ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS


 , ,
, ,  and
and 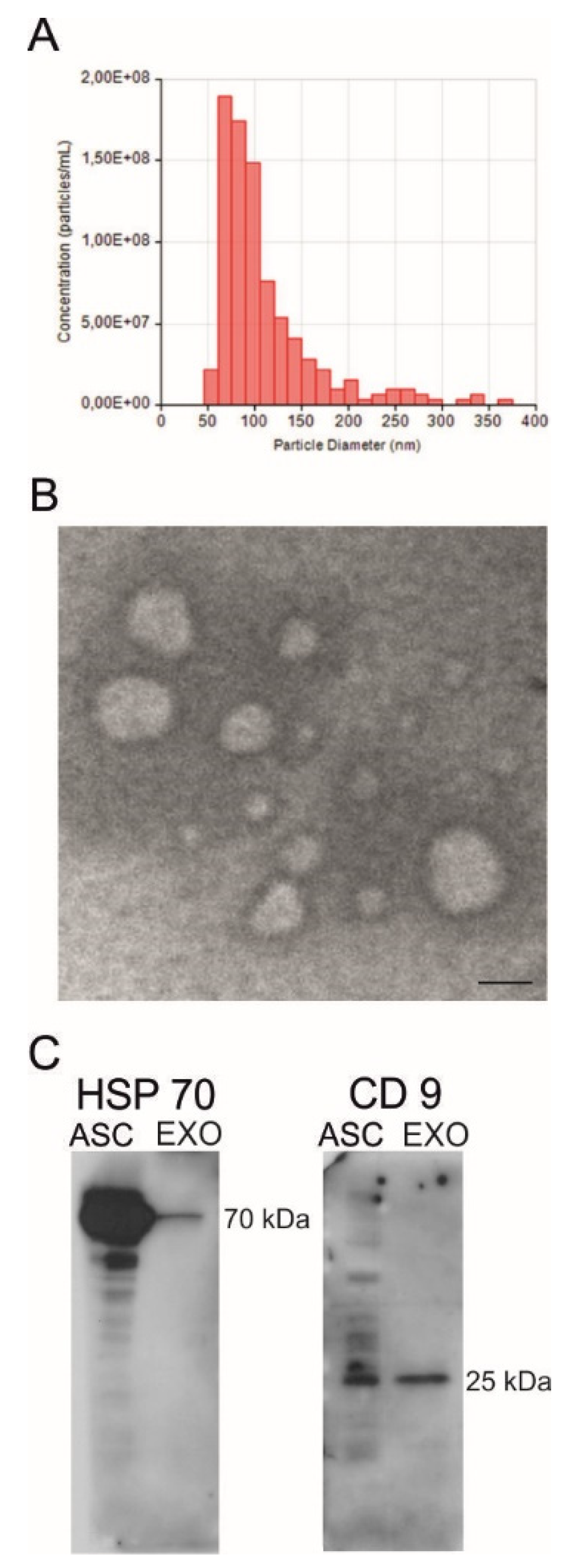{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Isolation and Characterization of ASC-Exosomes
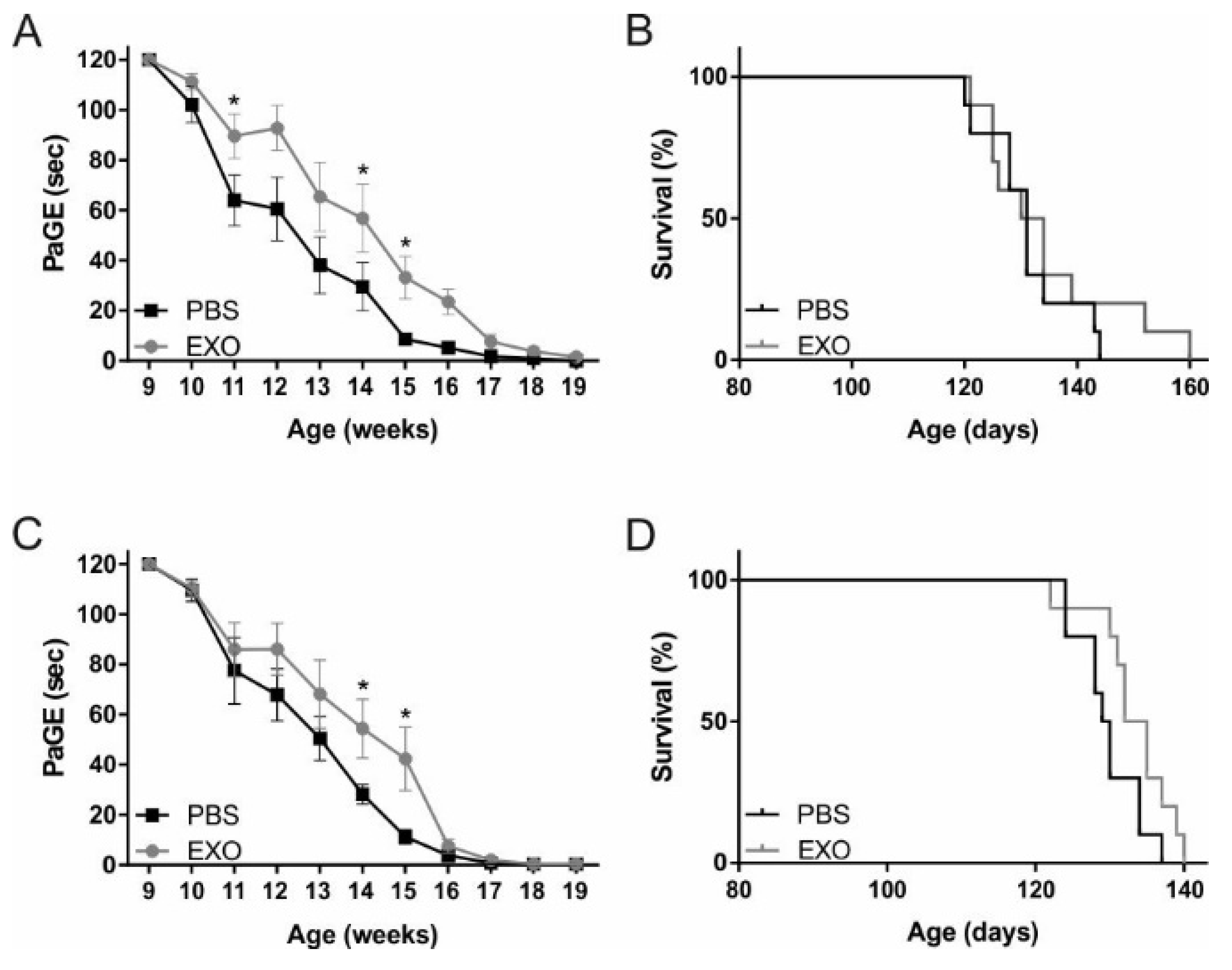
2.2. ASC-Exosomes Administration Improves Motor Performance of SOD1(G93A) Mice
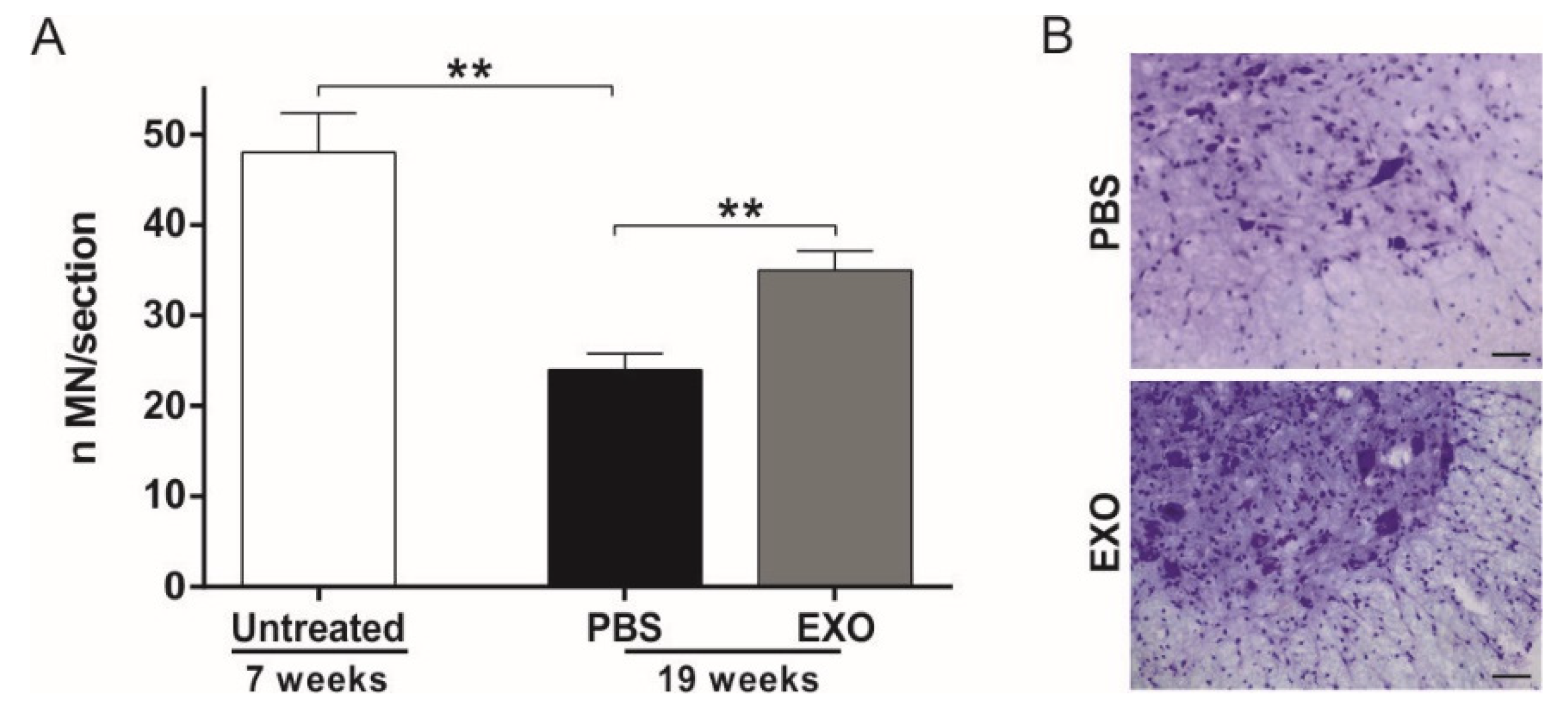
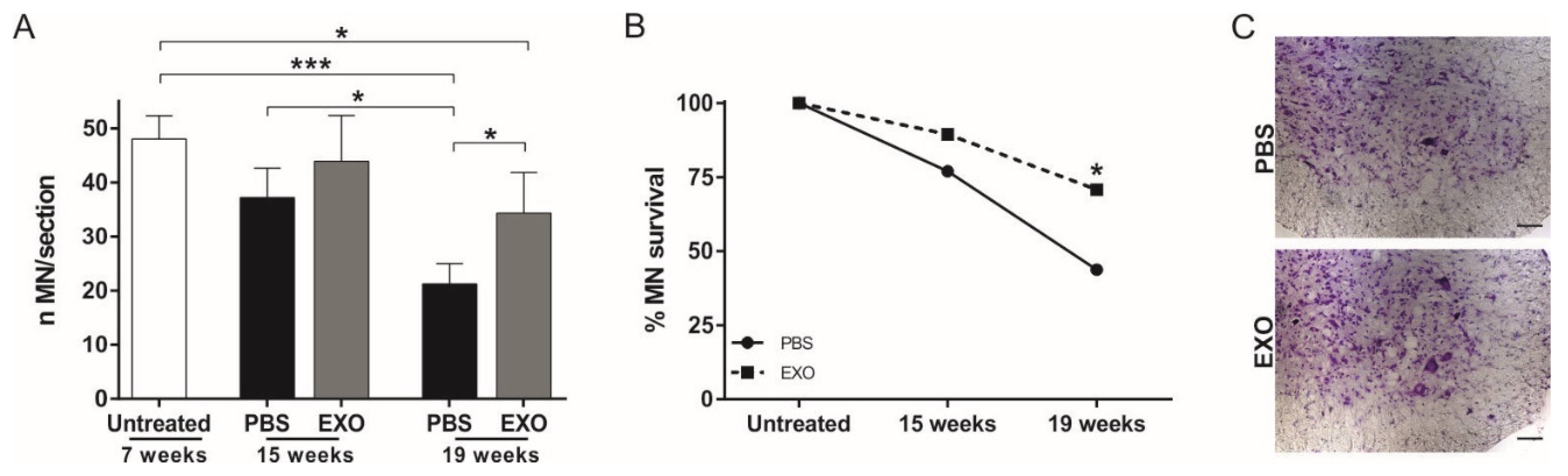
2.3. ASC-Exosomes Administration Protects Lumbar Spinal Cord MN from Neurodegeneration
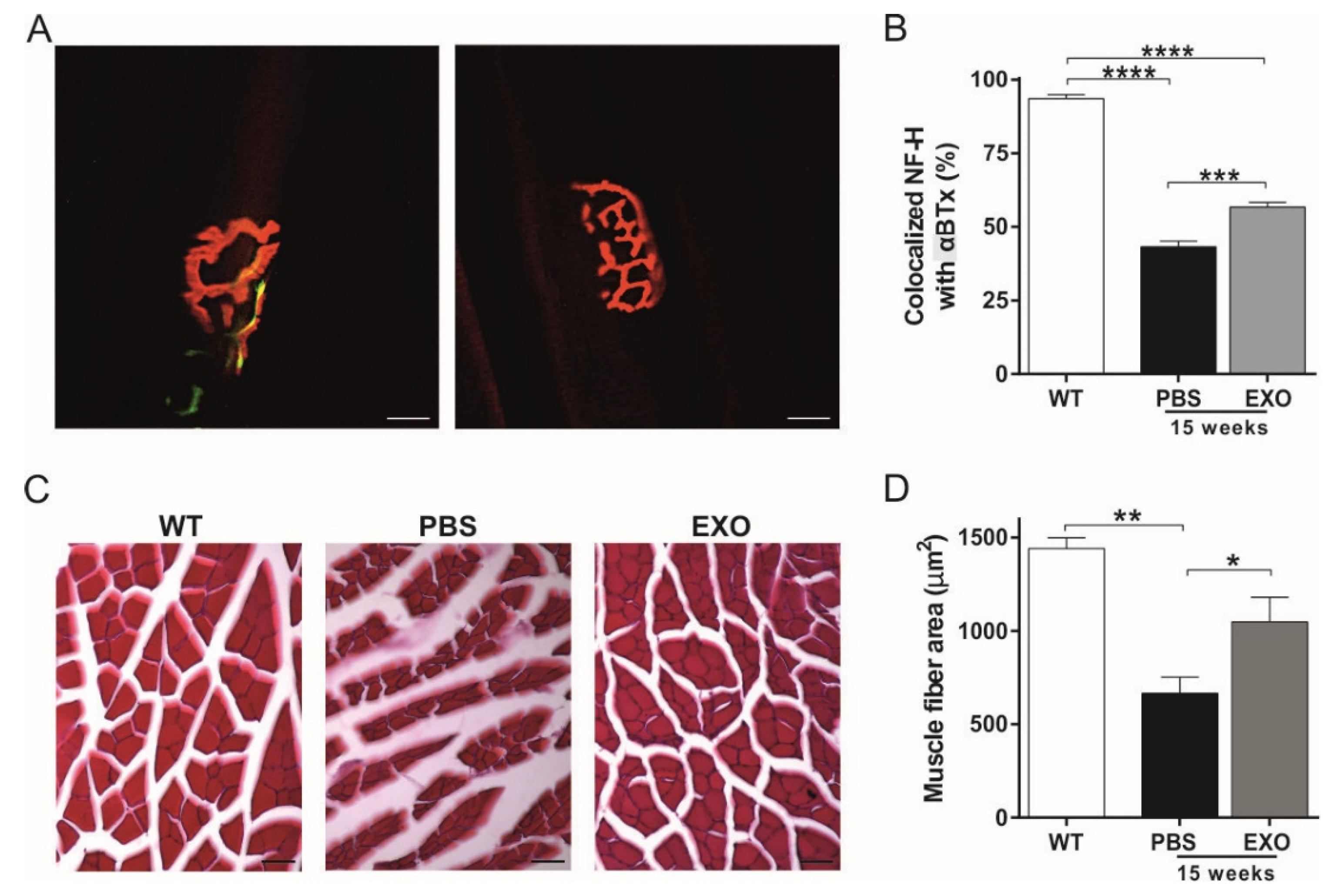
2.4. ASC-Exosomes Administration Preserves Neuromuscular Junction Functionality and Skeletal Muscle Fiber Morphology
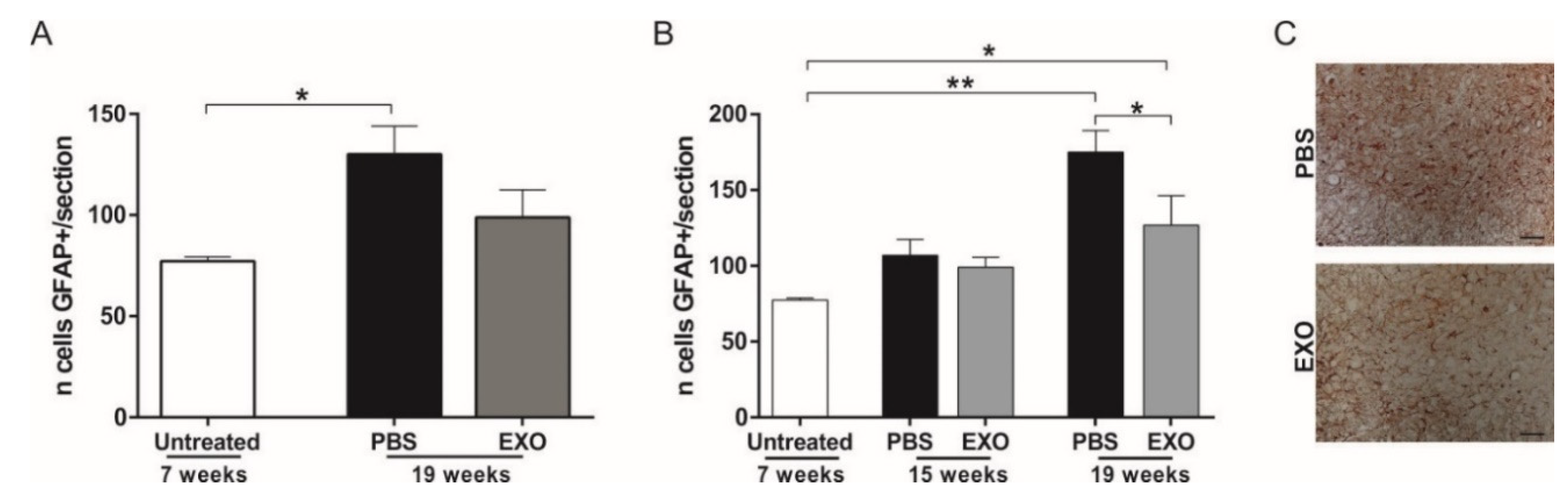
2.5. Effect of ASC-Exosomes Administration on Glial Cells
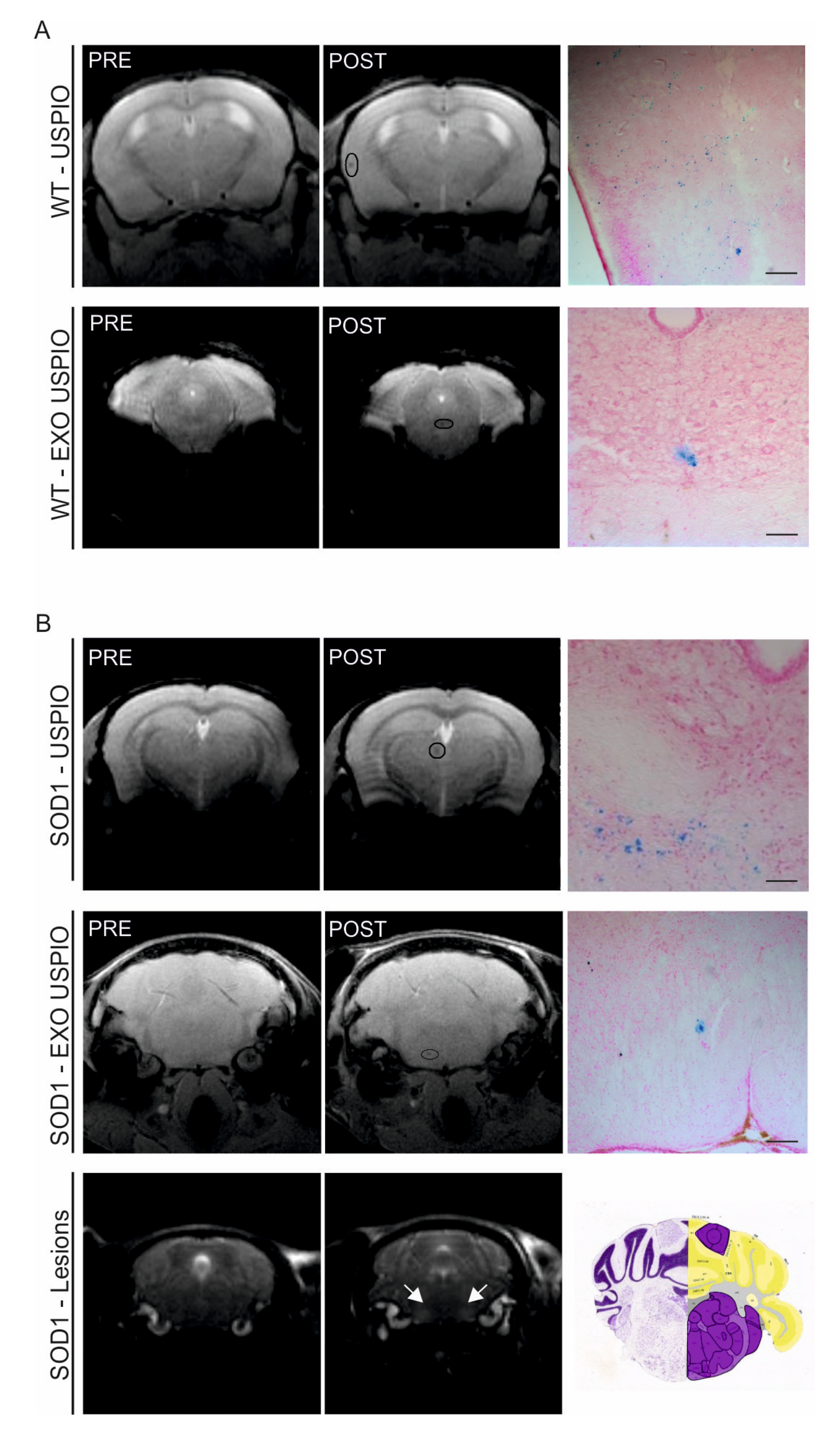
2.6. ASC-Exosomes Selectively Reach the Lesioned Area of the Brain in SOD1(G93A) Mice
3. Discussion
4. Materials and Methods
4.1. ASC Cell Cultures
4.2. ASC-Exosomes
4.3. Electron Microscopy and Western Blot
4.4. Animals
4.5. Motor Tests
4.6. Animal Treatments
4.7. Lumbar Spinal Cord Motoneurons Stereological Count
4.8. Immunohistochemistry of Lumbar Spinal Cord
4.9. Immunohistochemistry of Neuromuscular Junction and Hematoxylin-eosin Staining
4.10. Statistical Analysis
4.11. Exosomes-USPIO or USPIO Administration
4.12. In Vivo MRI
4.13. Histological Analysis after Exosomes-USPIO Administration
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| ASC | Adipose-derived stem cells |
| ASC-exosomes | Exosomes isolated from ASC |
| αBTx | α-bungarotoxin |
| Exosomes-USPIO | Exosomes labeled with ultra-small superparamagnetic iron oxide nanoparticles |
