediated intestinal absorption of water-soluble vitamins. Annu. Rev. Physiol. 2004, 66, 419–446. [Google Scholar] [CrossRef]
- Jang, Y.M.; Kim, D.W.; Kang, T.C.; Won, M.H.; Baek, N.I.; Moon, B.J.; Choi, S.Y.; Kwon, O.S. Human pyridoxal phosphatase. Molecular cloning, functional expression, and tissue distribution. J. Biol. Chem. 2003, 278, 50040–50046. [Google Scholar] [CrossRef] [Green Version]
- Cravo, M.L.; Camilo, M.E. Hyperhomocysteinemia in chronic alcoholism: Relations to folic acid and vitamins B(6) and B(12) status. Nutrition 2000, 16, 296–302. [Google Scholar] [CrossRef]
- Ferro, Y.; Carè, I.; Mazza, E.; Provenzano, F.; Colica, C.; Torti, C.; Romeo, S.; Pujia, A.; Montalcini, T. Protein and vitamin B6 intake are associated with liver steatosis assessed by transient elastography, especially in obese individuals. Clin. Mol. Hepatol. 2017, 23, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, A.H., Jr.; Henderson, J.M. Diseases associated with defects in vitamin B6 metabolism or utilization. Annu. Rev. Nutr. 1987, 7, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Kowlessar, O.D.; Haeffner, L.J.; Benson, G.D. Abnormal tryptophan metabolism in patients with adult celiac disease, with evidence for deficiency of vitamin B6. J. Clin. Investig. 1964, 43, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, E.P.; Selhub, J.; Bagley, P.J.; Dallal, G.; Roubenoff, R. Pyridoxine supplementation corrects vitamin B6 deficiency but does not improve inflammation in patients with rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1404-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biehl, J.P.; Vilter, R.W. Effect of isoniazid on vitamin B6 metabolism; its possible significance in producing isoniazid neuritis. Proc. Soc. Exp. Biol. Med. 1954, 85, 389–392. [Google Scholar] [CrossRef]
- Jaffe, I.A.; Altman, K.; Merryman, P. The antipyridoxine effect of penicillamine in man. J. Clin. Investig. 1964, 43, 1869–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Sawaki, S.; Hayami, S. Inhibitory effect of cycloserine on some enzymic activities related to vitamin B6. J. Vitaminol. 1957, 3, 68–72. [Google Scholar] [CrossRef]
- Lussana, F.; Zighetti, M.L.; Bucciarelli, P.; Cugno, M.; Cattaneo, M. Blood levels of homocysteine, folate, vitamin B6 and B12 in women using oral contraceptives compared to non-users. Thromb. Res. 2003, 112, 37–41. [Google Scholar] [CrossRef]
- Oxenkrug, G.F. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: The serotonin hypothesis revisited 40 years later. Isr. J. Psychiatry Relat. Sci. 2010, 47, 56–63. [Google Scholar]
- Midttun, O.; Ulvik, A.; Ringdal Pedersen, E.; Ebbing, M.; Bleie, O.; Schartum-Hansen, H.; Nilsen, R.M.; Nygård, O.; Ueland, P.M. Low plasma vitamin B-6 status affects metabolism through the kynurenine pathway in cardiovascular patients with systemic inflammation. J. Nutr. 2011, 141, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Di Salvo, M.L.; Safo, M.K.; Contestabile, R. Biomedical aspects of pyridoxal 5′-phosphate availability. Front. Biosci. 2012, 4, 897–913. [Google Scholar]
- Merigliano, C.; Mascolo, E.; Burla, R.; Saggio, I.; Vernì, F. The Relationship Between Vitamin B6, Diabetes and Cancer. Front. Genet. 2018, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, R.; di Salvo, M.L.; Bunik, V.; Tramonti, A.; Vernì, F. The multifaceted role of vitamin B(6) in cancer: Drosophila as a model system to investigate DNA damage. Open Biol. 2020, 10, 200034. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. 1), S67–S74. [Google Scholar]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef] [Green Version]
- Leklem, J.E. Vitamin B-6: A status report. J. Nutr. 1990, 120 (Suppl. 11), 1503–1507. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Balakrishna, N.; Pitla, S.; Reddy, P.Y.; Mudili, S.; Lopamudra, P.; Suryanarayana, P.; Viswanath, K.; Ayyagari, R.; Reddy, G.B. Status of B-vitamins and homocysteine in diabetic retinopathy: Association with vitamin-B12 deficiency and hyperhomocysteinemia. PLoS ONE 2011, 6, e26747. [Google Scholar] [CrossRef]
- Ahn, H.J.; Min, K.W.; Cho, Y.O. Assessment of vitamin B(6) status in Korean patients with newly diagnosed type 2 diabetes. Nutr. Res. Pract. 2011, 5, 34–39. [Google Scholar] [CrossRef]
- Nix, W.A.; Zirwes, R.; Bangert, V.; Kaiser, R.P.; Schilling, M.; Hostalek, U.; Obeid, R. Vitamin B status in patients with type 2 diabetes mellitus with and without incipient nephropathy. Diabetes Res. Clin. Pract. 2015, 107, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Iwakawa, H.; Nakamura, Y.; Fukui, T.; Fukuwatari, T.; Ugi, S.; Maegawa, H.; Doi, Y.; Shibata, K. Concentrations of Water-Soluble Vitamins in Blood and Urinary Excretion in Patients with Diabetes Mellitus. Nutr. Metab. Insights 2016, 9, 85–92. [Google Scholar] [CrossRef]
- Rogers, K.S.; Higgins, E.S.; Kline, E.S. Experimental diabetes causes mitochondrial loss and cytoplasmic enrichment of pyridoxal phosphate and aspartate aminotransferase activity. Biochem. Med. Metab. Biol. 1986, 36, 91–97. [Google Scholar] [CrossRef]
- Okada, M.; Shibuya, M.; Yamamoto, E.; Murakami, Y. Effect of diabetes on vitamin B6 requirement in experimental animals. Diabetes Obes. Metab. 1999, 1, 221–225. [Google Scholar] [CrossRef]
- Bennink, H.J.; Schreurs, W.H. Improvement of oral glucose tolerance in gestational diabetes by pyridoxine. Br. Med. J. 1975, 3, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Spellacy, W.N.; Buhi, W.C.; Birk, S.A. Vitamin B6 treatment of gestational diabetes mellitus: Studies of blood glucose and plasma insulin. Am. J. Obstet. Gynecol. 1977, 127, 599–602. [Google Scholar] [CrossRef]
- Nair, A.R.; Biju, M.P.; Paulose, C.S. Effect of pyridoxine and insulin administration on brain glutamate dehydrogenase activity and blood glucose control in streptozotocin-induced diabetic rats. Biochim. Biophys. Acta 1998, 1381, 351–354. [Google Scholar] [CrossRef]
- Solomon, L.R.; Cohen, K. Erythrocyte O2 transport and metabolism and effects of vitamin B6 therapy in type II diabetes mellitus. Diabetes 1989, 38, 881–886. [Google Scholar] [CrossRef]
- Kim, H.H.; Kang, Y.R.; Lee, J.Y.; Chang, H.B.; Lee, K.W.; Apostolidis, E.; Kwon, Y.I. The Postprandial Anti-Hyperglycemic Effect of Pyridoxine and Its Derivatives Using In Vitro and In Vivo Animal Models. Nutrients 2018, 10, 285. [Google Scholar] [CrossRef] [Green Version]
- Leklem, J.E.; Hollenbeck, C.B. Acute ingestion of glucose decreases plasma pyridoxal 5′-phosphate and total vitamin B-6 concentration. Am. J. Clin. Nutr. 1990, 51, 832–836. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef] [Green Version]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.; Ueland, P.M.; Selhub, J. Mechanistic perspective on the relationship between pyridoxal 5′-phosphate and inflammation. Nutr. Rev. 2013, 71, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Toyota, T.; Kai, Y.; Kakizaki, M.; Ohtsuka, H.; Shibata, Y.; Goto, Y. The endocrine pancreas in pyridoxine deficient rats. Tohoku J. Exp. Med. 1981, 134, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubí, B. Pyridoxal 5′-phosphate (PLP) deficiency might contribute to the onset of type I diabetes. Med. Hypotheses 2012, 78, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Marzio, A.; Merigliano, C.; Gatti, M.; Vernì, F. Sugar and chromosome stability: Clastogenic effects of sugars in vitamin B6-deficient cells. PLoS Genet. 