| i.n. | Intranasal |
| i.v. | Intravenous |
| MN | Motoneurons |
| MNJ | Neuromuscular junction |
| MRI | Magnetic resonance imaging |
| MSC | Mesenchymal stem cells |
| PaGE | Paw grip endurance test |
| SOD1 | Cu2+/Zn2+ superoxide dismutase |
| SOD1(G93A) | Human SOD1 gene with a G93A mutation |
| USPIO | Ultra-small superparamagnetic iron oxide nanoparticles |
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; de Carvalho, M. Emerging molecular biomarker targets for amyotrophic lateral sclerosis. Clin. Chim. Acta 2016, 455, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Bonafede, R.; Mariotti, R. ALS Pathogenesis and Therapeutic Approaches: The Role of Mesenchymal Stem Cells and Extracellular Vesicles. Front. Cell. Neurosci. 2017, 11, 157. [Google Scholar] [CrossRef] [PubMed]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Moore, V.D.G.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; E Milligan, C. Characterization of early pathogenesis in the SOD1G93A mouse model of ALS: Part I, background and methods. Brain Behav. 2013, 3, 335–350. [Google Scholar] [CrossRef]
- Benatar, M. Lost in translation: Treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 2007, 26, 1–13. [Google Scholar] [CrossRef]
- Meamar, R.; Nasr-Esfahani, M.H.; Mousavi, S.A.; Basiri, K. Stem cell therapy in amyotrophic lateral sclerosis. J. Clin. Neurosci. 2013, 20, 1659–1663. [Google Scholar] [CrossRef]