2014, 10, e1004199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascolo, E.; Amoroso, N.; Saggio, I.; Merigliano, C.; Vernì, F. Pyridoxine/pyridoxamine 5′-phosphate oxidase (Sgll/PNPO) is important for DNA integrity and glucose homeostasis maintenance in Drosophila. J. Cell. Physiol. 2020, 235, 504–512. [Google Scholar] [CrossRef]
- Cipressa, F.; Romano, S.; Centonze, S.; zur Lage, P.I.; Vernì, F.; Dimitri, P.; Gatti, M.; Cenci, G. Effete, a Drosophila chromatin-associated ubiquitin-conjugating enzyme that affects telomeric and heterochromatic position effect variegation. Genetics 2013, 195, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Musselman, L.P.; Fink, J.L.; Narzinski, K.; Ramachandran, P.V.; Hathiramani, S.S.; Cagan, R.L.; Baranski, T.J. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis. Models Mech. 2011, 4, 842–849. [Google Scholar] [CrossRef] [Green Version]
- Alfa, R.W.; Kim, S.K. Using Drosophila to discover mechanisms underlying type 2 diabetes. Dis. Models Mech. 2016, 9, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Graham, P.; Pick, L. Drosophila as a Model for Diabetes and Diseases of Insulin Resistance. Curr. Top. Dev. Biol. 2017, 121, 397–419. [Google Scholar]
- Merigliano, C.; Mascolo, E.; La Torre, M.; Saggio, I.; Vernì, F. Protective role of vitamin B6 (PLP) against DNA damage in Drosophila models of type 2 diabetes. Sci. Rep. 2018, 8, 11432. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Jove, M.; Ortega, F.; Xifra, G.; Ricart, W.; Obis, È.; Pamplona, R.; Portero-Otin, M.; Fernández-Real, J.M. Metabolomics uncovers the role of adipose tissue PDXK in adipogenesis and systemic insulin sensitivity. Diabetologia 2016, 59, 822–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascolo, E.; Barile, A.; Mecarelli, L.S.; Amoroso, N.; Merigliano, C.; Massimi, A.; Saggio, I.; Hansen, T.; Tramonti, A.; Di Salvo, M.L.; et al. The expression of four pyridoxal kinase (PDXK) human variants in Drosophila impacts on genome integrity. Sci. Rep. 2019, 9, 14188. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Genetic and hormonal regulation of tryptophan kynurenine metabolism: Implications for vascular cognitive impairment, major depressive disorder, and aging. Ann. N. Y. Acad. Sci. 2007, 1122, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Van de Kamp, J.L.; Smolen, A. Response of kynurenine pathway enzymes to pregnancy and dietary level of vitamin B-6. Pharmacol. Biochem. Behav. 1995, 51, 753–758. [Google Scholar] [CrossRef]
- Bender, D.A.; Njagi, E.N.; Danielian, P.S. Tryptophan metabolism in vitamin B6-deficient mice. Br. J. Nutr. 1990, 63, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Rios-Avila, L.; Nijhout, H.F.; Reed, M.C.; Sitren, H.S.; Gregory, J.F., 3rd. A mathematical model of tryptophan metabolism via the kynurenine pathway provides insights into the effects of vitamin B-6 deficiency, tryptophan loading, and induction of tryptophan 2,3-dioxygenase on tryptophan metabolites. J. Nutr. 2013, 143, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Yess, N.; Price, J.M.; Brown, R.R.; Swan, P.B.; Linkswiler, H. Vitamin B6 depletion in man: urinary excretion of tryptophan metabolites. J. Nutr. 1964, 84, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, F.; Tsubouchi, R.; Izuta, S.; Shibata, Y. Kynurenine metabolism and xanthurenic acid formation in vitamin B6-deficient rat after tryptophan injection. J. Nutr. Sci. Vitaminol. 1989, 35, 111–122. [Google Scholar] [CrossRef]
- Connick, J.H.; Stone, T.W. The role of kynurenines in diabetes mellitus. Med. Hypotheses 1985, 18, 371–376. [Google Scholar] [CrossRef]
- Hattori, M.; Kotake, Y.; Kotake, Y. Studies on the urinary excretion of xanthurenic acid in diabetics. Acta Vitaminol. Enzymol. 1984, 6, 221–228. [Google Scholar] [PubMed]
- Ikeda, S.; Kotake, Y. Urinary excretion of xanthurenic acid and zinc in diabetes: (3). Occurrence of xanthurenic acid-Zn2+ complex in urine of diabetic patients and of experimentally-diabetic rats. Ital. J. Biochem. 1986, 35, 232–241. [Google Scholar] [PubMed]
- Akarte, N.R.; Shastri, N.V. Studies on tryptophan-niacin metabolism in streptozotocin diabetic rats. Diabetes 1974, 23, 977–981. [Google Scholar] [CrossRef]
- Patterson, A.D.; Bonzo, J.A.; Li, F.; Krausz, K.W.; Eichler, G.S.; Aslam, S.; Tigno, X.; Weinstein, J.N.; Hansen, B.C.; Idle, J.R.; et al. Metabolomics reveals attenuation of the SLC6A20 kidney transporter in nonhuman primate and mouse models of type 2 diabetes mellitus. J. Biol. Chem. 2011, 286, 19511–19522. [Google Scholar] [CrossRef] [Green Version]
- Favennec, M.; Hennart, B.; Caiazzo, R.; Leloire, A.; Yengo, L.; Verbanck, M.; Arredouani, A.; Marre, M.; Pigeyre, M.; Bessede, A.; et al. The kynurenine pathway is activated in human obesity and shifted toward kynurenine monooxygenase activation. Obesity 2015, 23, 2066–2074. [Google Scholar] [CrossRef]
- Manusadzhian, V.G.; Kniazev Iu, A.; Vakhrusheva, L.L. [Mass spectrometric identification of xanthurenic acid in pre-diabetes]. Vopr. Meditsinskoi Khimii 1974, 20, 95–97. [Google Scholar]
- Oxenkrug, G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y. Xanthurenic acid, an abnormal metabolite of tryptophan and the diabetic symptoms caused in albino rats by its production. J. Vitaminol. 1955, 1, 73–87. [Google Scholar] [CrossRef]
- Kotake, Y.; Ueda, T.; Mori, T.; Igaki, S.; Hattori, M. Abnormal tryptophan metabolism and experimental diabetes by xanthurenic acid (XA). Acta Vitaminol. Enzymol. 1975, 29, 236–239. [Google Scholar]
- Meyramov, G.; Korchin, V.; Kocheryzkina, N. Diabetogenic activity of xanturenic acid determined by its chelating properties? Transplant. Proc. 1998, 30, 2682–2684. [Google Scholar] [CrossRef]
- Rogers, K.S.; Evangelista, S.J. 3-Hydroxykynurenine, 3-hydroxyanthranilic acid, and o-aminophenol inhibit leucine-stimulated insulin release from rat pancreatic islets. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1985, 178, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Malina, H.Z.; Richter, C.; Mehl, M.; Hess, O.M. Pathological apoptosis by xanthurenic acid, a tryptophan metabolite: Activation of cell caspases but not cytoskeleton breakdown. BMC Physiol. 2001, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Munipally, P.K.; Agraharm, S.G.; Valavala, V.K.; Gundae, S.; Turlapati, N.R. Evaluation of indoleamine 2,3-dioxygenase expression and kynurenine pathway metabolites levels in serum samples of diabetic retinopathy patients. Arch. Physiol. Biochem. 2011, 117, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Velagapudi, C.; Redus, L.; Thameem, F.; Kasinath, B.; Hura, C.E.; Lorenzo, C.; Abboud, H.E.; O’Connor, J.C. Tryptophan Metabolism in Patients with Chronic Kidney Disease Secondary to Type 2 Diabetes: Relationship to Inflammatory Markers. Int. J. Tryptophan Res. 2017, 10, 1178646917694600. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M. Fatty acids and glucolipotoxicity in the pathogenesis of Type 2 diabetes. Biochem. Soc. Trans. 2008, 36, 348–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Dubois, S.G.; Heilbronn, L.K.; Smith, S.R.; Albu, J.B.; Kelley, D.E.; Ravussin, E. Decreased expression of adipogenic genes in obese subjects with type 2 diabetes. Obesity 2006, 14, 1543–1552. [Google Scholar] [CrossRef]