- Boucherie, C.; Schäfer, S.; Lavand’Homme, P.; Maloteaux, J.-M.; Hermans, E. Chimerization of astroglial population in the lumbar spinal cord after mesenchymal stem cell transplantation prolongs survival in a rat model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2009, 87, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- Marconi, S.; Bonaconsa, M.; Scambi, I.; Squintani, G.; Rui, W.; Turano, E.; Ungaro, D.; D’Agostino, S.; Barbieri, F.; Angiari, S.; et al. Systemic treatment with adipose-derived mesenchymal stem cells ameliorates clinical and pathological features in the amyotrophic lateral sclerosis murine model. Neuroscience 2013, 248, 333–343. [Google Scholar] [CrossRef]
- De Munter, J.P.; Shafarevich, I.; Liundup, A.; Pavlov, D.; Wolters, E.C.; Gorlova, A.; Veniaminova, E.; Umriukhin, A.; Kalueff, A.V.; Svistunov, A.; et al. Neuro-Cells therapy improves motor outcomes and suppresses inflammation during experimental syndrome of amyotrophic lateral sclerosis in mice. CNS Neurosci. Ther. 2019, 26, 504–517. [Google Scholar] [CrossRef]
- Martínez-Muriana, A.; Pastor, D.; Mancuso, R.; Rando, A.; Osta, R.; Martínez, S.; López-Vales, R.; Navarro, X. Combined intramuscular and intraspinal transplant of bone marrow cells improves neuromuscular function in the SOD1G93A mice. Stem Cell Res. Ther. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, M.; Tian, W. Physiological and pathological impact of exosomes of adipose tissue. Cell Prolif. 2016, 49, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Yeo, R.W.Y.; Lim, S.K. Mesenchymal stem cell exosomes. Semin. Cell Dev. Biol. 2015, 40, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Tyagi, N. Exosomes in neurological disease, neuroprotection, repair and therapeutics: Problems and