- Huber, A.M.; Gershoff, S.N.; Hegsted, D.M. Carbohydrate and fat metabolism and response to insulin in vitamin B6-deficient rats. J. Nutr. 1964, 82, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Ribaya, J.D.; Gershoff, S.N. Effects of vitamin B6 deficiency on liver, kidney, and adipose tissue enzymes associated with carbohydrate and lipid metabolism, and on glucose uptake by rat epididymal adipose tissue. J. Nutr. 1977, 107, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnamurty, R.; Angel, J.F.; Sabry, Z.I. Response of lipogenesis to repletion in the pyridoxine-deficient rat. J. Nutr. 1968, 95, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanaka, N.; Kanda, M.; Toya, K.; Suehiro, H.; Kato, N. Vitamin B6 regulates mRNA expression of peroxisome proliferator-activated receptor-γ target genes. Exp. Ther. Med. 2011, 2, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, Y.; Kumoto, T.; Suehiro, H.; Nishimura, F.; Kato, N.; Hata, Y.; Sorisky, A.; Yanaka, N. RASSF6 expression in adipocytes is down-regulated by interaction with macrophages. PLoS ONE 2013, 8, e61931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, Y.; Kumoto, T.; Suehiro, H.; Yamamoto, T.; Nishimura, F.; Kato, N.; Yanaka, N. IκB kinase epsilon expression in adipocytes is upregulated by interaction with macrophages. Biosci. Biotechnol. Biochem. 2014, 78, 1357–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasheim, E.T.; Hofsø, D.; Hjelmesaeth, J.; Birkeland, K.I.; Bøhmer, T. Vitamin status in morbidly obese patients: A cross-sectional study. Am. J. Clin. Nutr. 2008, 87, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Huq, M.D.; Tsai, N.P.; Lin, Y.P.; Higgins, L.; Wei, L.N. Vitamin B6 conjugation to nuclear corepressor RIP140 and its role in gene regulation. Nat. Chem. Biol. 2007, 3, 161–165. [Google Scholar] [CrossRef]
- Bird, R.P. The Emerging Role of Vitamin B6 in Inflammation and Carcinogenesis. Adv. Food Nutr. Res. 2018, 83, 151–194. [Google Scholar]
- Nilsson, E.; Jansson, P.A.; Perfilyev, A.; Volkov, P.; Pedersen, M.; Svensson, M.K.; Poulsen, P.; Ribel-Madsen, R.; Pedersen, N.L.; Almgren, P.; et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014, 63, 2962–2976. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, P.; Zhao, Z.H.; Zhang, Y.; Ma, Z.M.; Wang, S.X. Vitamin B6 Prevents Endothelial Dysfunction, Insulin Resistance, and Hepatic Lipid Accumulation in Apoe (−/−) Mice Fed with High-Fat Diet. J. Diabetes Res. 2016, 2016, 1748065. [Google Scholar] [CrossRef] [Green Version]
- Meigs, J.B.; Jacques, P.F.; Selhub, J.; Singer, D.E.; Nathan, D.M.; Rifai, N.; D’Agostino, R.B., Sr.; Wilson, P.W. Fasting plasma homocysteine levels in the insulin resistance syndrome: The Framingham offspring study. Diabetes Care 2001, 24, 1403–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ala, O.A.; Akintunde, A.A.; Ikem, R.T.; Kolawole, B.A.; Ala, O.O.; Adedeji, T.A. Association between insulin resistance and total plasma homocysteine levels in type 2 diabetes mellitus patients in south west Nigeria. Diabetes Metab. Syndr. 2017, 11 (Suppl. 2), S803–S809. [Google Scholar] [CrossRef]