perspectives. Neural Regen. Res. 2015, 10, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Marzola, P.; Busato, A.; Bonafede, R.; Bontempi, P.; Scambi, I.; Schiaffino, L.; Benati, D.; Malatesta, M.; Sbarbati, A.; Mariotti, R. Magnetic resonance imaging of ultrasmall superparamagnetic iron oxide-labeled exosomes from stem cells: A new method to obtain labeled exosomes. Int. J. Nanomed. 2016, 11, 2481–2490. [Google Scholar] [CrossRef]
- Angenstein, F.; Niessen, H.G.; Goldschmidt, J.; Vielhaber, S.; Ludolph, A.C.; Scheich, H. Age-dependent changes in MRI of motor brain stem nuclei in a mouse model of ALS. NeuroReport 2004, 15, 2271–2274. [Google Scholar] [CrossRef]
- Bontempi, P.; Busato, A.; Bonafede, R.; Schiaffino, L.; Scambi, I.; Sbarbati, A.; Mariotti, R.; Marzola, P. MRI reveals therapeutical efficacy of stem cells: An experimental study on the SOD1(G93A) animal model. Magn. Reson. Med. 2017, 79, 459–469. [Google Scholar] [CrossRef]
- Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS-A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. [Google Scholar] [CrossRef]
- Abati, E.; Bresolin, N.; Comi, G.; Corti, S. Advances, Challenges, and Perspectives in Translational Stem Cell Therapy for Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 6703–6715. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2018, 39, 733–748. [Google Scholar] [CrossRef]
- Phinney, D.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Steam Cells 2017, 35, 851–858. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Kiaie, N.; Barreto, G.E.; Read, M.I.; Tafti, H.A.; Sahebkar, A. The Therapeutic Potential of Mesenchymal Stem Cell–Derived Exosomes in Treatment of Neurodegenerative Diseases. Mol. Neurobiol. 2019, 56, 8157–8167. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar] [CrossRef] [PubMed]