- Azzini, E.; Ruggeri, S.; Polito, A. Homocysteine: Its Possible Emerging Role in At-Risk Population Groups. Int. J. Mol. Sci. 2020, 21, 1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, T.; Ben Ya’acov, A.; Shabat, Y.; Zolotarovya, L.; Snir, R.; Ilan, Y. Altered distribution of regulatory lymphocytes by oral administration of soy-extracts exerts a hepatoprotective effect alleviating immune mediated liver injury, non-alcoholic steatohepatitis and insulin resistance. World J. Gastroenterol. 2015, 21, 7443–7456. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Zheng, S.R.; Wang, D.M. Adrenomedullin: An important participant in neurological diseases. Neural Regen. Res. 2020, 15, 1199–1207. [Google Scholar] [PubMed]
- Zhao, M.; Lamers, Y.; Ralat, M.A.; Coats, B.S.; Chi, Y.Y.; Muller, K.E.; Bain, J.R.; Shankar, M.N.; Newgard, C.B.; Stacpoole, P.W.; et al. Marginal vitamin B-6 deficiency decreases plasma (n-3) and (n-6) PUFA concentrations in healthy men and women. J. Nutr. 2012, 142, 1791–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Feng, J.; Peng, Q.; Liu, X.; Fan, Z. Advanced Glycation End Products: Potential Mechanism and Therapeutic Target in Cardiovascular Complications under Diabetes. Oxidative Med. Cell. Longev. 2019, 2019, 9570616. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.M.S.; Nunes, E.A.; Lago, L.; Pedron, C.N.; Manieri, T.M.; Sato, R.H.; Oliveira, V.X.J.; Cerchiaro, G. Generation of Advanced Glycation End-Products (AGEs) by glycoxidation mediated by copper and ROS in a human serum albumin (HSA) model peptide: Reaction mechanism and damage in motor neuron cells. Mutat. Res. 2017, 824, 42–51. [Google Scholar] [CrossRef]
- Deo, P.; McCullough, C.L.; Almond, T.; Jaunay, E.L.; Donnellan, L.; Dhillon, V.S.; Fenech, M. Dietary sugars and related endogenous advanced glycation end-products increase chromosomal DNA damage in WIL2-NS cells, measured using cytokinesis-block micronucleus cytome assay. Mutagenesis 2020, 35, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.M.; Folkers, K.; Minadeo, M.; VanBuskirk, R.; Xia, L.J.; Tamagawa, H. A deficiency of vitamin B6 is a plausible molecular basis of the retinopathy of patients with diabetes mellitus. Biochem. Biophys. Res. Commun. 1991, 179, 615–619. [Google Scholar] [CrossRef]
- Cohen, K.L.; Gorecki, G.A.; Silverstein, S.B.; Ebersole, J.S.; Solomon, L.R. Effect of pyridoxine (vitamin B6) on diabetic patients with peripheral neuropathy. J. Am. Podiatry Assoc. 1984, 74, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Li, H.; Adijiang, A.; Pischetsrieder, M.; Niwa, T. Pyridoxal phosphate prevents progression of diabetic nephropathy. Nephrol. Dial. Transplant. 2007, 22, 2165–2174. [Google Scholar] [CrossRef] [Green Version]
- Elbarbary, N.S.; Ismail, E.A.R.; Zaki, M.A.; Darwish, Y.W.; Ibrahim, M.Z.; El-Hamamsy, M. Vitamin B complex supplementation as a homocysteine-lowering therapy for early stage diabetic nephropathy in pediatric patients with type 1 diabetes: A randomized controlled trial. Clin. Nutr. 2020, 39, 49–56. [Google Scholar] [CrossRef]
- Horikawa, C.; Aida, R.; Kamada, C.; Fujihara, K.; Tanaka, S.; Tanaka, S.; Araki, A.; Yoshimura, Y.; Moriya, T.; Akanuma, Y.; et al. Vitamin B6 intake and incidence of diabetic retinopathy in Japanese patients with type 2 diabetes: Analysis of data from the Japan Diabetes Complications Study (JDCS). Eur. J. Nutr. 2019. [Google Scholar] [CrossRef]