- Jarmalavičiūtė, A.; Tunaitis, V.; Pivoraitė, U.; Venalis, A.; Pivoriunas, A. Exosomes from dental pulp stem cells rescue human dopaminergic neurons from 6-hydroxy-dopamine–induced apoptosis. Cytotherapy 2015, 17, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Bonafede, R.; Scambi, I.; Peroni, D.; Potrich, V.; Boschi, F.; Benati, D.; Bonetti, B.; Mariotti, R. Exosome derived from murine adipose-derived stromal cells: Neuroprotective effect on in vitro model of amyotrophic lateral sclerosis. Exp. Cell Res. 2016, 340, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Bonafede, R.; Brandi, J.; Manfredi, M.; Scambi, I.; Schiaffino, L.; Merigo, F.; Turano, E.; Bonetti, B.; Marengo, E.; Cecconi, D.; et al. The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis. Cells 2019, 8, 1087. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Li, Y.; Cui, Y.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic Administration of Exosomes Released from Mesenchymal Stromal Cells Promote Functional Recovery and Neurovascular Plasticity After Stroke in Rats. Br. J. Pharmacol. 2013, 33, 1711–1715. [Google Scholar] [CrossRef]
- Zhang, Y.; Chopp, M.; Meng, Y.; Katakowski, M.; Xin, H.; Mahmood, A.; Xiong, Y. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J. Neurosurg. 2015, 122, 856–867. [Google Scholar] [CrossRef]
- Farinazzo, A.; Angiari, S.; Turano, E.; Bistaffa, E.; Dusi, S.; Ruggieri, S.; Bonafede, R.; Mariotti, R.; Constantin, G.; Bonetti, B. Nanovesicles from adipose-derived mesenchymal stem cells inhibit T lymphocyte trafficking and ameliorate chronic experimental autoimmune encephalomyelitis. Sci. Rep. 2018, 8, 7473. [Google Scholar] [CrossRef]
- Long, Q.; Upadhya, D.; Hattiangady, B.; Kim, D.-K.; An, S.Y.; Shuai, B.; Prockop, D.J.; Shetty, A.K. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc. Natl. Acad. Sci. USA 2017, 114, 3536–3545. [Google Scholar] [CrossRef]
- Ding, M.; Shen, Y.; Wang, P.; Xie, Z.; Xu, S.; Zhu, Z.; Wang, Y.; Lyu, Y.; Wang, D.; Xu, L.; et al. Exosomes Isolated From Human Umbilical Cord Mesenchymal Stem Cells Alleviate Neuroinflammation and Reduce Amyloid-Beta Deposition by Modulating Microglial Activation in Alzheimer’s Disease. Neurochem. Res. 2018, 43, 2165–2177. [Google Scholar] [CrossRef]
- Sun, X.; Jung, J.-H.; Arvola, O.; Santoso, M.R.; Giffard, R.G.; Yang, P.C.; Stary, C.M. Stem Cell-Derived Exosomes Protect Astrocyte Cultures From in vitro Ischemia and Decrease Injury as Post-stroke Intravenous Therapy. Front. Cell. Neurosci. 2019, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.-H.; Wei, J.; Lu, M.-Q.; Jin, M.-Y.; Geng, H.-L. Protective effect of human umbilical cord mesenchymal stem cell exosomes on preserving the morphology and angiogenesis of placenta in rats with preeclampsia. Biomed. Pharmacother. 2018, 105, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Zhang, L.; Duan, L.; Wang, X.; Min, Y.; Yu, H. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J. Mol. Med. 2013, 92, 387–397. [Google Scholar] [CrossRef]