- Stitt, A.; Gardiner, T.A.; Alderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 2002, 51, 2826–2832. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiazza, F.; Cento, A.S.; Collotta, D.; Nigro, D.; Rosa, G.; Baratta, F.; Bitonto, V.; Cutrin, J.C.; Aragno, M.; Mastrocola, R.; et al. Protective Effects of Pyridoxamine Supplementation in the Early Stages of Diet-Induced Kidney Dysfunction. Biomed. Res. Int. 2017, 2017, 2682861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muellenbach, E.A.; Diehl, C.J.; Teachey, M.K.; Lindborg, K.A.; Archuleta, T.L.; Harrell, N.B.; Andersen, G.; Somoza, V.; Hasselwander, O.; Matuschek, M.; et al. Interactions of the advanced glycation end product inhibitor pyridoxamine and the antioxidant alpha-lipoic acid on insulin resistance in the obese Zucker rat. Metabolism 2008, 57, 1465–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, K.M.; Abul Qais, F.; Hasan, H.; Naseem, I. Anti-diabetic study of vitamin B6 on hyperglycaemia induced protein carbonylation, DNA damage and ROS production in alloxan induced diabetic rats. Toxicol. Res. 2019, 8, 568–579. [Google Scholar] [CrossRef]
- Nakamura, S.; Niwa, T. Pyridoxal phosphate and hepatocyte growth factor prevent dialysate-induced peritoneal damage. J. Am. Soc. Nephrol. 2005, 16, 144–150. [Google Scholar] [CrossRef]
- Adrover, M.; Vilanova, B.; Frau, J.; Muñoz, F.; Donoso, J. The pyridoxamine action on Amadori compounds: A reexamination of its scavenging capacity and chelating effect. Bioorganic Med. Chem. 2008, 16, 5557–5569. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Adrover, M.; Frau, J.; Salvà, A.; Donoso, J.; Muñoz, F. DFT studies on Schiff base formation of vitamin B6 analogues. Reaction between a pyridoxamine-analogue and carbonyl compounds. J. Phys. Chem. A 2010, 114, 4634–4640. [Google Scholar] [CrossRef]
- Ramis, R.; Ortega-Castro, J.; Caballero, C.; Casasnovas, R.; Cerrillo, A.; Vilanova, B.; Adrover, M.; Frau, J. How Does Pyridoxamine Inhibit the Formation of Advanced Glycation End Products? The Role of Its Primary Antioxidant Activity. Antioxidants 2019, 8, 344. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.J.; Boyd, A.C.; O’Harte, F.P.; McKillop, A.M.; Wiggam, M.I.; Mooney, M.H.; McCluskey, J.T.; Lindsay, J.R.; Ennis, C.N.; Gamble, R.; et al. Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes 2003, 52, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [Green Version]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravi, M.C.; Armiento, A.; Laurenti, O.; Cassone-Faldetta, M.; De Luca, O.; Moretti, A.; De Mattia, G. Insulin decreases intracellular oxidative stress in patients with type 2 diabetes mellitus. Metabolism 2006, 55, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Arabski, M.; Krupa, R.; Wozniak, K.; Zadrozny, M.; Kasznicki, J.; Zurawska, M.; Drzewoski, J. DNA damage and repair in type 2 diabetes mellitus. Mutat. Res. 2004, 554, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Zhong, A.; Chang, M.; Yu, T.; Gau, R.; Riley, D.J.; Chen, Y.; Chen, P.L. Aberrant DNA damage response and DNA repair pathway in high glucose conditions. J. Cancer Res. Updates 2018, 7, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, M.T.; Navidi, A.A.; Rezaei, M.; Babahmadi-Rezaei, H. Oxidative damage to DNA and lipids: Correlation with protein glycation in patients with type 1 diabetes. J. Clin. Lab. Anal. 2010, 24, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Tatsch, E.; Bochi, G.V.; Piva, S.J.; De Carvalho, J.A.; Kober, H.; Torbitz, V.D.; Duarte, T.; Signor, C.; Coelho, A.C.; Duarte, M.M.; et al. Association between DNA strand breakage and oxidative, inflammatory and endothelial biomarkers in type 2 diabetes. Mutat. Res. 2012, 732, 16–20. [Google Scholar] [CrossRef]