- Nojima, H.; Freeman, C.M.; Schuster, R.M.; Japtok, L.; Kleuser, B.; Edwards, M.J.; Gulbins, E.; Lentsch, A.B. Hepatocyte exosomes mediate liver repair and regeneration via sphingosine-1-phosphate. J. Hepatol. 2015, 64, 60–68. [Google Scholar] [CrossRef]
- Dai, J.; Lin, W.; Zheng, M.; Liu, Q.; He, B.; Luo, C.; Lu, X.; Pei, Z.; Su, H.; Yao, X. Alterations in AQP4 expression and polarization in the course of motor neuron degeneration in SOD1G93A mice. Mol. Med. Rep. 2017, 16, 1739–1746. [Google Scholar] [CrossRef]
- Nagai, M.; Re, D.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef]
- Wang, L.; Pei, S.; Han, L.; Guo, B.; Li, Y.; Duan, R.; Yao, Y.; Xue, B.; Chen, X.; Jia, Y. Mesenchymal Stem Cell-Derived Exosomes Reduce A1 Astrocytes via Downregulation of Phosphorylated NFκB P65 Subunit in Spinal Cord Injury. Cell. Physiol. Biochem. 2018, 50, 1535–1559. [Google Scholar] [CrossRef]
- Xian, P.; Hei, Y.; Wang, R.; Wang, T.; Yang, J.; Li, J.; Di, Z.; Liu, Z.; Baskys, A.; Liu, W.; et al. Mesenchymal stem cell-derived exosomes as a nanotherapeutic agent for amelioration of inflammation-induced astrocyte alterations in mice. Theranostics 2019, 9, 5956–5975. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Perets, N.; Betzer, O.; Shapira, R.; Brenstein, S.; Angel, A.; Sadan, T.; Ashery, U.; Popovtzer, R.; Offen, D. Golden Exosomes Selectively Target Brain Pathologies in Neurodegenerative and Neurodevelopmental Disorders. Nano Lett. 2019, 19, 3422–3431. [Google Scholar] [CrossRef] [PubMed]
- Peroni, D.; Scambi, I.; Pasini, A.; Lisi, V.; Bifari, F.; Krampera, M.; Rigotti, G.; Sbarbati, A.; Galie’, M. Stem molecular signature of adipose-derived stromal cells. Exp. Cell Res. 2008, 314, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Busato, A.; Bonafede, R.; Bontempi, P.; Scambi, I.; Schiaffino, L.; Benati, N.; Malatesta, M.; Sbarbati, A.; Marzola, P.; Mariotti, R. Labeling and Magnetic Resonance Imaging of Exosomes Isolated from Adipose Stem Cells. Curr. Protoc. Cell Boil. 2017, 75, 3441–34415. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonafede, R.; Turano, E.; Scambi, I.; Busato, A.; Bontempi, P.; Virla, F.; Schiaffino, L.; Marzola, P.; Bonetti, B.; Mariotti, R. ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS. Int. J. Mol. Sci. 2020, 21, 3651. https://doi.org/10.3390/ijms21103651
Bonafede R, Turano E, Scambi I, Busato A, Bontempi P, Virla F, Schiaffino L, Marzola P, Bonetti B, Mariotti R. ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS. International Journal of Molecular Sciences. 2020; 21(10):3651. https://doi.org/10.3390/ijms21103651
Chicago/Turabian StyleBonafede, Roberta, Ermanna Turano, Ilaria Scambi, Alice Busato, Pietro Bontempi, Federica Virla, Lorenzo Schiaffino, Pasquina Marzola, Bruno Bonetti, and Raffaella Mariotti. 2020. "ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS" International Journal of Molecular Sciences 21, no. 10: 3651. https://doi.org/10.3390/ijms21103651
APA StyleBonafede, R., Turano, E., Scambi, I., Busato, A., Bontempi, P., Virla, F., Schiaffino, L., Marzola, P., Bonetti, B., & Mariotti, R. (2020). ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS. International Journal of Molecular Sciences, 21(10), 3651. https://doi.org/10.3390/ijms21103651