- Umemura, T.; Sai, K.; Takagi, A.; Hasegawa, R.; Kurokawa, Y. Formation of 8-hydroxydeoxyguanosine (8-OH-dG) in rat kidney DNA after intraperitoneal administration of ferric nitrilotriacetate (Fe-NTA). Carcinogenesis 1990, 11, 345–347. [Google Scholar] [CrossRef]
- Dandona, P.; Thusu, K.; Cook, S.; Snyder, B.; Makowski, J.; Armstrong, D.; Nicotera, T. Oxidative damage to DNA in diabetes mellitus. Lancet 1996, 347, 444–445. [Google Scholar] [CrossRef]
- Hinokio, Y.; Suzuki, S.; Hirai, M.; Chiba, M.; Hirai, A.; Toyota, T. Oxidative DNA damage in diabetes mellitus: Its association with diabetic complications. Diabetologia 1999, 42, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Binici, D.N.; Karaman, A.; Coşkun, M.; Oğlu, A.U.; Uçar, F. Genomic damage in patients with type-2 diabetes mellitus. Genet. Couns. 2013, 24, 149–156. [Google Scholar]
- Boehm, B.O.; Möller, P.; Högel, J.; Winkelmann, B.R.; Renner, W.; Rosinger, S.; Seelhorst, U.; Wellnitz, B.; März, W.; Melzner, J.; et al. Lymphocytes of type 2 diabetic women carry a high load of stable chromosomal aberrations: A novel risk factor for disease-related early death. Diabetes 2008, 57, 2950–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Pérez, L.M.; Cerda-Flores, R.M.; Gallegos-Cabriales, E.C.; Dávila-Rodríguez, M.I.; Ibarra-Costilla, E.; Cortés-Gutiérrez, E.I. Frequency of micronuclei in Mexicans with type 2 diabetes mellitus. Prague Med. Rep. 2007, 108, 248–255. [Google Scholar] [PubMed]
- Grindel, A.; Brath, H.; Nersesyan, A.; Knasmueller, S.; Wagner, K.H. Association of Genomic Instability with HbA1c levels and Medication in Diabetic Patients. Sci. Rep. 2017, 7, 41985. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S.; Briarava, M.; Pilati, P. Vitamin B6 and Cancer Risk: A Field Synopsis and Meta-Analysis. J. Natl. Cancer Inst. 2017, 109, 1–9. [Google Scholar] [CrossRef]
- Zuo, H.; Ueland, P.M.; Midttun, Ø.; Tell, G.S.; Fanidi, A.; Zheng, W.; Shu, X.; Xiang, Y.; Wu, J.; Prentice, R.; et al. Vitamin B6 catabolism and lung cancer risk: Results from the Lung Cancer Cohort Consortium (LC3). Ann. Oncol. 2019, 30, 478–485. [Google Scholar] [CrossRef]
- Gylling, B.; Myte, R.; Schneede, J.; Hallmans, G.; Häggström, J.; Johansson, I.; Ulvik, A.; Ueland, P.M.; Van Guelpen, B.; Palmqvist, R. Vitamin B-6 and colorectal cancer risk: A prospective population-based study using 3 distinct plasma markers of vitamin B-6 status. Am. J. Clin. Nutr. 2017, 105, 897–904. [Google Scholar] [CrossRef]
- Vigneri, P.; Frasca, F.; Sciacca, L.; Pandini, G.; Vigneri, R. Diabetes and cancer. Endocr. Relat. Cancer 2009, 16, 1103–1123. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascolo, E.; Vernì, F. Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 3669. https://doi.org/10.3390/ijms21103669
Mascolo E, Vernì F. Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms. International Journal of Molecular Sciences. 2020; 21(10):3669. https://doi.org/10.3390/ijms21103669
Chicago/Turabian StyleMascolo, Elisa, and Fiammetta Vernì. 2020. "Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms" International Journal of Molecular Sciences 21, no. 10: 3669. https://doi.org/10.3390/ijms21103669
APA StyleMascolo, E., & Vernì, F. (2020). Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms. International Journal of Molecular Sciences, 21(10), 3669. https://doi.org/10.3390/ijms21103669